1. Introduction
Macroautophagy is an important catabolic process for maintaining cellular homeostasis. Misfolded, old, or damaged proteins and organelles are captured in autophagosomes—vesicles with a double membrane—and are subsequently degraded by fusion with lysosomes [
1,
2]. The degraded products like amino acids or fatty acids can further be metabolized to generate energy, or they can be recycled to protein or membrane biogenesis. Autophagy is particularly important and upregulated during metabolic stress, including glucose reduction and other bioenergetic deficiencies [
3]. Impaired autophagy plays a major role in aging and various human disorders, including many neurodegenerative disorders [
4,
5].
Autophagy is a highly regulated process involving the controlled expression of autophagy-related genes (atg). The formation of autophagosomes starts with the generation of a double lipid membrane, which subsequently matures from an early phagophore into a mature autophagosome containing lipidated ATG microtubule-associated proteins 1A/1B light chain 3 proteins (MAP1LC3 proteins, LC3 proteins). Three members of the LC3 family occur in humans: LC3A, LC3B, and LC3C, of which LC3B is widely used as a marker for autophagy and a tool to assess general autophagic flux [
6]. LC3A and LC3C are suggested to have similar functions to LC3B, but might also be involved in autophagy-unrelated phenomena such as signal transduction or viral replication [
7,
8,
9]. Their precise molecular mode of action remains unknown.
The members of the LC3 protein family are generated from different mRNA transcripts that are derived from distinct genomic locations, but they share high sequence similarity [
9]. LC3B is localized to the nucleus as well as to the cytoplasm and has been shown to cycle between these cellular compartments. This process is regulated by lysine acetylation and deacetylation dependent on the class III histone deacetylase (HDAC), sirtuin 1 (SIRT1) [
10]. Moreover, LC3B is present in a non-lipidated (LC3B-I) and a lipidated (LC3B-II) form. In the cytoplasm, deacetylated LC3B-I is lipidated by conjugation with phosphatidylethanolamine and thereby converted to LC3B-II, which in turn is recruited by autophagosomes. Whether LC3A and LC3C are regulated in a similar manner is still unknown. Besides the LC3s, three further members of the ATG8 protein family exist in humans: GABARAP, GABARAP-L1, and GABARAP-L2, which are thought to be mainly involved in late steps of autophagy, such as autophagosome elongation and autophagosome-to-lysosome fusion [
11,
12].
To gain further insight into the cellular roles of the different LC3s under near-physiological conditions, we analyzed their distribution (nuclear versus cytosolic) and their degree of co-localization with sequestosome1 (p62), a major ubiquitin and autophagy receptor, in response to an experimentally induced decapacitation of LC3B, the canonical and best-characterized LC3. LC3B decapacitation was achieved (i) with a selective SIRT1 inhibitor (Ex527) under normal and starvation conditions; trichostatin A (TSA), an inhibitor of non-sirtuin HDACs, was studied as an alternative pro-acetylation agent. Beyond these pharmacological treatments, we used (ii) siRNA-mediated knock-down experiments to reduce the total expression of SIRT1. Finally, (iii) the effects of siRNA-mediated knock-down of all three LC3 paralogues were investigated. Primary human diploid fibroblasts were chosen for these investigations since they offer the study of physiological regulatory events in a genetically undisturbed, human setting of unreduced complexity.
In all cases, we subsequently studied the cellular localization and expression of LC3A, LC3B and LC3C under Bafilomycin A1 (BafA1) treatment. BafA1 is an inhibitor of V-ATPase that prevents autophagosome-lysosome fusion and autophagolysosome acidification. The observed differential behavior of the ATG8 protein family implies a substantial, although incomplete mutual functional compensation of the three LC3 family members. Comparison of conserved regions, binding sites, and recognized LIR motifs supported the idea that a sub- or neo-functionalization of the LC3 protein family has occurred during evolution.
2. Materials and Methods
2.1. Cell Culture
Cell culture media and supplements, including high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM), Phosphate-Buffered Saline (PBS) and Fetal Calf Serum (FCS) were obtained from Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA). Laboratory chemicals including Ex527 (6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide) and trichostatin A were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA) at the highest purity available. Bafilomycin A1 (BafA1) was from Biozol (Eching, Germany).
Primary human fibroblasts (IMR90 cells) were obtained from the Coriell Institute for Medical Research (Camden, NJ, USA) and cultivated as described previously [
13]. In brief, the cells were maintained in phenol red-containing DMEM with high glucose (4.5 g/L) supplemented with 10% heat-inactivated FCS, 1 mM sodium pyruvate, and 1x antibiotic/antimycotic mixture (order number LS-B5909, LSBio, Eching, Germany). Starvation experiments were performed by replacing the standard medium with Earle’s balanced salt solution (Sigma-Aldrich, St. Louis, MO, USA; order number E2888) for 4 h preceding the analysis. For routine culture, cells were grown on 10 cm dishes, maintained at 37 °C in a humidified atmosphere containing 5% CO
2, and were passaged twice a week. Experiments were carried out with cells at population doublings (PDLs) between 20 and 33. For Western blotting experiments, cells were plated at a density of 0.4 × 10
6 cells per 10 cm dish and grown in full medium for 24 h. The cells were subsequently washed with pre-warmed PBS and new medium was added (10 mL), followed by treatment for 48 h with 10 nM TSA (in DMSO), 100 nM Ex527 (in DMSO), or 10 µL DMSO as vehicle control. Four hours prior to harvest, 2 µM BafA1 was added where appropriate. All treatment groups were visually inspected using light microscopy by two independent researchers to avoid the analysis of morphologically damaged cells.
2.2. Western Blotting
Treated cells were washed with ice-cold PBS and harvested, with brief sonication, in lysis buffer (50 mM Tris-HCl, pH 6.8; 2% sodium dodecyl sulfate (SDS); 10% sucrose; and EDTA-free protease inhibitor cocktail (order number 5056489001 from Roche, Basel, Switzerland)). Protein concentration was determined using a BCA kit (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol. An amount of 10 μg protein was loaded on a 12–15% SDS gel and separated by electrophoresis. Afterwards, the proteins were transferred onto a nitrocellulose membrane and blocked with 4% non-fat milk in PBST. Individual proteins were detected with the following primary antibodies: rabbit anti-LC3A (1:1000, from Cell Signaling, Danvers, MA, USA; order number 4599), rabbit anti-LC3B (1:1000, from Sigma-Aldrich, St. Louis, MO, USA; order number L7543), rabbit anti-LC3C (1:1000, from Abcam, Cambridge, UK; order number ab150367), mouse anti-GABARAP (1:1000, from MBL, Woburn, MA, USA; order number M135-3), rabbit anti-GABARAP-L1 (1:1000, from Cell Signaling, Danvers, MA, USA; order number 26632), rabbit anti-GABARAP-L2 (1:1000, from Cell Signaling, Danvers, MA, USA; order number 14256), mouse anti-SIRT1 (1:1000, from Cell Signaling, Danvers, MA, USA; order number 8469), guinea pig anti-p62 (1:1000, from Progen, Heidelberg, Germany; order number GP62-C), mouse anti-α-tubulin (1:1000, from Sigma-Aldrich, St. Louis, MO, USA; order number T9026). The immunoreactive signal of α-tubulin was used as control for equal protein loading. Primary antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (1:5000, from Jackson Immunoresearch, West Grove, PA, USA). Immunoreactive bands were developed using commercial kits (Enhanced Chemiluminescence Plus from Amersham Pharmacia Biotech, Piscataway, NJ, USA) and scanned with an Amersham Imager 600. The densitometric analyses of the developed bands were done with Aida Image Analysis Software (Raytest, Straubenhardt, Germany).
