Cannabinoid-Induced Autophagy and Heme Oxygenase-1 Determine the Fate of Adipose Tissue-Derived Mesenchymal Stem Cells under Stressful Conditions


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Metabolic Activity Assay
2.4. Trypan Blue Staining
2.5. siRNA Transfection
2.6. Quantitative RT-PCR Analysis
2.7. Western Blot Analysis
2.8. Statistics
3. Results
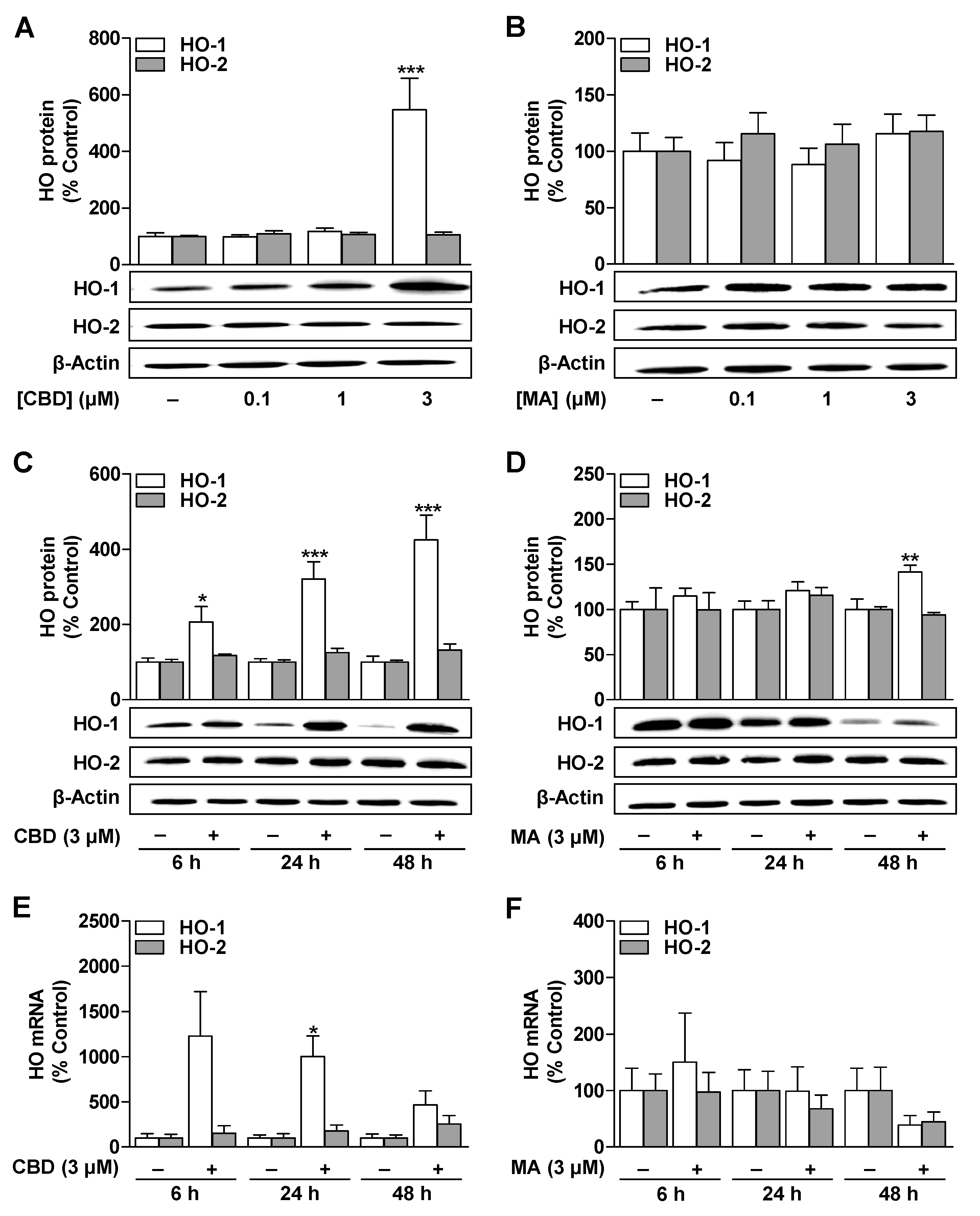
3.1. CBD Induces HO-1 mRNA and Protein Expression in ADMSCs under Serum-Free Conditions
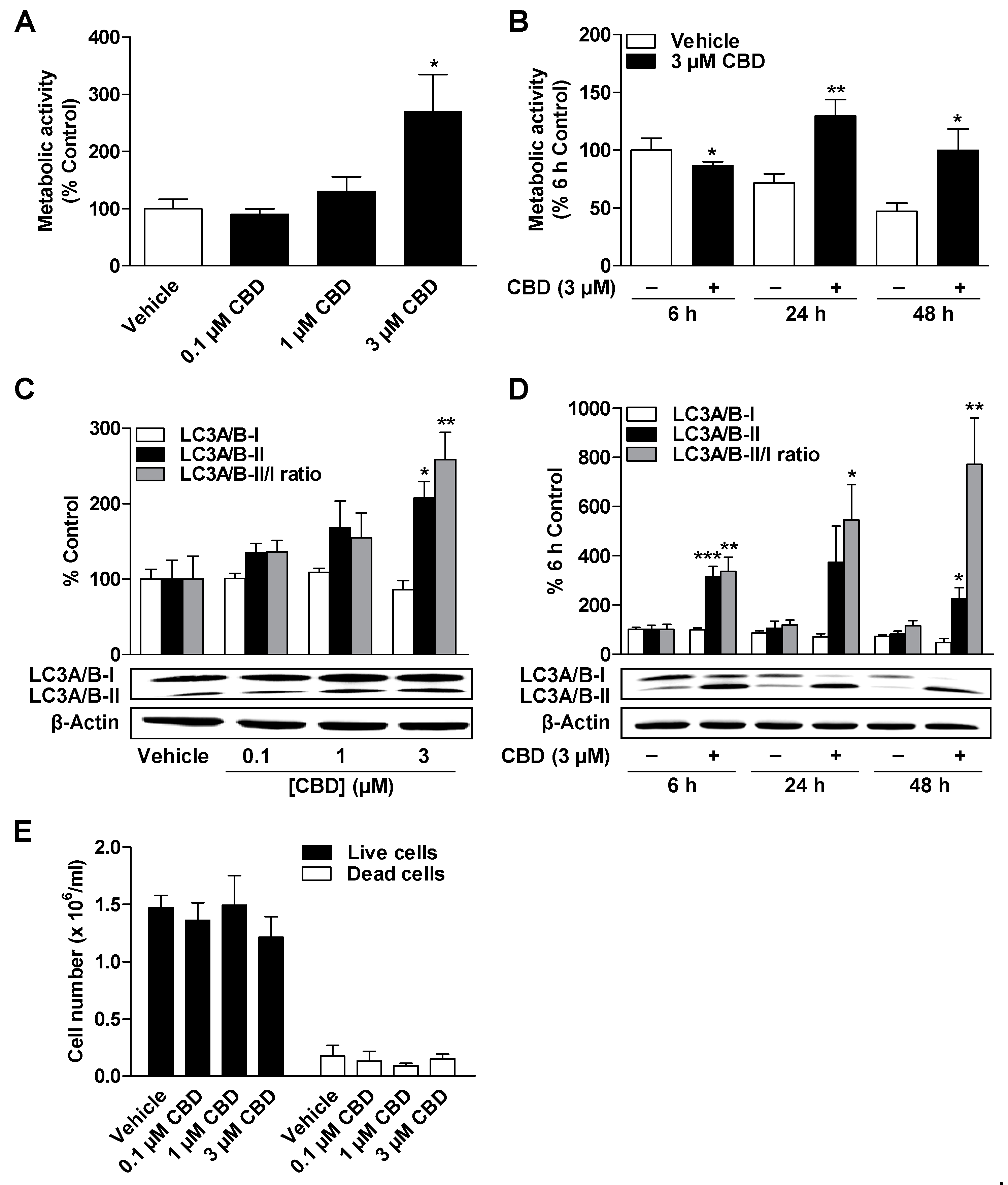
