Uremic Toxins Affecting Cardiovascular Calcification: A Systematic Review

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources and Searches
2.2. Study Selection Criteria
3. Results
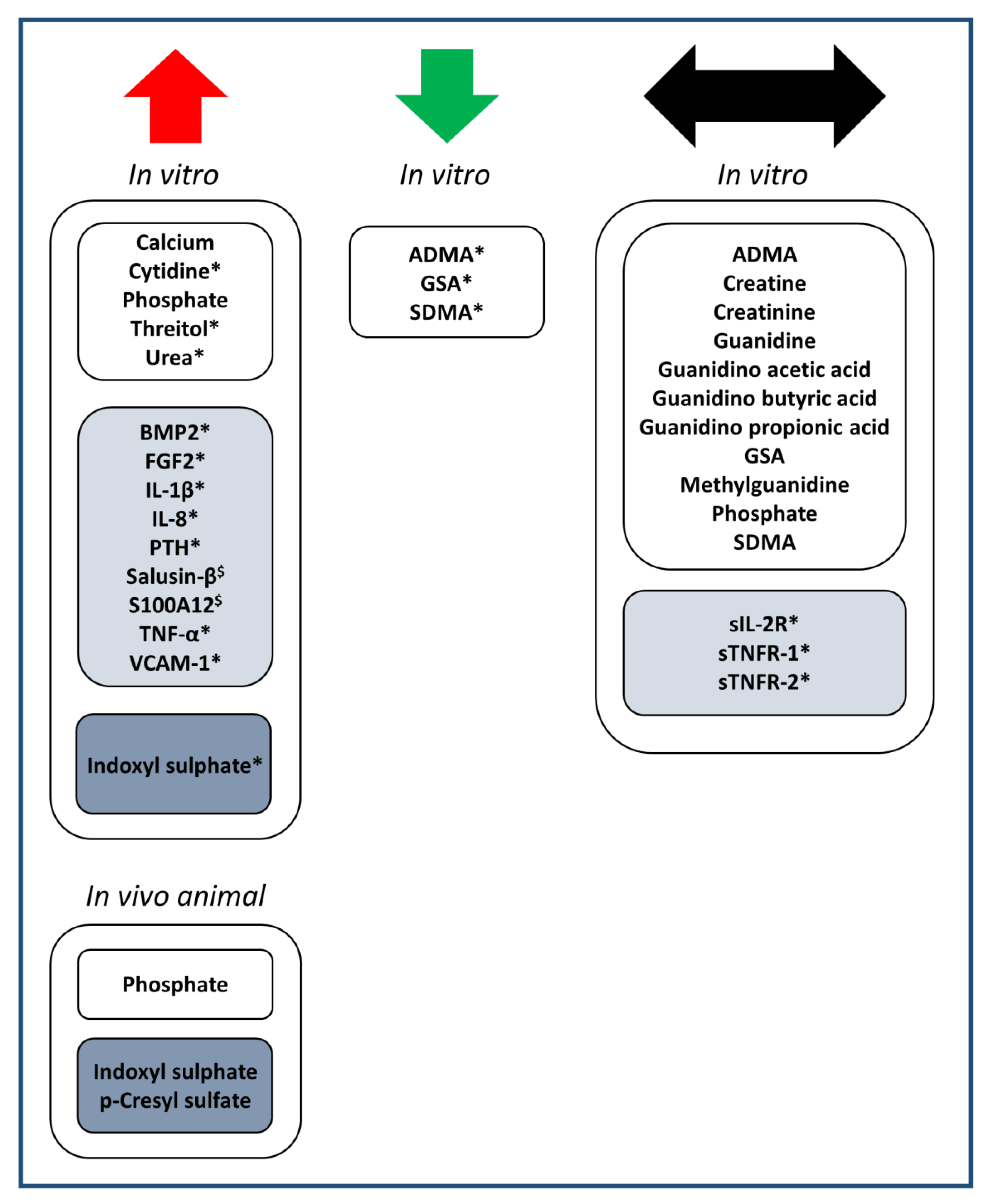
3.1. Identification of Uremic Toxins Studied in the Context of Uremia and Cardiovascular Calcification
3.2. Effect of the Identified Substances on Cardiovascular Calcification
3.2.1. Low Molecular Weight Substances, Increased in Blood in CKD (Table 1)
3.2.2. Middle Molecular Weight Substances, Increased in Blood in CKD (Table 2)
3.2.3. Protein-Bound Substances, Increased in Blood in CKD (Table 3)
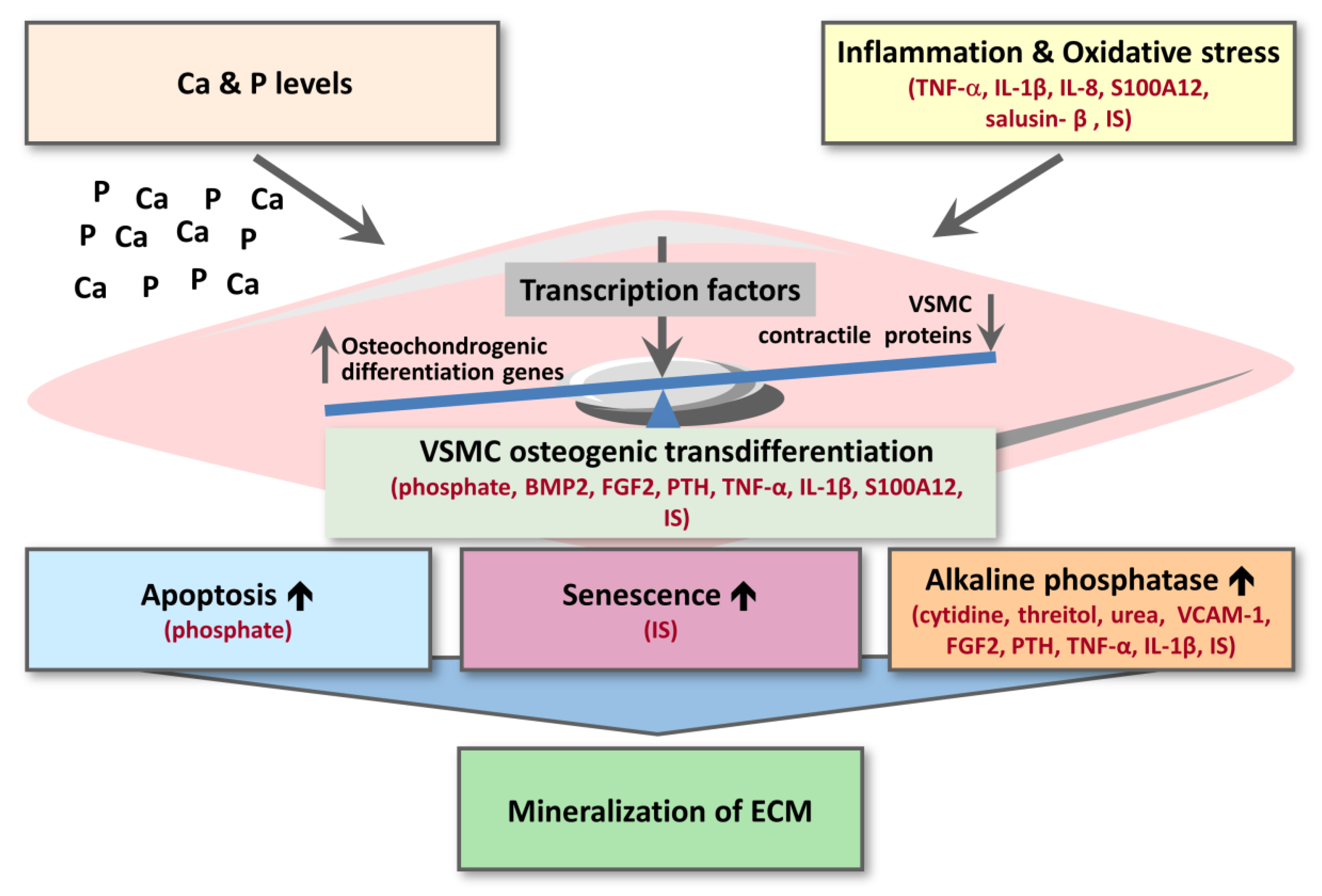
3.3. Mechanisms and Signalling Pathways Associating Uremic Toxins with Calcification
3.3.1. Inflammation and Oxidative Stress
3.3.2. VSMC Osteogenic Transdifferentiation
3.3.3. VSMC Apoptosis and Senescence
3.3.4. Alkaline Phosphatase
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M.; Alberta Kidney Disease, N. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.W.; Kauppila, L.I.; O’Donnell, C.J.; Kiel, D.P.; Hannan, M.; Polak, J.M.; Cupples, L.A. Abdominal aortic calcific deposits are an important predictor of vascular morbidity and mortality. Circulation 2001, 103, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Wayhs, R.; Zelinger, A.; Raggi, P. High coronary artery calcium scores pose an extremely elevated risk for hard events. J. Am. Coll. Cardiol. 2002, 39, 225–230. [Google Scholar] [CrossRef]
- Otto, C.M.; Lind, B.K.; Kitzman, D.W.; Gersh, B.J.; Siscovick, D.S. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N. Engl. J. Med. 1999, 341, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, I.; Holt, S.G.; Hewitson, T.D.; Smith, E.R.; Toussaint, N.D. Current and potential therapeutic strategies for the management of vascular calcification in patients with chronic kidney disease including those on dialysis. Semin Dial. 2018, 31, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Blacher, J.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; London, G.M. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 2001, 38, 938–942. [Google Scholar] [CrossRef]
- Kraus, M.A.; Kalra, P.A.; Hunter, J.; Menoyo, J.; Stankus, N. The prevalence of vascular calcification in patients with end-stage renal disease on hemodialysis: A cross-sectional observational study. Adv. Chronic. Dis. 2015, 6, 84–96. [Google Scholar] [CrossRef]
- Goodman, W.G.; Goldin, J.; Kuizon, B.D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.; et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N. Engl. J. Med. 2000, 342, 1478–1483. [Google Scholar] [CrossRef]
- Masuda, C.; Dohi, K.; Sakurai, Y.; Bessho, Y.; Fukuda, H.; Fujii, S.; Sugimoto, T.; Tanabe, M.; Onishi, K.; Shiraki, K.; et al. Impact of chronic kidney disease on the presence and severity of aortic stenosis in patients at high risk for coronary artery disease. Cardiovasc. Ultrasound. 2011, 9, 31. [Google Scholar] [CrossRef]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular calcification: The killer of patients with chronic kidney disease. J. Am. Soc. Nephrol. 2009, 20, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Raggi, P. Cardiovascular disease: Coronary artery calcification predicts risk of CVD in patients with CKD. Nat. Rev. Nephrol. 2017, 13, 324–326. [Google Scholar] [CrossRef]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutelingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transplant. 2016, 31, 31–39. [Google Scholar] [CrossRef]