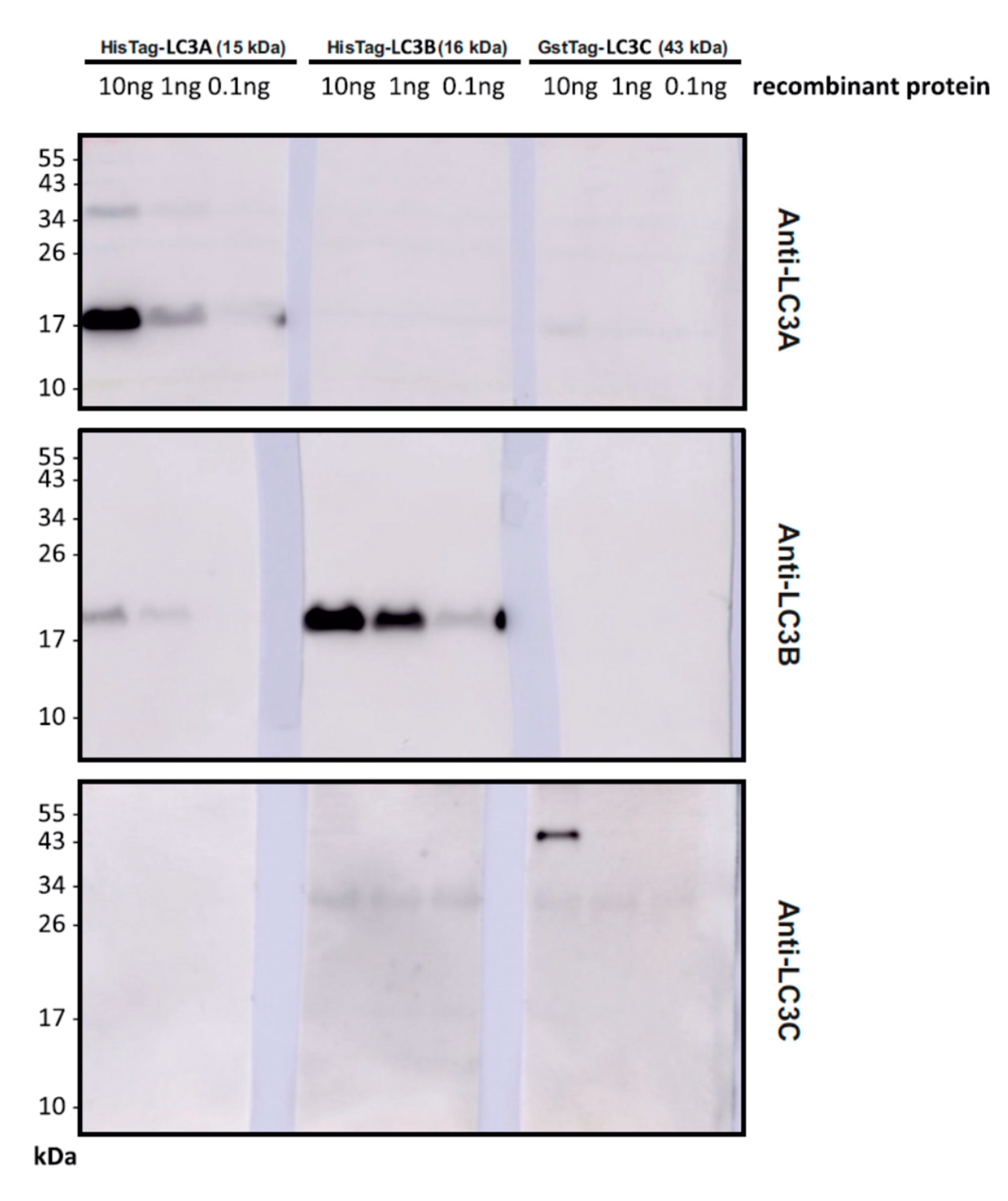
2.3. Antibody Validation
Recombinant His-tagged LC3A, untagged LC3B and GST-tagged LC3C were purchased from Novus Biologicals (Centennial, CO, USA). The recombinant human LC3A protein (order number NBP1-45308), fused to a His-tag at the C-terminus, and the recombinant human LC3B protein (order number NBP2-50960) was expressed in E. coli. The recombinant human LC3C protein (order number H00440738-P01) with a GST-tag at the N-terminus had been expressed in wheat germ. The LC3 proteins were diluted in Western blotting lysis buffer to contain a total amount of 10 ng, 1 ng or 0.1 ng protein together with 20 µg milk proteins. Samples were supplemented with loading buffer and loaded several times onto a gel without boiling. Western blotting was performed in the same manner as described in 2.2. After blotting, the membranes were cut vertically to allow parallel incubation with antibodies against LC3A, LC3B, and LC3C using rabbit anti-LC3A (1:1000, from Cell Signaling, Danvers, MA, USA; order number 4599), rabbit anti-LC3B (1:1000, Sigma-Aldrich, St. Louis, MO, USA; order number L7543), rabbit anti-LC3C (1:1000, from Abcam, Cambridge, UK; order number ab150367). The membrane strips carrying identical LC3s were then developed in parallel using enhanced chemiluminescence as before.
2.4. Immunoprecipitation
Cells cultivated in 15 cm dishes were treated as appropriate, washed with ice-cold PBS and lysed in IP lysis buffer (50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 2 mM EDTA; 0.5 mM EGTA; 1% Triton X-100; 10% glycerol; protease and phosphatase inhibitor cocktail; 1 µM TSA; 20 mM nicotinamide; 1 mM dithiothreitol) with brief sonication. Protein A/G magnetic beads (order number 88,803 from Thermo Fisher Scientific, Waltham, MA, USA) were washed three times with IP lysis buffer in low retention tubes (Kisker Biotech, Steinfurt, Germany) before adding 1 mg protein (per 25 µL beads), followed by 1 h incubation at 4 °C while rotating. Afterwards, the beads were removed and the samples were incubated on a tube rotator at 4 °C overnight with 1 µg rabbit anti-LC3A (Cell Signaling, Danvers, MA, USA; order number 4599), rabbit anti-LC3B (Sigma-Aldrich, St. Louis, MO, USA; order number L7543), or anti-LC3C (Abcam, Cambridge, UK; order number ab150367). Thereafter, magnetic beads reconstituted in IP lysis buffer were applied to the samples and incubated for 1 h on a tube rotator at 4 °C. The beads were washed five times with IP washing buffer (50 mM Tris-HCl, pH 7.4; 200 mM NaCl; 2 mM EDTA; 0.5 mM EGTA; 1% Triton X-100; protease and phosphatase inhibitor cocktail; 1 µM TSA; 20 mM nicotinamide; 1 mM dithiothreitol). Then, the beads were stripped in loading buffer (150 mM Tris-HCl, pH 6.8; 15% glycerol; 10% mercaptoethanol; 3% SDS; 0.003% bromophenol blue) for 10 min on a thermomixer at 36 °C before brief centrifugation and bead removal. The isolated proteins were boiled for 5 min at 95 °C, loaded onto a 15% SDS gel and separated by standard PAGE-Western blotting. To quantify the immunoprecipitated LC3 proteins, the same antibodies were used as for the precipitation itself; acetylated lysines were visualized with rabbit anti-acetylated lysine antibody (1:1000, Cell Signaling, Danvers, MA, USA; order number 9441).
2.5. Immunocytochemistry
Primary human fibroblasts (IMR90 cells) were grown on glass cover slips, treated as appropriate, and fixed with 4% paraformaldehyde. Unspecific epitopes were blocked with 3% BSA before permeabilization with 0.1% Triton X-100 in PBS. The fixed cells were then incubated overnight with primary antibodies diluted in PBS containing 1% BSA: rabbit anti-LC3A (1:200, from Cell Signaling, Danvers, MA, USA; order number 4599), mouse anti-LC3B (1:200, from NanoTools, Teningen, Germany; order number 0260-100), rabbit anti-LC3C (1:200, from Cell Signaling, Danvers, MA, USA; order number 14736), guinea pig anti-p62 (1:400, from Progen, Heidelberg, Germany; order number GP62-C), rabbit anti-LAMP2 (1:1000, from BioCat, Heidelberg, Germany; order number WA-ABV10697.100). The cells were subsequently incubated with Cy2-, Cy3- and Cy5-coupled secondary antibodies (1:500, from Jackson Immunoresearch, West Grove, PA, USA) for 2 h at room temperature. Cell nuclei were counterstained with 1 µg/mL 4,6-diamidino-2-phenylindole (DAPI, from Sigma-Aldrich, St. Louis, MO, USA). The cells were then analyzed and photographed using a confocal laser-scanning microscope (TCS SP5 from Leica Microsystems, Wetzlar, Germany). All pictures were taken with a 63x objective and 2x zoom at the highest possible resolution (2048 × 2048 pixels).