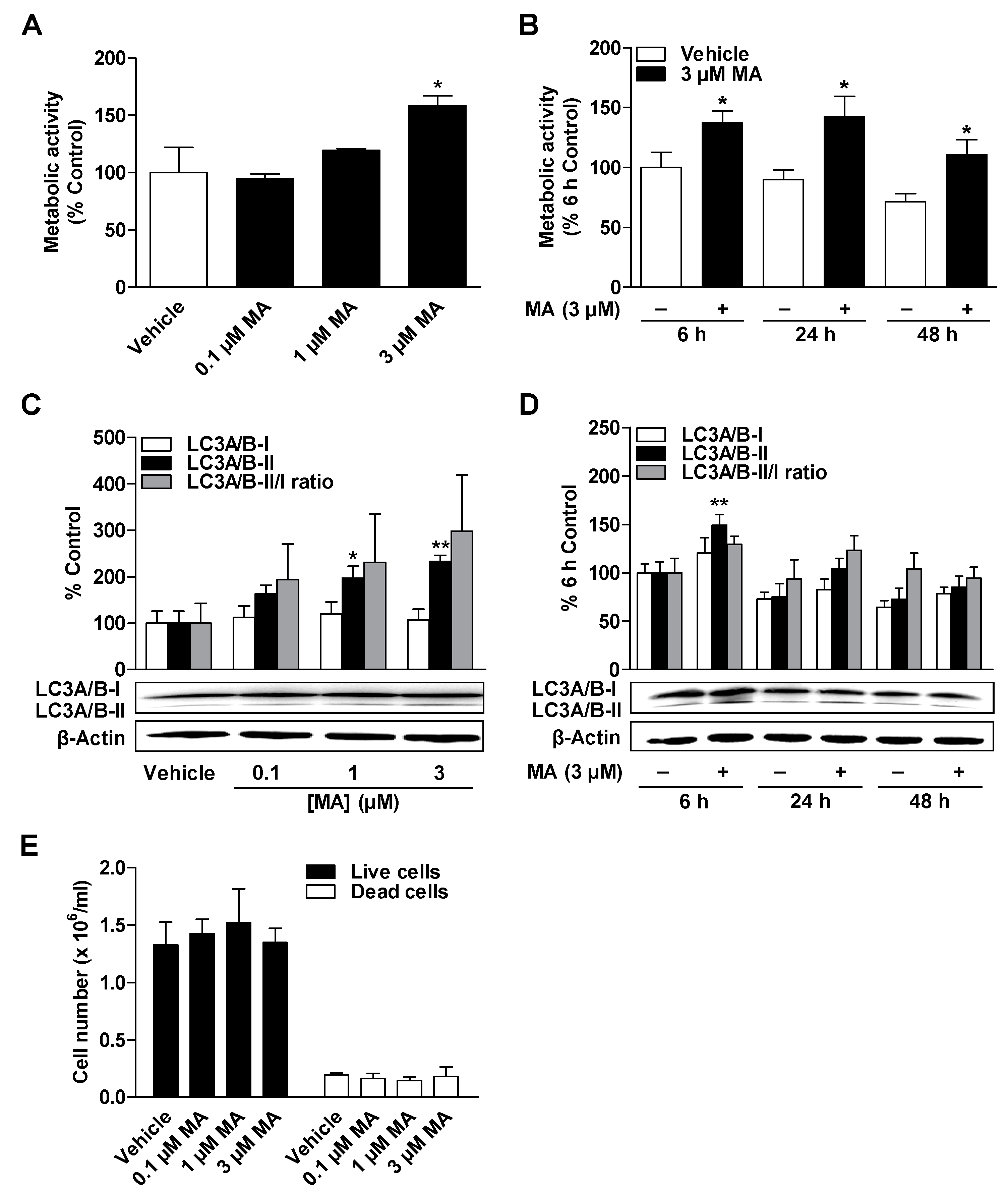
3.2. CBD and MA Increase Metabolic Activity and Autophagy of ADMSCs under Serum-Free Conditions
3.3. Cannabinoid-Binding Receptors Mediate neither the