- Drueke, T.B. The new Kidney Disease: Improving Global Outcomes (KDIGO) guideline for the mineral and bone disorder associated with chronic kidney disease (MBD-CKD). Nephrol. Ther. 2010, 6, 149–150. [Google Scholar] [CrossRef]
- Shioi, A.; Nishizawa, Y. Roles of hyperphosphatemia in vascular calcification. Clin. Calcium. 2009, 19, 180–185. [Google Scholar]
- Shao, J.S.; Cheng, S.L.; Sadhu, J.; Towler, D.A. Inflammation and the osteogenic regulation of vascular calcification: A review and perspective. Hypertension 2010, 55, 579–592. [Google Scholar] [CrossRef]
- Furmanik, M.; Chatrou, M.; van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; van Eys, G.; Bochaton-Piallat, M.L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef]
- Hosaka, N.; Mizobuchi, M.; Ogata, H.; Kumata, C.; Kondo, F.; Koiwa, F.; Kinugasa, E.; Akizawa, T. Elastin degradation accelerates phosphate-induced mineralization of vascular smooth muscle cells. Calcif. Tissue Int. 2009, 85, 523–529. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work, G. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef]
- Hung, S.C.; Kuo, K.L.; Wu, C.C.; Tarng, D.C. Indoxyl Sulfate: A Novel Cardiovascular Risk Factor in Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Bammens, B.; Evenepoel, P.; Keuleers, H.; Verbeke, K.; Vanrenterghem, Y. Free serum concentrations of the protein-bound retention solute p-cresol predict mortality in hemodialysis patients. Kidney Int. 2006, 69, 1081–1087. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transpl. 2010, 25, 1183–1191. [Google Scholar] [CrossRef]
- Lai, Y.H.; Wang, C.H.; Kuo, C.H.; Lin, Y.L.; Tsai, J.P.; Hsu, B.G. Serum P-Cresyl Sulfate Is a Predictor of Central Arterial Stiffness in Patients on Maintenance Hemodialysis. Toxins 2019, 12, 10. [Google Scholar] [CrossRef]
- Ketteler, M.; Rothe, H.; Kruger, T.; Biggar, P.H.; Schlieper, G. Mechanisms and treatment of extraosseous calcification in chronic kidney disease. Nat. Rev. Nephrol. 2011, 7, 509–516. [Google Scholar] [CrossRef]
- Available online: http://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42016051604 (accessed on 26 May 2020).
- Schepers, E.; Glorieux, G.; Dou, L.; Cerini, C.; Gayrard, N.; Louvet, L.; Maugard, C.; Preus, P.; Rodriguez-Ortiz, M.; Argiles, A.; et al. Guanidino compounds as cause of cardiovascular damage in chronic kidney disease: An in vitro evaluation. Blood Purif. 2010, 30, 277–287. [Google Scholar] [CrossRef]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial cells exposed to phosphate and indoxyl sulphate promote vascular calcification through interleukin-8 secretion. Nephrol. Dial. Transpl. 2019, 34, 1125–1134. [Google Scholar] [CrossRef]
- Tertti, R.; Harmoinen, A.; Leskinen, Y.; Metsarinne, K.P.; Saha, H. Comparison of calcium phosphate product values using measurement of plasma total calcium and serum ionized calcium. Hemodial. Int. 2007, 11, 411–416. [Google Scholar] [CrossRef]
- Cazana-Perez, V.; Cidad, P.; Donate-Correa, J.; Martin-Nunez, E.; Lopez-Lopez, J.R.; Perez-Garcia, M.T.; Giraldez, T.; Navarro-Gonzalez, J.F.; Alvarez de la Rosa, D. Phenotypic Modulation of Cultured Primary Human Aortic Vascular Smooth Muscle Cells by Uremic Serum. Front. Physiol. 2018, 9, 89. [Google Scholar] [CrossRef]
- Guerrero, F.; Montes de Oca, A.; Aguilera-Tejero, E.; Zafra, R.; Rodriguez, M.; Lopez, I. The effect of vitamin D derivatives on vascular calcification associated with inflammation. Nephrol. Dial. Transpl. 2012, 27, 2206–2212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shigematsu, T.; Kono, T.; Satoh, K.; Yokoyama, K.; Yoshida, T.; Hosoya, T.; Shirai, K. Phosphate overload accelerates vascular calcium deposition in end-stage renal disease patients. Nephrol. Dial. Transpl. 2003, 18 (Suppl. 3), iii86–iii89. [Google Scholar] [CrossRef]