2.6. Subcellular Fractionation
IMR90 cells were cultivated on 10 cm dishes, treated as described, and lysed in 500 µL fractionation buffer (20 mM HEPES, pH 7.4; 250 mM sucrose; 10 mM KCl; 1.5 mM MgCl
2; 1 mM EDTA; 1 mM EGTA; protease inhibitor cocktail (Roche, Basel, Switzerland)). The lysates were then passed 10 times through a 25-gauge needle using a 1 mL syringe and left on ice for 20 min. Nuclei were isolated through centrifugation at 720 g for 10 min at 4 °C. The supernatant was removed and centrifuged again at 10,000 g for 10 min at 4 °C. The pellet discarded, and 2% SDS were added to the cytosolic supernatant. The nuclear fraction was washed by adding 500 µL fresh fractionation buffer and repeating the steps described above. The pellet was then reconstituted in 100 µL fractionation buffer with 2% SDS. Protein concentration was determined using a BCA kit (Thermo Fisher Scientific, Waltham, MA, USA) as per the manufacturer’s protocol. An amount of 10 μg protein was loaded on a 15% SDS gel and separated by electrophoresis. The proteins were transferred onto a nitrocellulose membrane, blocked with 4% non-fat milk in PBST, and detected with the following primary antibodies: rabbit anti-LC3A (1:1000, Cell Signaling, Danvers, MA, USA; order number 4599), rabbit anti-LC3B (1:1000, Sigma-Aldrich, St. Louis, MO, USA; order number L7543), rabbit anti-LC3C (1:1000, Abcam, Cambridge, UK; order number ab150367), mouse anti-histone H3 (1:1000, Cell Signaling, Danvers, MA, USA; order number 14269), mouse anti-α-tubulin (1:1000, Sigma-Aldrich, St. Louis, MO, USA; order number T9026). The detection by secondary antibodies was done as described in
Section 2.2.
2.7. Knock-Down Experiments
IMR90 cells were transfected by electroporation (Nucleoffector 2b, Lonza, Basel, Switzerland; program: U-24). A number of 8000 cells per transfection were reconstituted in 400 µL electroporation buffer (10 mM HEPES, pH 7.8; 135 mM KCl; 2 mM MgCl2; 200 µM CaCl2; 2 mM EGTA; 25% FCS) with 30 µg siRNA (small interfering RNA) or scrRNA (scrambled RNA) and transferred to an electroporation cuvette (Sigma-Aldrich, St. Louis, MO, USA; order number Z706094-50EA). After electroporation, the cells were left for 8 min at 37 °C in an incubator containing 5% CO2. Thereafter, 600 µL culture medium was added, and the cells were seeded in a 24-well plate with coverslips adding an additional 1000 µL culture medium. One day later, the cells were treated with 100 nM Ex527 or vehicle (DMSO). After 24 h, the medium was replaced, and the cells harvested by fixation after an additional 24 h. Four hours prior to fixation, the cells were starved for 4 h using Earle’s balanced salt solution and/or treated with 2 µM BafA1 where appropriate.
2.8. Bioinformatic Analysis
Protein sequences from
H. sapiens LC3A, LC3B, LC3C, GABARAP, GABARAP-L1, GABARAP-L2 and were obtained from Ensembl [
14] and aligned using Clustal Omega from EMBL-EBI. A phylogenetic tree based on DNA data from Ensembl was also constructed with Clustal Omega from EMBL-EBI and the Interactive Tree Of Life software [
15] to incorporate
H. sapiens LC3A, LC3B, LC3C, GABARAP, GABARAP-L1, GABARAP-L2;
D. melanogaster ATG8A and ATG8B; and
S. cerevisiae ATG8 as out-group. ATG7 binding sites and lipidation sites were extracted from previous studies revealing their exact amino acid sequences [
16,
17]. Specific interaction partners of human Atg8 family proteins were assembled from previously published data [
18]. Their sequences were then retrieved from Ensembl and analyzed in the following manner: the protein sequences of interaction partners unique to a specific ATG8 were analyzed for putative LIR domains that would follow the established LIR sequence pattern: Y/F/W–X–X–V/I/L (X = any amino acid) [
19]. Finally, the relative occurrence of all amino acids on the four LIR positions was calculated.
2.9. Statistical Analysis