CBD-Increased HO-1 Expression nor the CBD- and MA-Increased Metabolic Activity of ADMSCs
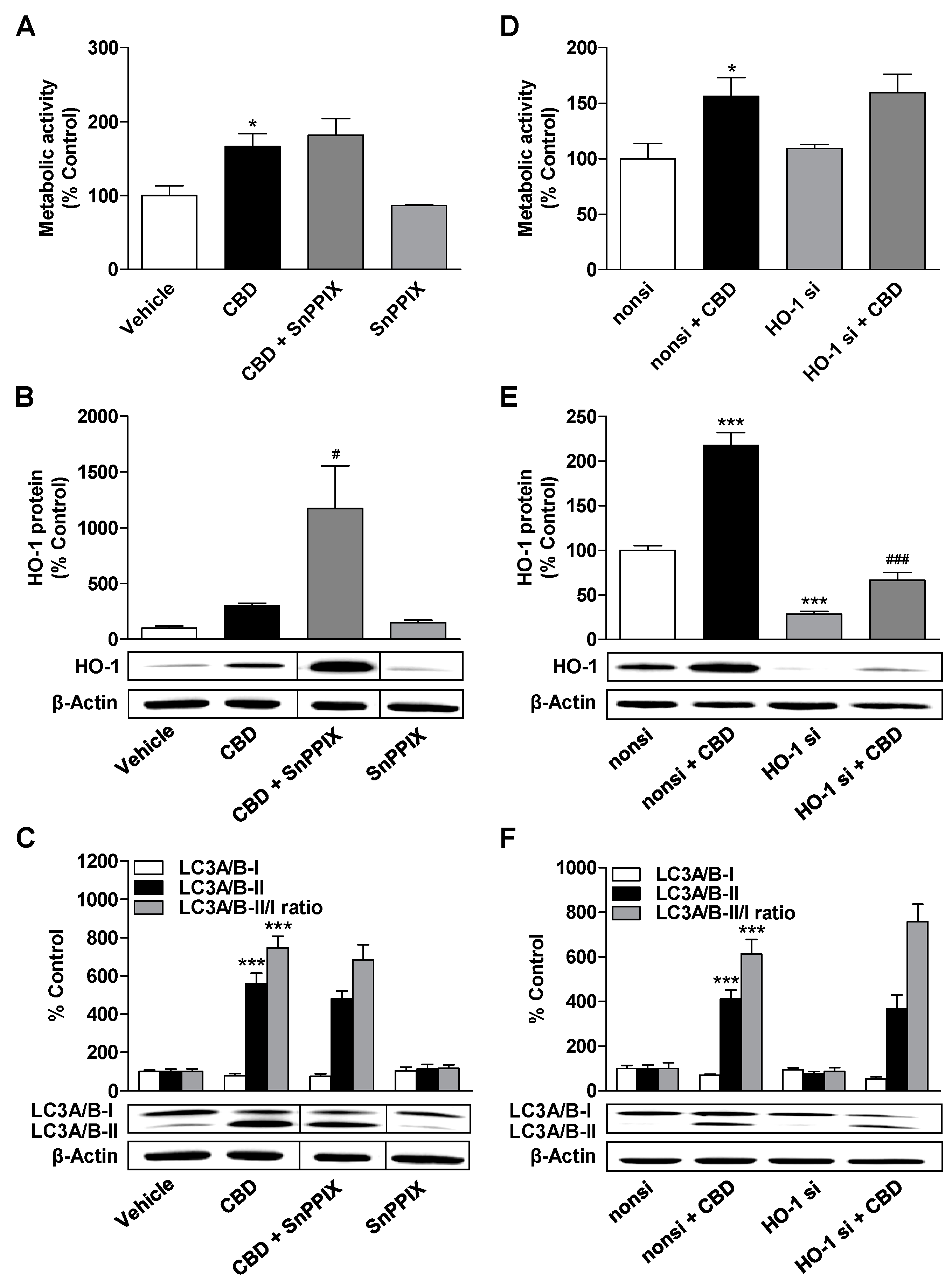
3.4. HO-1 Does not Mediate the CBD-Induced Increase in