- Shibata, M.; Shigematsu, T.; Hatamura, I.; Saji, F.; Mune, S.; Kunimoto, K.; Hanba, Y.; Shiizaki, K.; Sakaguchi, T.; Negi, S. Reduced expression of perlecan in the aorta of secondary hyperparathyroidism model rats with medial calcification. Ren. Fail. 2010, 32, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Belmokhtar, K.; Ortillon, J.; Jaisson, S.; Massy, Z.A.; Boulagnon Rombi, C.; Doue, M.; Maurice, P.; Fritz, G.; Gillery, P.; Schmidt, A.M.; et al. Receptor for advanced glycation end products: A key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter PIT-1 expression. Nephrol. Dial. Transpl. 2019, 34, 2018–2030. [Google Scholar] [CrossRef]
- Sage, A.P.; Lu, J.; Tintut, Y.; Demer, L.L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011, 79, 414–422. [Google Scholar] [CrossRef]
- Azpiazu, D.; Gonzalez-Parra, E.; Ortiz, A.; Egido, J.; Villa-Bellosta, R. Impact of post-dialysis calcium level on ex vivo rat aortic wall calcification. PLoS ONE 2017, 12, e0183730. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M. Calcium as a cardiovascular toxin in CKD-MBD. Bone 2017, 100, 94–99. [Google Scholar] [CrossRef]
- Yang, H.; Curinga, G.; Giachelli, C.M. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 2004, 66, 2293–2299. [Google Scholar] [CrossRef]
- Hegner, B.; Schaub, T.; Janke, D.; Zickler, D.; Lange, C.; Girndt, M.; Jankowski, J.; Schindler, R.; Dragun, D. Targeting proinflammatory cytokines ameliorates calcifying phenotype conversion of vascular progenitors under uremic conditions in vitro. Sci. Rep. 2018, 8, 12087. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; Duan, D.; O’Neill, K.D.; Wolisi, G.O.; Koczman, J.J.; Laclair, R.; Moe, S.M. The mechanisms of uremic serum-induced expression of bone matrix proteins in bovine vascular smooth muscle cells. Kidney Int. 2006, 70, 1046–1053. [Google Scholar] [CrossRef]
- Rong, S.; Zhao, X.; Jin, X.; Zhang, Z.; Chen, L.; Zhu, Y.; Yuan, W. Vascular calcification in chronic kidney disease is induced by bone morphogenetic protein-2 via a mechanism involving the Wnt/beta-catenin pathway. Cell Physiol. Biochem. 2014, 34, 2049–2060. [Google Scholar] [CrossRef]
- Vianna, H.R.; Soares, C.M.; Silveira, K.D.; Elmiro, G.S.; Mendes, P.M.; de Sousa Tavares, M.; Teixeira, M.M.; Miranda, D.M.; Simoes, E.S.A.C. Cytokines in chronic kidney disease: Potential link of MCP-1 and dyslipidemia in glomerular diseases. Pediatr. Nephrol. 2013, 28, 463–469. [Google Scholar] [CrossRef]
- Isoyama, N.; Leurs, P.; Qureshi, A.R.; Bruchfeld, A.; Anderstam, B.; Heimburger, O.; Barany, P.; Stenvinkel, P.; Lindholm, B. Plasma S100A12 and soluble receptor of advanced glycation end product levels and mortality in chronic kidney disease Stage 5 patients. Nephrol. Dial. Transpl. 2015, 30, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, F.; Xu, Y.; Sun, S.; Wang, H.; Du, Q.; Gu, C.; Black, S.M.; Han, Y.; Tang, H. Salusin-beta Promotes Vascular Calcification via Nicotinamide Adenine Dinucleotide Phosphate/Reactive Oxygen Species-Mediated Klotho Downregulation. Antioxid. Redox Signal. 2019, 31, 1352–1370. [Google Scholar] [CrossRef]
- Sipahi, S.; Genc, A.B.; Acikgoz, S.B.; Yildirim, M.; Aksoy, Y.E.; Vatan, M.B.; Dheir, H.; Altindis, M. Relationship of salusin-alpha and salusin-beta levels with atherosclerosis in patients undergoing haemodialysis. Singap. Med. J. 2019, 60, 210–215. [Google Scholar] [CrossRef]