Depending on the structure of the data, either one-way or two-way analysis of variance (ANOVA) was performed, followed by Benjamini and Hochberg adjustment for multiple comparisons. In the case of two-way ANOVA analysis, variable #1 was usually the presence or absence of TSA or Ex527, while variable #2 was the identity of the LC3s to be compared. Statistical evaluation of the autophagic fluxes was performed by Kruskal-Wallis one-way ANOVA (on Ranks) since the data did not always pass the normality test implemented in the employed software GraphPad Prism 7.03. Post-hoc test significances with smaller p-values than 0.05 are indicated by asterisks or hash signs in the figures. Quantitative results are generally presented as arithmetic mean and standard deviation (SD).
4. Discussion
Autophagy is a key degradative pathway for maintaining cellular homeostasis [
4,
5]. Although autophagy has historically been described as a non-selective process, more recent evidence demonstrates that selective and thus cargo-specific forms of autophagy exist [
2]. The microtubule-associated protein 1A/1B light chain 3B (LC3B) is arguably the most widely used marker of autophagic activity and autophagic flux, as it exhibits a posttranslationally modified, lipidated from that can be related to ongoing phagophore formation [
2,
6]. In humans, there are three paralogues of the LC3 gene family: LC3A, LC3B, and LC3C [
7]. Despite the established importance of LC3B for the autophagic cascade, only a small number of studies have explicitly focused on LC3A and LC3C in terms of their cellular localization and regulation [
26]. LC3C has been proposed to link the secretory and the autophagic pathway [
27]. It has been further implicated in xenophagy and the specific targeting of mitochondria for mitophagy [
28,
29]. LC3A also seems to be involved in mitophagy since both proteins, LC3A and LC3C, have been related to Parkin-independent mitophagy pathways [
30]. While LC3C has been described to target mitochondria through the outer mitochondrial membrane (OMM) protein metaxin 1, LC3A acts through binding to another OMM protein, FK506-binding protein 8 [
31]. Autophagosomal cargo profiling has confirmed that upon BafA1 treatment, LC3A-, LC3B-, and LC3C-containing autophagosomes showed specificity for their cargo [
29]. Besides the LC3 subfamily, three other members of the ATG8 family called GABARAPs have been described to play a role in different forms of autophagy that may occur independently of the LC3s [
11,
32,
33].
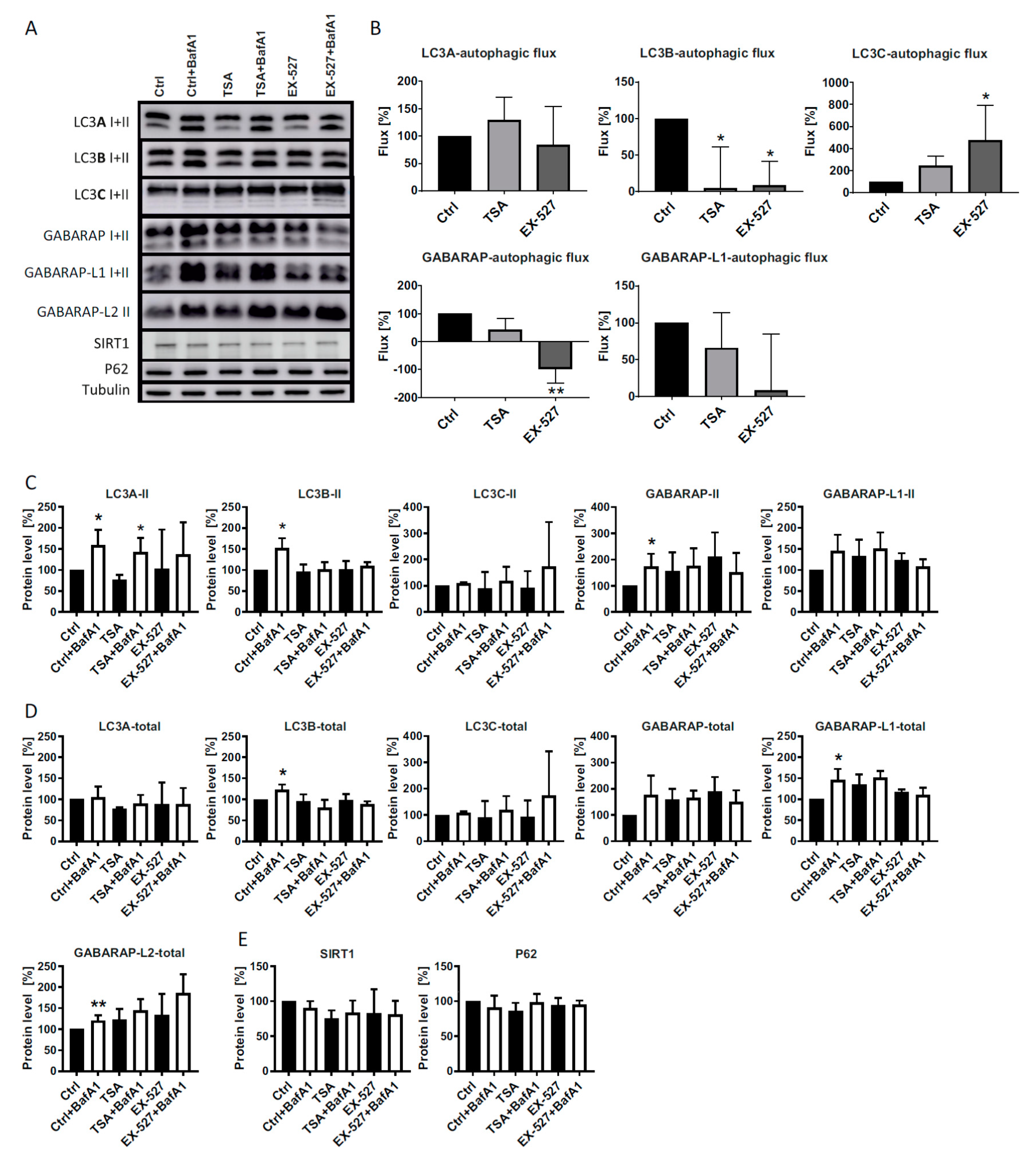
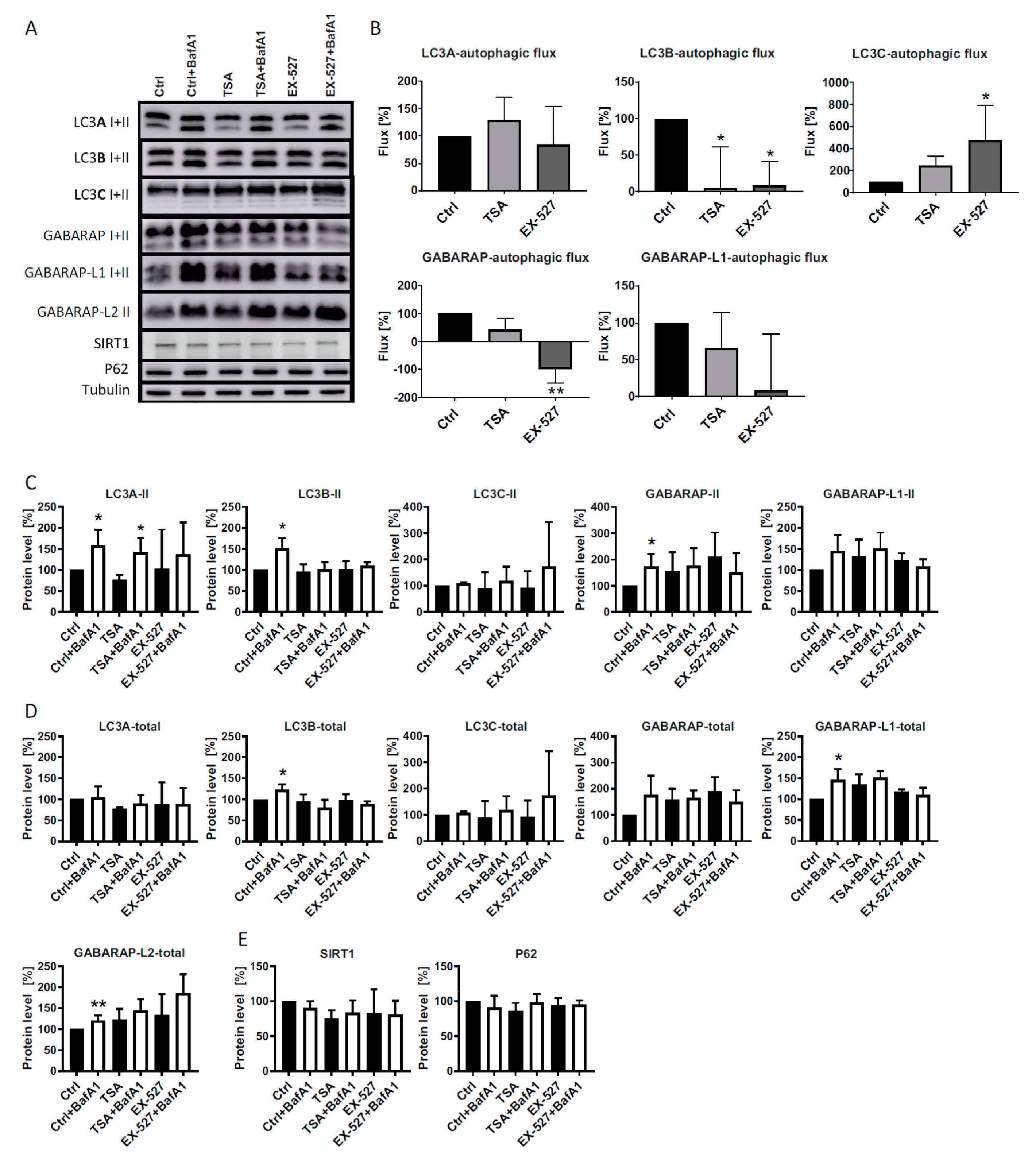
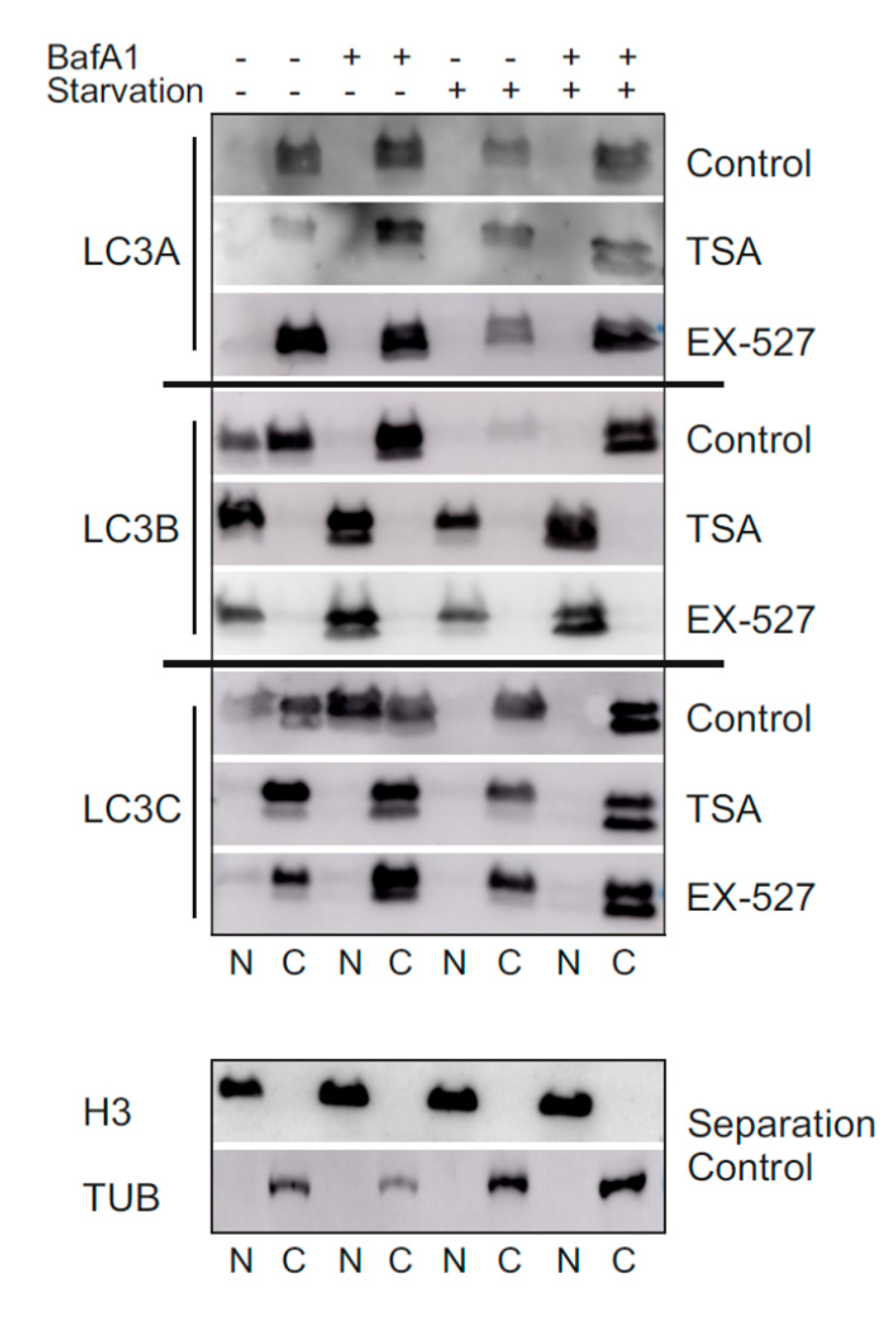
In the present study, we analyzed LC3 lipidation as a marker for autophagic flux in primary human cells (diploid fibroblasts) in the presence of Ex527 and TSA, two deacetylase inhibitors known to act selectively on SIRT1 and non-sirtuin HDACs, respectively. These deacetylase inhibitors have been reported to show clear effects on LC3′s translocation from the nucleus into the cytosol [
10], an effect that was recapitulated in this study (
Figure 3,
Figure 4 and
Figure 7). With the exception of GABARAP-L2, two distinct bands could be resolved and quantified by Western blotting for each ATG8 protein. In analyzing the BafA1-induced difference in lipidation of the remaining five proteins as described [
6], it was found that “LC3B-mediated autophagic flux” was severely compromised, as expected from LC3B’s intracellular distribution (
Figure 1A,B). The same phenomenon was measured for GABARAP and GABARAP-L1. In contrast, “LC3A-mediated autophagic flux” was unaffected by both treatments trapping LC3B in the nucleus, while “LC3C-mediated autophagic flux” actually increased significantly in cells treated with Ex527 (
Figure 1B). These changes were not brought about by a selective loss of the unlipidated proteins, but rather in the presence of stable (LC3A) or increased (LC3C) levels of the lipidated proteins (
Figure 1). Based on these results, we conclude that the physiological regulatory mechanisms for LC3A and LC3C differ substantially from LC3B regulation, and that this differential response to acetylation might potentially serve as a buffering (LC3A) or compensatory (LC3C) mechanism to survive (local) LC3B insufficiency. The latter hypothesis was explored in detail by quantitative microscopic analyses summarized in the following.