Metabolic Activity and Autophagy of ADMSCs
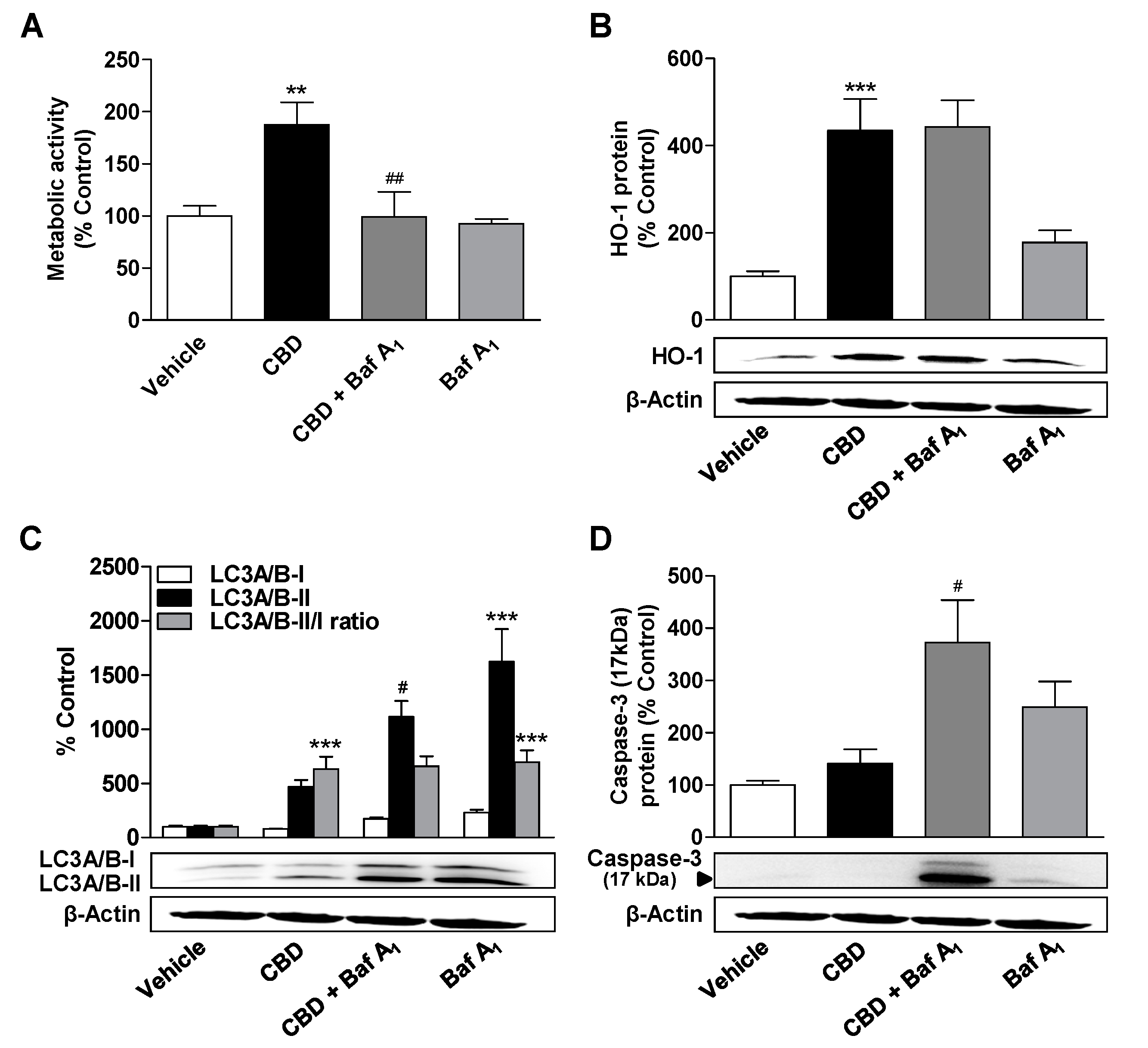
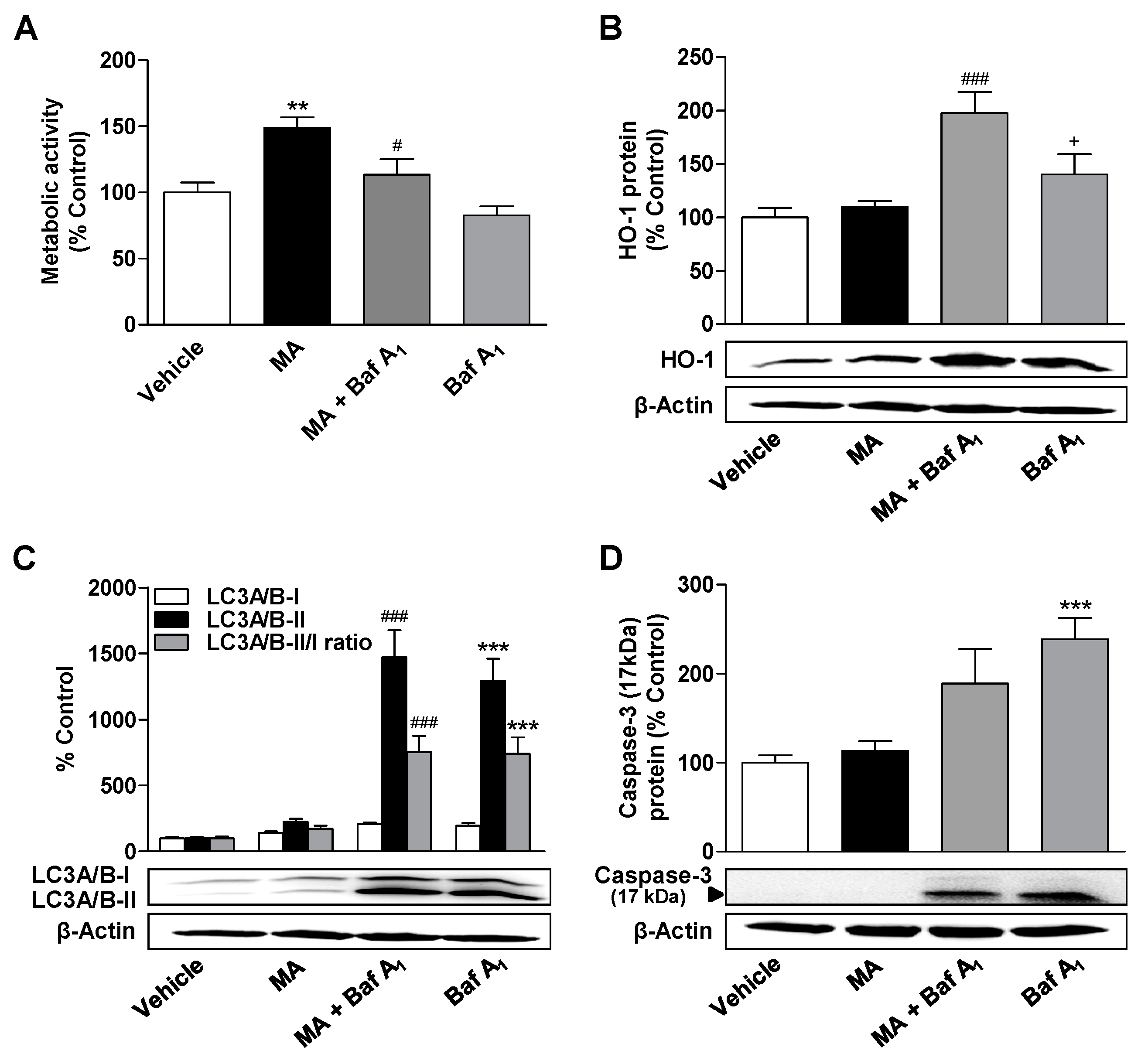
3.5. Inhibition of Autophagy Leads to a Decrease in the