- Zickler, D.; Willy, K.; Girndt, M.; Fiedler, R.; Martus, P.; Storr, M.; Schindler, R. High cut-off dialysis in chronic haemodialysis patients reduces serum procalcific activity. Nephrol. Dial. Transpl. 2016, 31, 1706–1712. [Google Scholar] [CrossRef]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transpl. 2018, 33, 574–585. [Google Scholar] [CrossRef]
- Vaccaro, F.; Mule, G.; Cottone, S.; Soresi, M.; Giannitrapani, L.; Vadala, A.; Sparacino, V.; Calabrese, S.; Picone, F.P.; Montalto, G.; et al. Circulating levels of adhesion molecules in chronic kidney disease correlate with the stage of renal disease and with C-reactive protein. Arch. Med. Res. 2007, 38, 534–538. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease. Int. J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, J.; Shen, Z.; Gu, Y.; Xu, L.; Hu, J.; Zhang, X.; Ding, X. Indoxyl sulfate accelerates vascular smooth muscle cell calcification via microRNA-29b dependent regulation of Wnt/beta-catenin signaling. Toxicol. Lett. 2018, 284, 29–36. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Y.; Zhang, H.; Ning, Y.; Song, N.; Hu, J.; Cai, J.; Shi, Y.; Ding, X.; Zhang, X. Amelioration of Uremic Toxin Indoxyl Sulfate-Induced Osteoblastic Calcification by SET Domain Containing Lysine Methyltransferase 7/9 Protein. Nephron 2019, 141, 287–294. [Google Scholar] [CrossRef]
- He, X.; Jiang, H.; Gao, F.; Liang, S.; Wei, M.; Chen, L. Indoxyl sulfate-induced calcification of vascular smooth muscle cells via the PI3K/Akt/NF-kappaB signaling pathway. Microsc Res. Tech. 2019, 82, 2000–2006. [Google Scholar] [CrossRef]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol. Cell Physiol. 2012, 303, C126–C134. [Google Scholar] [CrossRef]
- Ochi, A.; Mori, K.; Nakatani, S.; Emoto, M.; Morioka, T.; Motoyama, K.; Fukumoto, S.; Imanishi, Y.; Shoji, T.; Ishimura, E.; et al. Indoxyl sulfate suppresses hepatic fetuin-A expression via the aryl hydrocarbon receptor in HepG2 cells. Nephrol. Dial. Transpl. 2015, 30, 1683–1692. [Google Scholar] [CrossRef]
- Adijiang, A.; Goto, S.; Uramoto, S.; Nishijima, F.; Niwa, T. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol. Dial. Transpl. 2008, 23, 1892–1901. [Google Scholar] [CrossRef]
- Adijiang, A.; Higuchi, Y.; Nishijima, F.; Shimizu, H.; Niwa, T. Indoxyl sulfate, a uremic toxin, promotes cell senescence in aorta of hypertensive rats. Biochem. Biophys Res. Commun. 2010, 399, 637–641. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Mare, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- Shroff, R.; Long, D.A.; Shanahan, C. Mechanistic insights into vascular calcification in CKD. J. Am. Soc Nephrol. 2013, 24, 179–189. [Google Scholar] [CrossRef]
- Tentori, F.; Blayney, M.J.; Albert, J.M.; Gillespie, B.W.; Kerr, P.G.; Bommer, J.; Young, E.W.; Akizawa, T.; Akiba, T.; Pisoni, R.L.; et al. Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: The Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 2008, 52, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Hulbert-Shearon, T.E.; Levin, N.W.; Port, F.K. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef]
- Janmaat, C.J.; van Diepen, M.; Gasparini, A.; Evans, M.; Qureshi, A.R.; Arnlov, J.; Barany, P.; Elinder, C.G.; Rotmans, J.I.; Vervloet, M.; et al. Lower serum calcium is independently associated with CKD progression. Sci. Rep. 2018, 8, 5148. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, W.C. Targeting serum calcium in chronic kidney disease and end-stage renal disease: Is normal too high? Kidney Int. 2016, 89, 40–45. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Joshi, F.R.; Rajani, N.K.; Abt, M.; Woodward, M.; Bucerius, J.; Mani, V.; Tawakol, A.; Kallend, D.; Fayad, Z.A.; Rudd, J.H. Does Vascular Calcification Accelerate Inflammation?: A Substudy of the dal-PLAQUE Trial. J. Am. Coll Cardiol. 2016, 67, 69–78. [Google Scholar] [CrossRef]