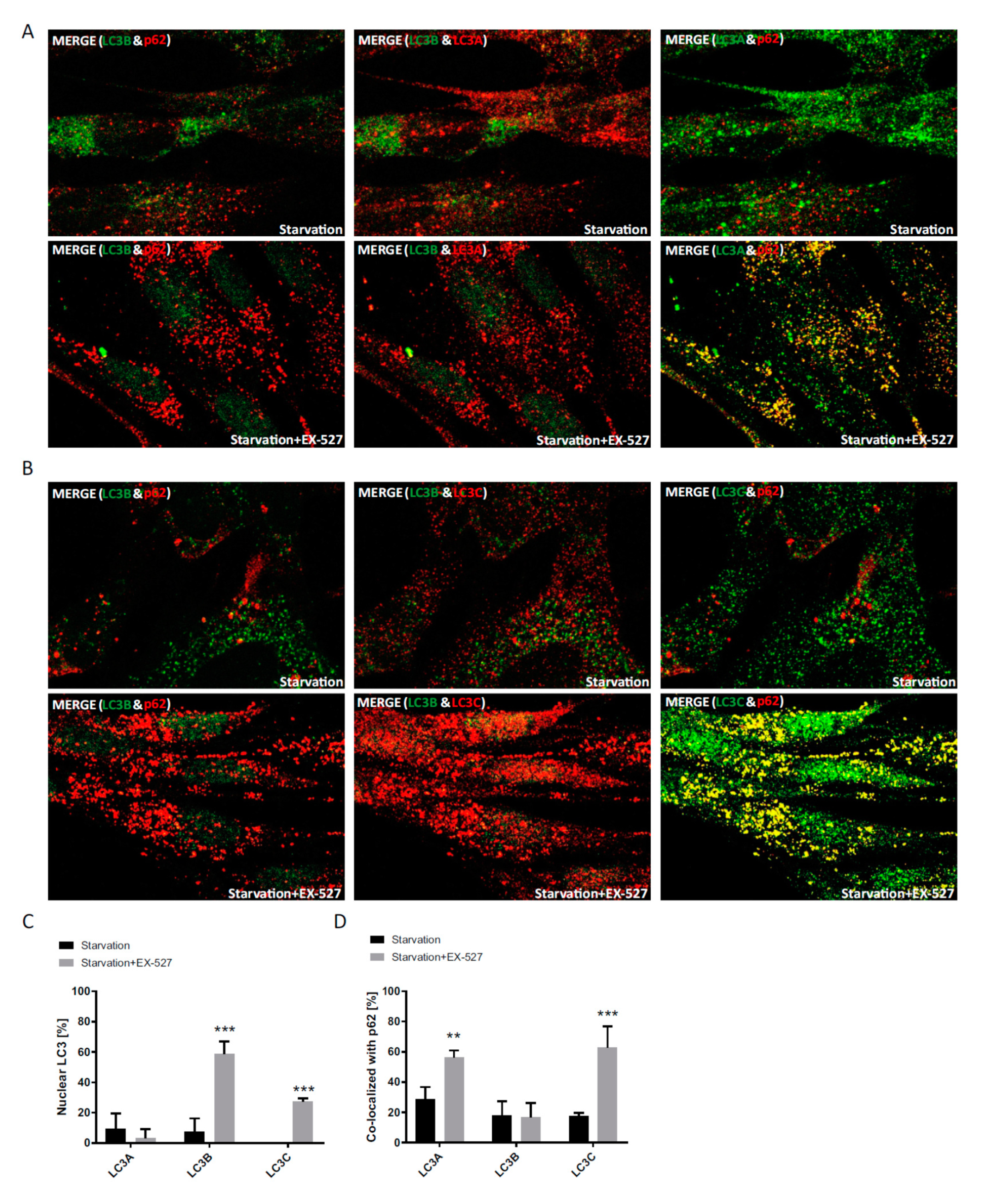
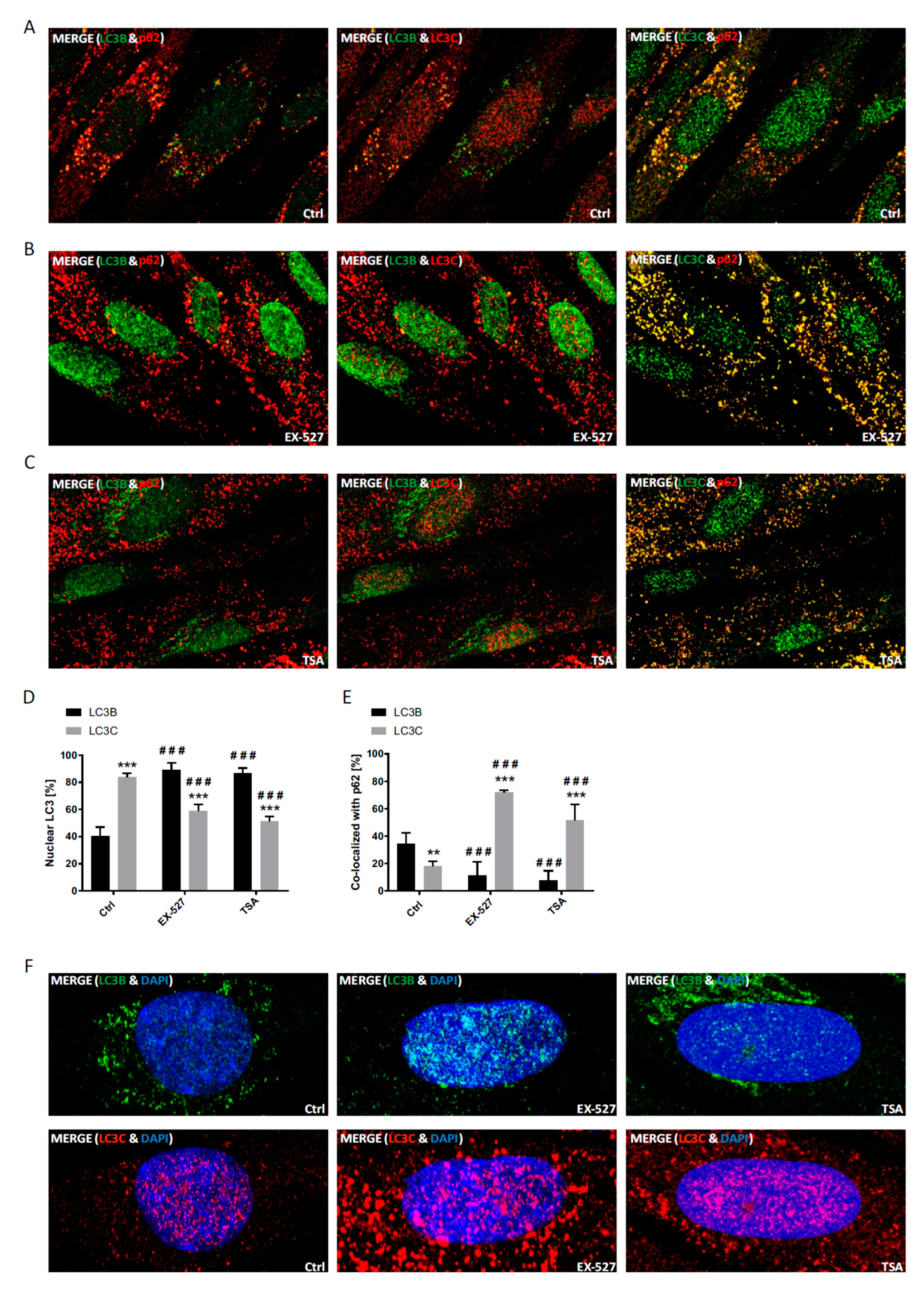
In immunostaining untreated primary human cells, we found that LC3B was mainly restricted to the nucleus, whereas LC3A (and p62) was primarily localized to the cytoplasm, while LC3C was stained in both compartments (
Supplementary Figure S1). Upon BafA1 treatment, the majority of LC3B appeared in the cytoplasm to co-localize with p62, which was visible as vesicular structures and likely demarcated autophagosomes. At the same time, p62 was also co-localized with LC3A, while LC3C was found to be mainly migrating into the opposite direction into the nucleus; it was thus poorly co-localized with p62 (
Figure 3 and
Figure 4). Hence, both LC3A and LC3B would be appropriately distributed to engage in cytosolic autophagy under current basal conditions (i.e., with BafA1 treatment), whereas such a role for LC3C under these conditions appears unlikely. Still, provided that LC3B might also have a relevant function in the nucleus [
10,
24], LC3C would be suitable to replace it there.