- Glorieux, G.L.; Dhondt, A.W.; Jacobs, P.; Van Langeraert, J.; Lameire, N.H.; De Deyn, P.P.; Vanholder, R.C. In vitro study of the potential role of guanidines in leukocyte functions related to atherogenesis and infection. Kidney Int. 2004, 65, 2184–2192. [Google Scholar] [CrossRef]
- Schibler, D.; Russell, R.G.; Fleisch, H. Inhibition by pyrophosphate and polyphosphate of aortic calcification induced by vitamin D3 in rats. Clin. Sci. 1968, 35, 363–372. [Google Scholar]
- Lomashvili, K.A.; Garg, P.; Narisawa, S.; Millan, J.L.; O’Neill, W.C. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: Potential mechanism for uremic vascular calcification. Kidney Int. 2008, 73, 1024–1030. [Google Scholar] [CrossRef]
- Eloot, S.; Ledebo, I.; Ward, R.A. Extracorporeal removal of uremic toxins: Can we still do better? Semin Nephrol. 2014, 34, 209–227. [Google Scholar] [CrossRef]
- Lesaffer, G.; De Smet, R.; Lameire, N.; Dhondt, A.; Duym, P.; Vanholder, R. Intradialytic removal of protein-bound uraemic toxins: Role of solute characteristics and of dialyser membrane. Nephrol. Dial. Transpl. 2000, 15, 50–57. [Google Scholar] [CrossRef]
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-cresol sulfate by hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436. [Google Scholar] [CrossRef]
- Sternkopf, M.; Thoröe-Boveleth, S.; Beck, T.; Oleschko, K.; Erlenkötter, A.; Tschulena, U.; Steppan, S.; Speer, T.; Goettsch, C.; Jankowski, V.; et al. A Bifunctional Adsorber Particle for the Removal of Hydrophobic Uremic Toxins from Whole Blood of Renal Failure Patients. Toxins 2019, 11, 389. [Google Scholar] [CrossRef]
- Kato, S.; Otake, K.-I.; Chen, H.; Akpinar, I.; Buru, C.T.; Islamoglu, T.; Snurr, R.Q.; Farha, O.K. Zirconium-Based Metal–Organic Frameworks for the Removal of Protein-Bound Uremic Toxin from Human Serum Albumin. J. Am. Chem. Soc. 2019, 141, 2568–2576. [Google Scholar] [CrossRef]
- Pavlenko, D.; van Geffen, E.; van Steenbergen, M.J.; Glorieux, G.; Vanholder, R.; Gerritsen, K.G.; Stamatialis, D. New low-flux mixed matrix membranes that offer superior removal of protein-bound toxins from human plasma. Sci. Rep. 2016, 6, 34429. [Google Scholar] [CrossRef]
- Sandeman, S.R.; Zheng, Y.; Ingavle, G.C.; Howell, C.A.; Mikhalovsky, S.V.; Basnayake, K.; Boyd, O.; Davenport, A.; Beaton, N.; Davies, N. A haemocompatible and scalable nanoporous adsorbent monolith synthesised using a novel lignin binder route to augment the adsorption of poorly removed uraemic toxins in haemodialysis. Biomed. Mater. 2017, 12, 035001. [Google Scholar] [CrossRef]
- Brettschneider, F.; Tölle, M.; von der Giet, M.; Passlick-Deetjen, J.; Steppan, S.; Peter, M.; Jankowski, V.; Krause, A.; Kühne, S.; Zidek, W.; et al. Removal of protein-bound, hydrophobic uremic toxins by a combined fractionated plasma separation and adsorption technique. Artif. Organs 2013, 37, 409–416. [Google Scholar] [CrossRef]
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of Protein-Bound Uremic Toxins during Hemodialysis Using a Binding Competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402. [Google Scholar] [CrossRef]
- Thongprayoon, C.; Kaewput, W.; Hatch, S.T.; Bathini, T.; Sharma, K.; Wijarnpreecha, K.; Ungprasert, P.; D’Costa, M.; Mao, M.A.; Cheungpasitporn, W. Effects of Probiotics on Inflammation and Uremic Toxins Among Patients on Dialysis: A Systematic Review and Meta-Analysis. Dig. Dis. Sci. 2019, 64, 469–479. [Google Scholar] [CrossRef]