To test the idea a potential autophagic involvement LC3 proteins beyond LC3B, this protein was trapped inside the nucleus by preventing its nuclear export pharmacologically [
10]. The according treatment with a selective SIRT1 inhibitor, Ex527, indeed led to a far-reaching loss of LC3B from the cytosol (
Figure 3,
Figure 4 and
Figure 7) and a proportional loss of co-localization with p62, which would functionally involve autophagic insufficiency. In contrast, LC3A’s distribution was only moderately affected, and its almost complete co-localization with p62 was unchanged. LC3C, in turn, relocated actively into the cytosol and was proportionally more co-localized with p62. As total protein levels of all three LC3 proteins were either unchanged or increased in BafA1- and Ex527-treated cells (
Figure 1), the described protein redistributions cannot be interpreted to have arisen from selective protein loss. For example, the 2.5-fold increased cytosolic fraction of LC3C in
Figure 4D (and its proportional increase in p62 co-localization in
Figure 4E) must have been accompanied by an absolute increase of LC3C protein in the cytosol since total protein levels of LC3C in parallel experiments analyzed by Western blotting were unchanged (
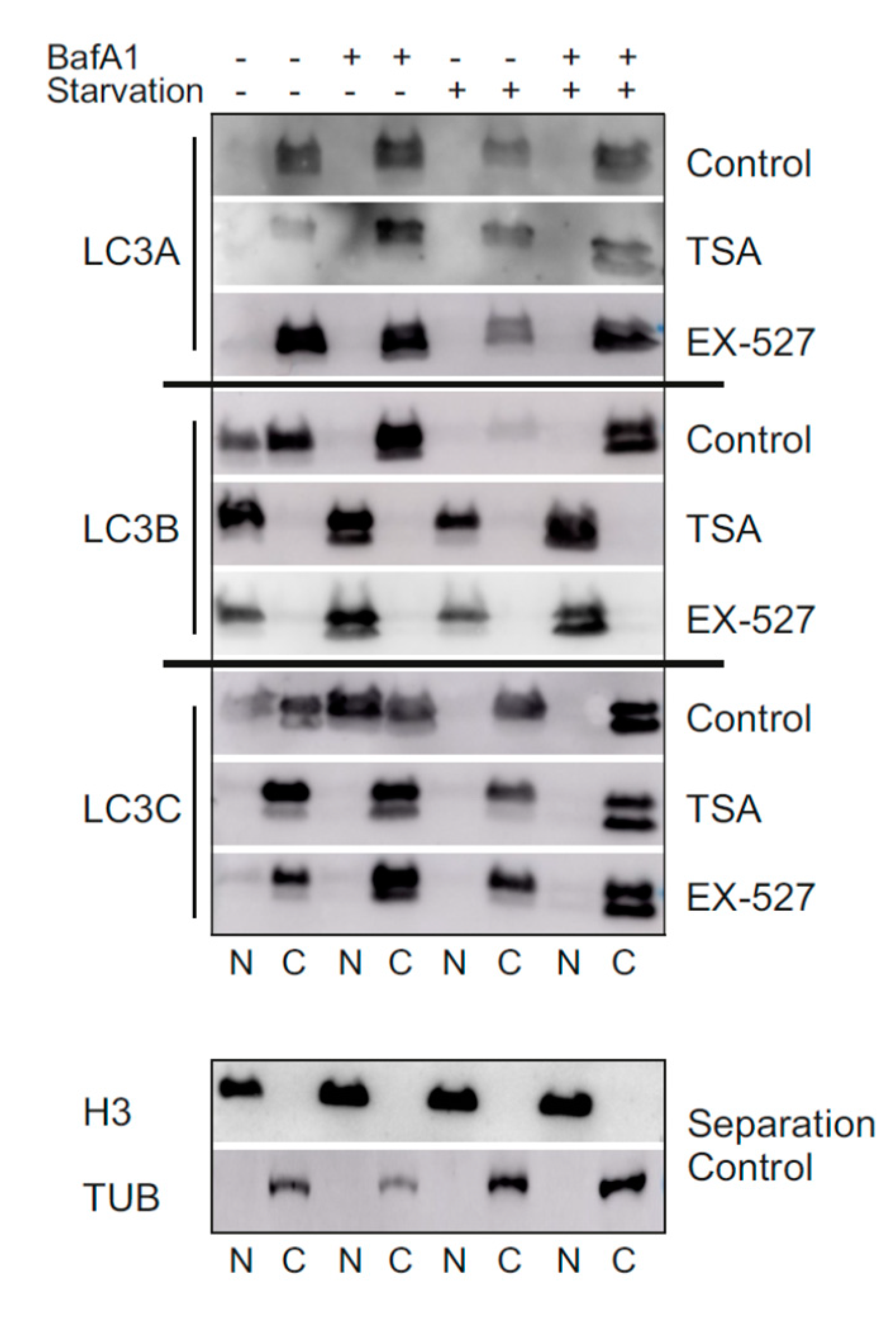
Figure 1D). Interestingly, we observed an overall very similar pattern of redistributions with TSA, a non-sirtuin HDAC deacetylase inhibitor. As per quantitative image analysis and subcellular fractionation, LC3B was trapped in the nuclear regions as pronouncedly as with the selective SIRT1 inhibitor, Ex527 (
Figure 3D,
Figure 4D and
Figure 7), even if the trapping may have been brought about by a different mechanism (suppressed dispersion in the cytoplasm through HDAC6 inhibition [
22]). Nevertheless, very similar responses by LC3A and LC3C were induced, supporting the conclusion that these responses were secondary to functional LC3B insufficiency. Additional validation of our conclusions from these pharmacological experiments comes for the analysis of sirtuin 1 knock-down experiments, which again trapped mostly LC3B in the nucleus and increased the cytosolic association of only LC3A and LC3C with p62 (
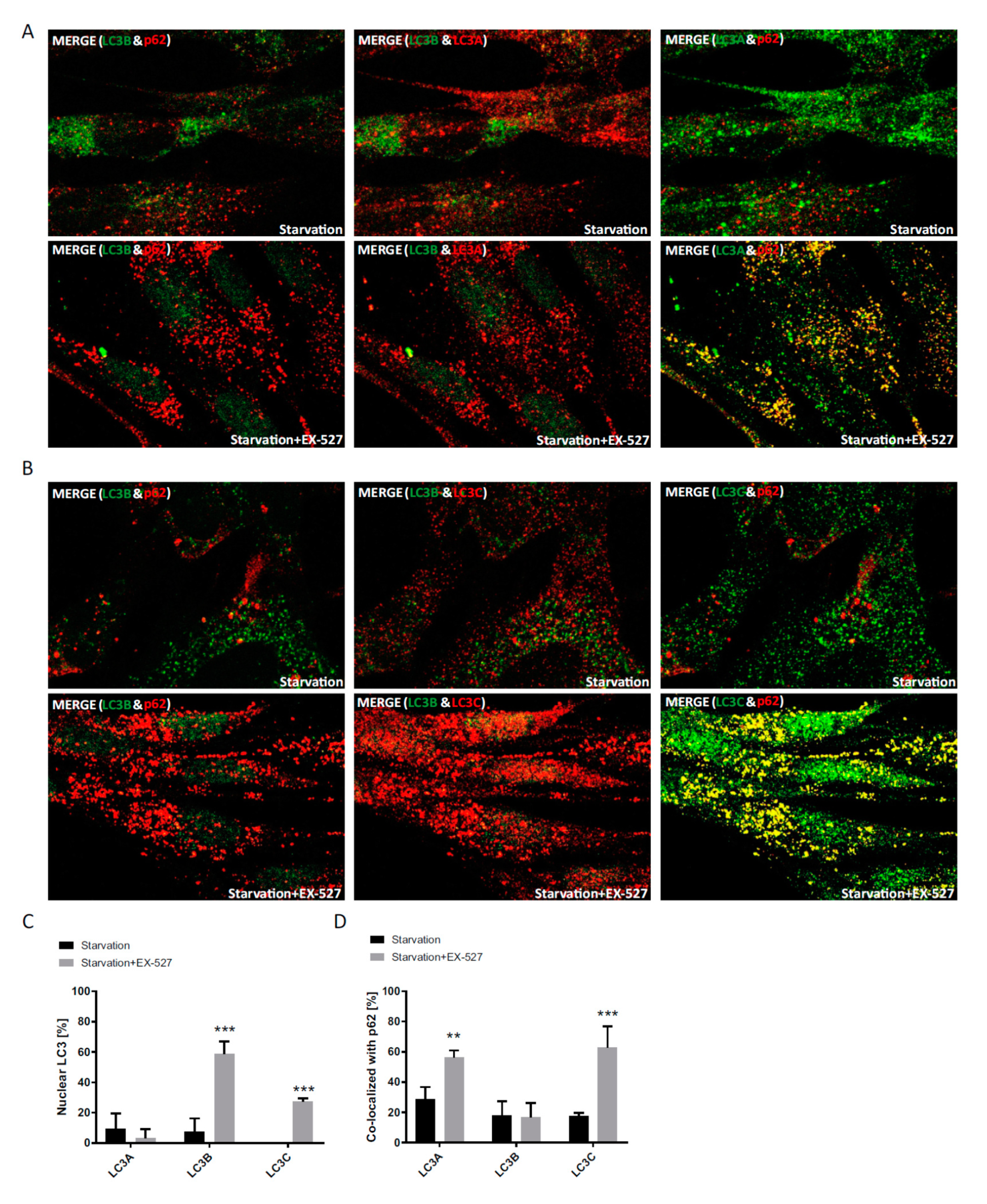
Figure 5). Moreover, the robustness and the LC3B-specificity of the acetylation regulatory mechanism were recapitulated under conditions of nutritional starvation (
Figure 6). Finally, the distributional patterns of all LC3s and their redistributions in response to deacetylase inhibitor treatment were also verified in biochemical experiments, i.e., subcellular fractionation followed by Western blotting (
Figure 7).
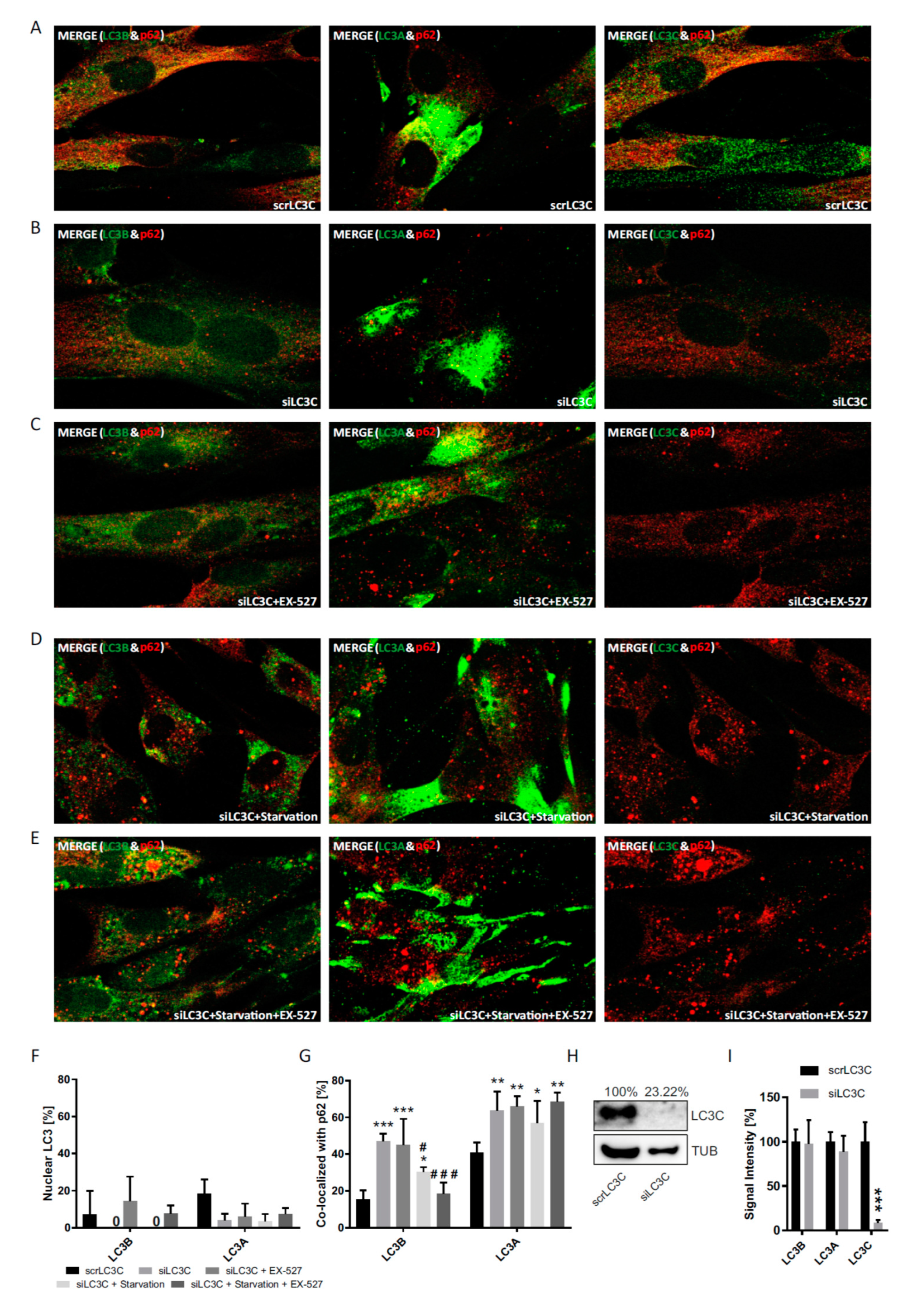
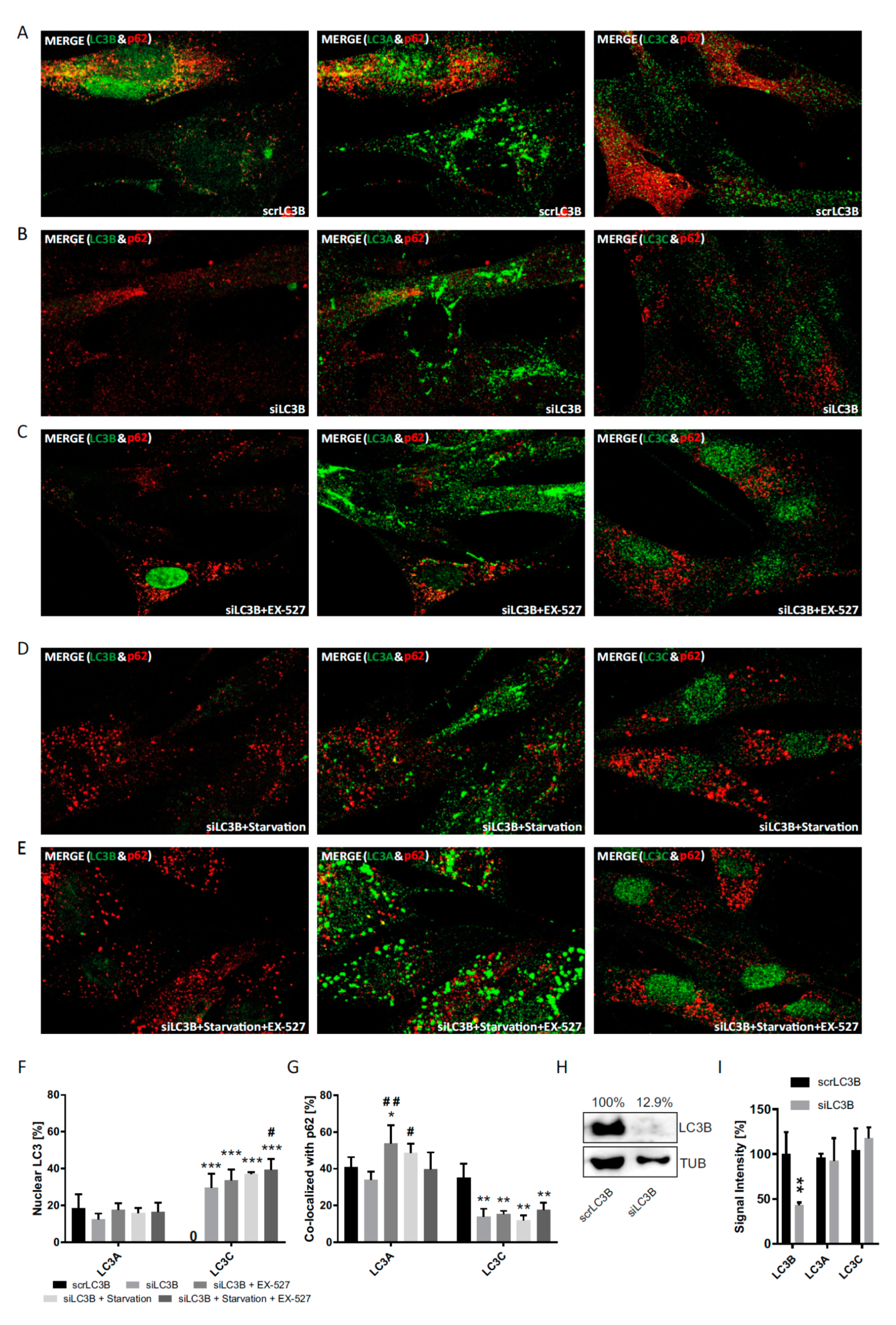
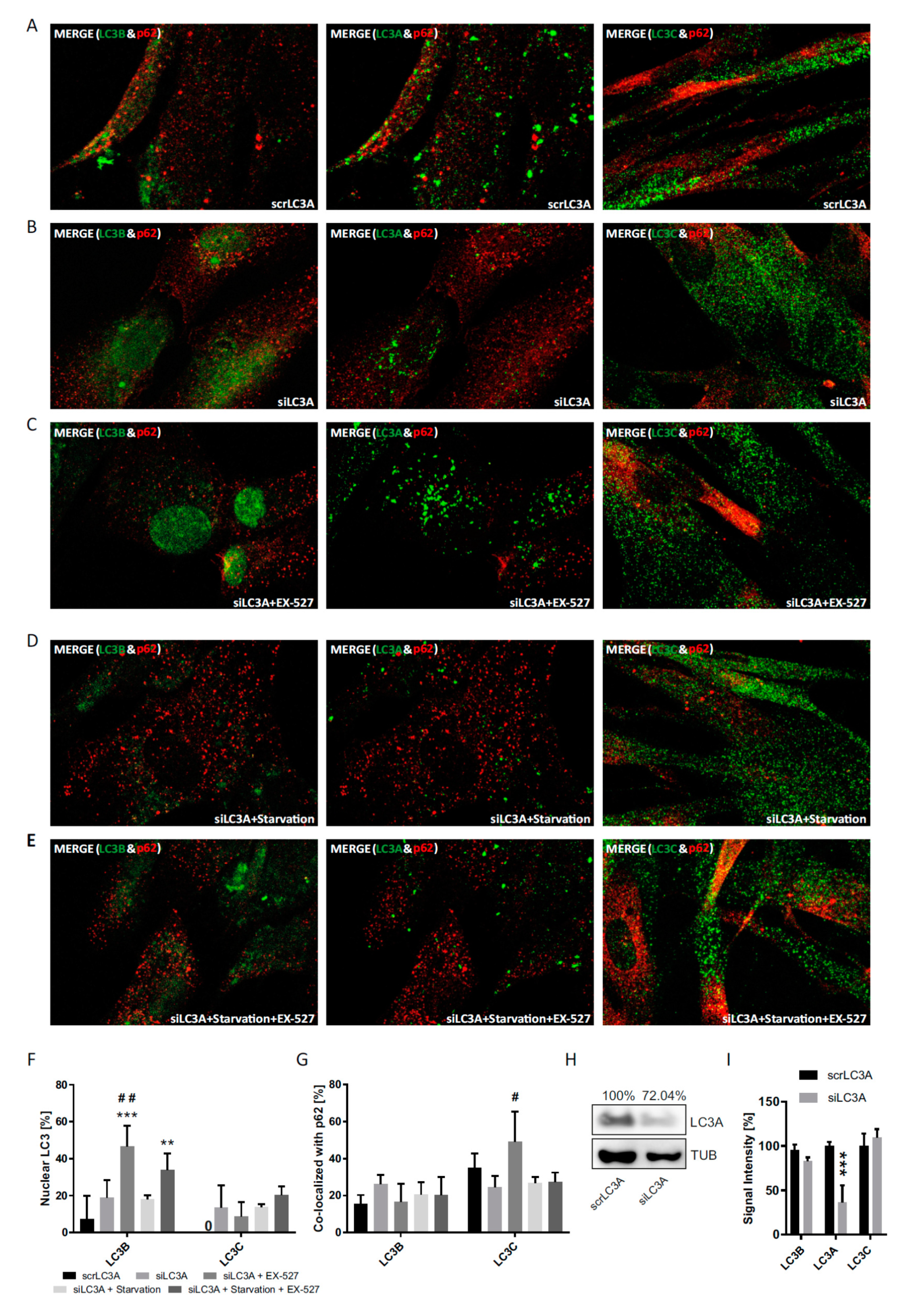
In a third approach, we also investigated the differential response behavior of the three LC3 proteins in response to the genetic suppression of each of them by transfection with specific siRNAs (
Figure 8,
Figure 9 and
Figure 10). Despite some technical interference (all proteins were less restricted to the nucleus at baseline even after scrambled RNA treatment), the fractional cytosolic association with p62 could be reliably quantified. The experiments confirmed the idea of a reciprocal and thus compensatory behavior of LC3B and LC3C under the assumption [
10,
24] that LC3B would have an essential function not only in the cytosol, but also in the nucleus.
A sequence and binding partner comparison of the six human ATG8 proteins lends further plausibility to this idea. These proteins were confirmed to be highly conserved including their ATG7 binding sites and their common lipidation site, which may point towards a sub-functionalization rather than a neo-functionalization, which would usually involve addition of entire new functional domains (e.g., continuous sequence stretches at the termini or in the middle of one of the family members). This was generally not the case, but with the exception of short sequence stretches at both termini of LC3C (