- Schulman, G.; Vanholder, R.; Niwa, T. AST-120 for the management of progression of chronic kidney disease. Int. J. Nephrol. Renov. Dis. 2014, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Maré, A.; D’Haese, P.C.; Verhulst, A. The Role of Sclerostin in Bone and Ectopic Calcification. Int. J. Mol. Sci. 2020, 21, 3199. [Google Scholar] [CrossRef]
- de Oliveira, R.A.; Barreto, F.C.; Mendes, M.; dos Reis, L.M.; Castro, J.H.; Britto, Z.M.; Marques, I.D.; Carvalho, A.B.; Moyses, R.M.; Jorgetti, V. Peritoneal dialysis per se is a risk factor for sclerostin-associated adynamic bone disease. Kidney Int. 2015, 87, 1039–1045. [Google Scholar] [CrossRef]
- Desjardins, L.; Liabeuf, S.; Oliveira, R.B.; Louvet, L.; Kamel, S.; Lemke, H.D.; Vanholder, R.; Choukroun, G.; Massy, Z.A.; European Uremic Toxin Work, G. Uremic toxicity and sclerostin in chronic kidney disease patients. Nephrol. Ther. 2014, 10, 463–470. [Google Scholar] [CrossRef]
- Brandenburg, V.M.; Verhulst, A.; Babler, A.; D’Haese, P.C.; Evenepoel, P.; Kaesler, N. Sclerostin in chronic kidney disease-mineral bone disorder think first before you block it! Nephrol. Dial. Transpl. 2019, 34, 408–414. [Google Scholar] [CrossRef]
- Holmar, J.F.I.; Uhlin, F.; Fernstrom, A.; Luman, M. Estimation of dialysis patients’ survival through combined approach of small molecule uremic markers. Proc. Est. Acad. Sci. 2014, 63, 227–233. [Google Scholar] [CrossRef]
- Holmar, J.; Noels, H.; Böhm, M.; Bhargava, S.; Jankowski, J.; Orth-Alampour, S. Development, establishment and validation of in vitro and ex vivo assays of vascular calcification. Biochem. Biophys Res. Commun. 2020, 530, 462–470. [Google Scholar] [CrossRef]
- Gajjala, P.R.; Fliser, D.; Speer, T.; Jankowski, V.; Jankowski, J. Emerging role of post-translational modifications in chronic kidney disease and cardiovascular disease. Nephrol. Dial. Transpl. 2015, 30, 1814–1824. [Google Scholar] [CrossRef]
- Kalim, S.; Karumanchi, S.A.; Thadhani, R.I.; Berg, H.A. Protein Carbamylation in Kidney Disease: Pathogenesis and Clinical Implications. Am. J. Kidney Dis. 2014, 64, 793–803. [Google Scholar] [CrossRef]
- Speer, T.; Owala, F.O.; Holy, E.W.; Zewinger, S.; Frenzel, F.L.; Stahli, B.E.; Razavi, M.; Triem, S.; Cvija, H.; Rohrer, L.; et al. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur. Heart J. 2014, 35, 3021–3032. [Google Scholar] [CrossRef]
- Mori, D.; Matsui, I.; Shimomura, A.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Oka, T.; Kubota, K.; Yonemoto, S.; et al. Protein carbamylation exacerbates vascular calcification. Kidney Int. 2018, 94, 72–90. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Effect | Substance | Species | Tissue or Cell Type | Type of Study | Analysis of Calcification or Calcification-Associated Process * by | Reference | Plasma Level in CKD | Classified as Uremic Toxin ** | |
|---|---|---|---|---|---|---|---|---|---|
| In Vitro | Animal | ||||||||
| INHIBITION | Asymmetric dimethylarginine (ADMA) | Human | VSMC | V $ | Alizarin red staining | Schepers [29] | ↑ [§] | V | |
| Symmetric dimethylarginine (SDMA) | Human | VSMC | V $ | Alizarin red staining | Schepers [29] | ↑ [§] | V | ||
| Guanidinosuccinic acid (GSA) | Human | VSMC | V $ | Alizarin red staining | Schepers [29] | ↑ [§] | V | ||
| INDUCTION | Phosphate (P) | Human | VSMC | V | Calcium content Alizarin red staining | Bouabdallah [30] | ↑ [31] | ||
| Human | VSMC | V | Alizarin red staining Upregulation of MSX2, SOX9 and OPN Increased apoptosis | Cazana-Perez [32] | |||||
| Human | VSMC | V | Calcium content Upregulation of CBFA1/RUNX2 and BMP2 | Guerrero [33] | |||||
| Human | VSMC | V | Alizarin red staining | Schepers [29] | |||||
| Human | VSMC | V $ | High binding affinity of calcium to ECM | Shigematsu [34] 1 | |||||
| Rat | VSMC | V | Von Kossa staining | Shibata [35] | |||||
| Rat | Aorta | V | Calcium content | Guerrero [33] | |||||
| Mouse | Aorta | V 2 | Von Kossa staining | Belmokhtar [36] | |||||
| Mouse | VSMC | V | Alizarin red staining Increased ROS generation Upregulation of PIT-1, BMP2 and CBFA1/RUNX2 | Belmokhtar [36] | |||||
| Mouse | VSMC | V | Upregulation of BMP2 and OPN | Sage [37] 1 | |||||
| Rat | Aorta | V 3 | Von Kossa staining | Shibata [35] | |||||
| Calcium | Rat | Aorta | V | Calcium content | Azpiazu [38] 1 | ↑ [39] | |||
| Human | VSMC | V | Calcium content | Yang [40] 1 | |||||
| Cytidine | Human | MSC | V $ | ALP activity Calcium content | Hegner [41] | ↑ [§] | V | ||
| Urea | Human | MSC | V $ | ALP activity Calcium content | Hegner [41] | ↑ [§] | V | ||
| Threitol | Human | MSC | V $ | ALP activity Calcium content | Hegner [41] | ↑ [§] | V | ||
| NO EFFECT | Guanidino compounds: | Human | VSMC | V | Alizarin red staining | Schepers [29] | |||
| ADMA | ↑ [§] | V | |||||||
| SDMA | ↑ [§] | V | |||||||
| GSA | ↑ [§] | V | |||||||
| Creatine | ↑ [§] | V | |||||||
| Creatinine | ↑ [§] | V | |||||||
| Guanidine | ↑ [§] | V | |||||||
| Guanidino acetic acid | ↑ [§] | V | |||||||
| Guanidino butyric acid | ↑ [§] | V | |||||||
| Guanidino propionic acid | ↑ [§] | V | |||||||
| Methylguanidine | ↑ [§] | V | |||||||
| Effect | Substance | Species | Tissue or Cell Type | Type of Study | Analysis of Calification or Calcification-Associated Process * by | Reference | Plasma level in CKD | Classified as Uremic Toxin ** | |
|---|---|---|---|---|---|---|---|---|---|
| In vitro | Animal | ||||||||
| INDUCTION | Bone morphogenetic protein (BMP2) | Bovine | VSMC | V $ | Calcium content Upregulation of CBFA1/RUNX2 | Chen [42] | ↑ [42] | ||
| Rat | VSMC | V $ | Upregulation of CBFA1/RUNX2, MSX2 and PIT-1 | Rong [43] 1 | |||||
| Fibroblast growth factor 2 (FGF2) | Human | MSC | V $ | ALP activity Alizarin red staining Calcium content Upregulation of CBFA1/RUNX2, OPN and osterix | Hegner [41] | ↑ [§] | |||
| Interleukin-1β (IL-1β) | Human | MSC | V $ | ALP activity Alizarin red staining Calcium content Upregulation of CBFA1/RUNX2, OPN and osterix | Hegner [41] | ↑ [§] | V | ||
| Interleukin-8 (IL-8) | Human | VSMC | V $ | Alizarin red staining Calcium content Downregulation of OPN | Bouabdallah [30] | ↑ [44] | |||
| Parathyroid hormone (PTH) | Human | MSC | V $ | ALP activity Alizarin red staining Calcium content Upregulation of CBFA1/RUNX2, OPN and osterix | Hegner [41] | ↑ [§] | V | ||
| S100A12 (RAGE ligand) | Mouse | VSMC | V | Alizarin red staining Increased ROS generation Upregulation of PIT-1, BMP2 and CBFA1/RUNX2 | Belmokhtar [36] | ↑ [45] | |||
| Salusin-β | Human | VSMC | V & | Calcium content ALP activity Alizarin red staining Increased ROS generation Upregulation of BMP2 and CBFA1/RUNX2 | Sun [46] | ↑ [47] | |||
| Rat | Aorta | V & | Calcium content ALP activity Von Kossa staining | Sun [46] | |||||
| Tumor necrosis factor alpha (TNF-α) | Human | MSC | V | ALP activity Alizarin red staining Calcium content Upregulation of CBFA1/RUNX2, OPN and osterix | Hegner [41] | ↑ [§] | V | ||
| Human | VSMC | V $ | Calcium content | Guerrero [33] | |||||
| Human | VSMC | V $ | ALP activity | Zickler [48] | |||||