Figure 11B). In parallel with its behavior in cell culture, LC3C also stood out to some extent regarding its inferred interaction partners (
Figure 11C), since the analyzed, putative LIR domains of its exclusive binding partners were distinguishable by their different occupation at AA positions 1 and 3. These slight, yet noticeable differences are well compatible with a functional differentiation of LC3C, albeit within the same paradigm, autophagy. Interestingly, many unique interaction partners of human ATG8s do not take part in autophagic processes, while many interaction partners that can bind to several different ATG8s do so. This could point to two things: first, the interaction with partners unrelated to autophagy may primarily drive their evolution. Second, ATG8 proteins are more likely to cross-compensate for the loss of another ATG8 with regard to autophagic processes rather than non-autophagic processes.
In conclusion, the present study reveals a differential cellular localization and distributional regulation of LC3A and LC3C as compared to LC3B. LC3A and LC3C might have important autophagy-related functions due to their buffering (LC3A) and compensating (LC3C) ability of a lack of LC3B function. In general, LC3A is equidistributed between nucleus and cytosol, does not tend to travel much, but can associate more heavily with (cytosolic) p62 upon demand. LC3C, in turn, is highly mobile, and it always tends to migrate where LC3B has been decapacitated or removed, be it the cytosol or the nucleus. This behavior is indicative of a functional replacement. A significant decrease in the activity of sirtuins and HDACs in age-related diseases is widely recognized [
34]. Hence, it may be a specific physiological task of LC3C to compensate for an LC3B loss-of-function in the cytosol due to age-associated sirtuin deficiency.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}