| Human | VSMC | V $ | Calcium content | Zickler [49] | |||||
| Rat | Aorta | V $ | Calcium content | Guerrero [33] | |||||
| Vascular cell adhesion molecule-1 (VCAM-1) | Human | VSMC | V $ | ALP activity Alizarin red staining | Zickler [48] | ↑ [50] | |||
| NO EFFECT | Soluble tumor necrosis factor receptor-1 (sTNFR-1) | Human | VSMC | V $ | ALP activity Alizarin red staining | Zickler [48] | ↑ [48] | ||
| Soluble tumor necrosis factor receptor-2 (sTNFR-2) | Human | VSMC | V $ | ALP activity Alizarin red staining | Zickler [48] | ↑ [48] | |||
| Soluble interleukin-2 receptor (sIL-2R) | Human | VSMC | V $ | ALP activity Alizarin red staining | Zickler [48] | ↑ [48] | |||
| Effect | Substance | Species | Tissue or Cell Type | Type of Study | Analysis of Calcification or Calcification-Associated Process * by | Reference | Plasma Level in CKD | Classified as Uremic Toxin ** | |
|---|---|---|---|---|---|---|---|---|---|
| In vitro | Animal | ||||||||
| INDUCTION | Indoxyl sulphate (IS) | Human | VSMC | V $ | Calcium content Alizarin red staining Upregulation of CBFA1/RUNX2, ALP, BMP2 and OPN Downregulation of α-SMA and SM22-α | Bouabdallah [30] | ↑ [§] | V | |
| Human | VSMC | V | Alizarin red staining Upregulation of CBFA1/RUNX2, ALP and OPN Downregulation of α-SMA and SMTN | Chen [51] | |||||
| Human | VSMC | V | Alizarin red staining Upregulation of CBFA1/RUNX2 and OPN | Zhang [52] | |||||
| Human | VSMC | V | Alizarin red staining Upregulation of CBFA1/RUNX2 and OPN | Chen [53] | |||||
| Human | VSMC | V | Alizarin red staining ALP activity Downregulation of α-SMA Upregulation of CBFA1/RUNX2 | He [54] | |||||
| Human | VSMC | V | Upregulation of p53, p21, and prelamin A | Muteliefu [55] | |||||
| Human | HepG2 | V | Downregulation of fetuin-A | Ochi [56] | |||||
| Rat | Aorta | V $ | Calcium content Alizarin red staining | Bouabdallah [30] | |||||
| Rat | Aorta | V 1 | Von Kossa staining Upregulation of OPN, CBFA1/RUNX2, ALP and osteocalcin | Adijiang [57] | |||||
| Rat | Aorta | V 2 | Von Kossa staining | Adijiang [58] | |||||
| Rat | Aorta | V 3 | Alizarin red staining Downregulation of α-SMA and SMTN Upregulation of CBFA1/RUNX2, ALP and OPN | Chen [51] | |||||
| Rat | Aorta | V 4 | Calcium content Von Kossa staining Activation of inflammation and coagulation pathways | Opdebeeck [59] | |||||
| Rat | Aorta | V 2 | Upregulation of 8-OHdG and MDA in the calcification area | Muteliefu [55] | |||||
| p-Cresyl sulphate | Rat | Aorta | V 4 | Calcium content Von Kossa staining Activation of inflammation and coagulation pathways | Opdebeeck [59] | ↑ [§] | V | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holmar, J.; de la Puente-Secades, S.; Floege, J.; Noels, H.; Jankowski, J.; Orth-Alampour, S. Uremic Toxins Affecting Cardiovascular Calcification: A Systematic Review. Cells 2020, 9, 2428. https://doi.org/10.3390/cells9112428
Holmar J, de la Puente-Secades S, Floege J, Noels H, Jankowski J, Orth-Alampour S. Uremic Toxins Affecting Cardiovascular Calcification: A Systematic Review. Cells. 2020; 9(11):2428. https://doi.org/10.3390/cells9112428
Chicago/Turabian StyleHolmar, Jana, Sofia de la Puente-Secades, Jürgen Floege, Heidi Noels, Joachim Jankowski, and Setareh Orth-Alampour. 2020. "Uremic Toxins Affecting Cardiovascular Calcification: A Systematic Review" Cells 9, no. 11: 2428. https://doi.org/10.3390/cells9112428
APA StyleHolmar, J., de la Puente-Secades, S., Floege, J., Noels, H., Jankowski, J., & Orth-Alampour, S. (2020). Uremic Toxins Affecting Cardiovascular Calcification: A Systematic Review. Cells, 9(11), 2428. https://doi.org/10.3390/cells9112428