Metabolic Activity Elevated by CBD and MA, to an Increase in Caspase-3 Cleavage, and to the Induction of HO-1 Expression in MA-Treated ADMSCs
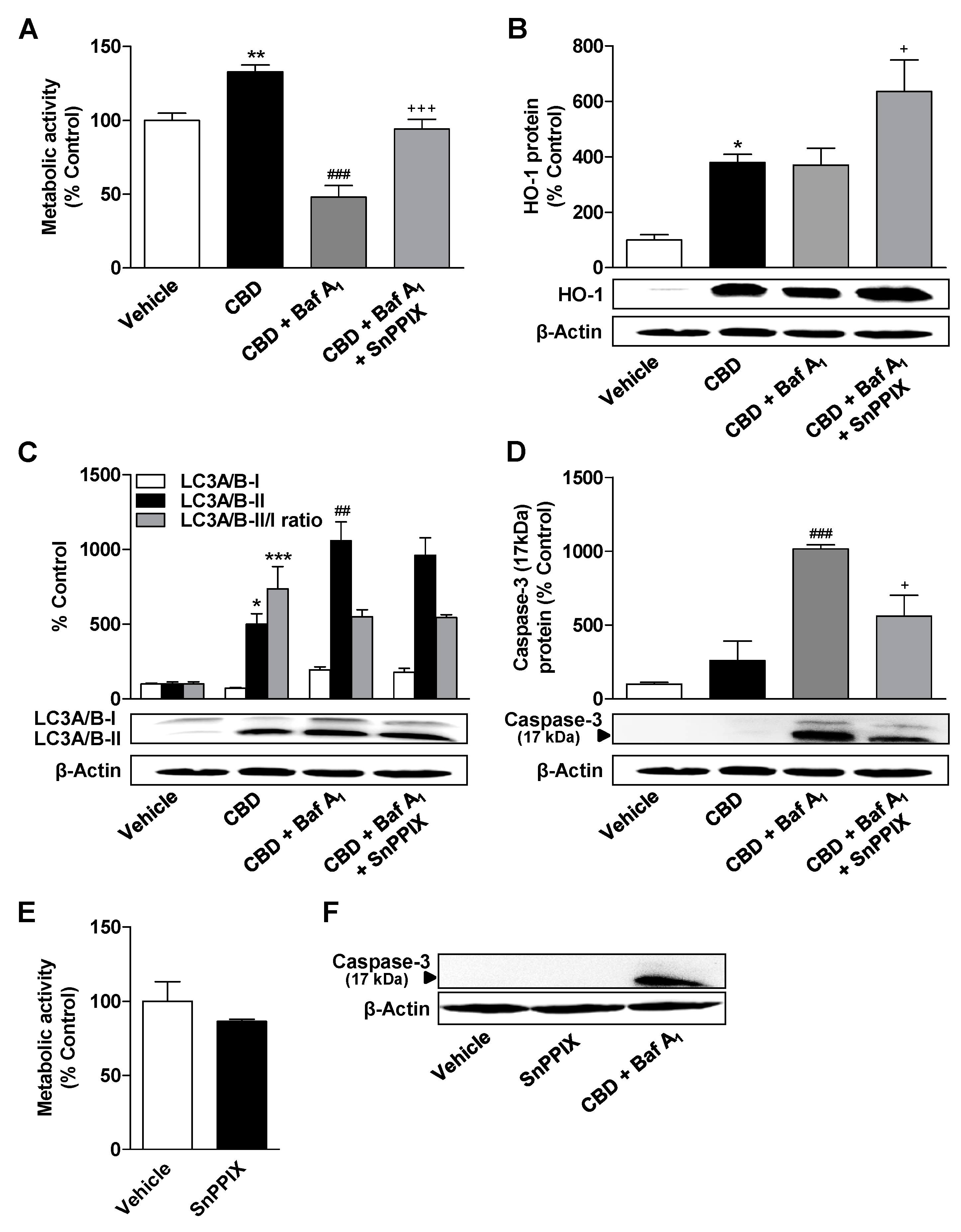
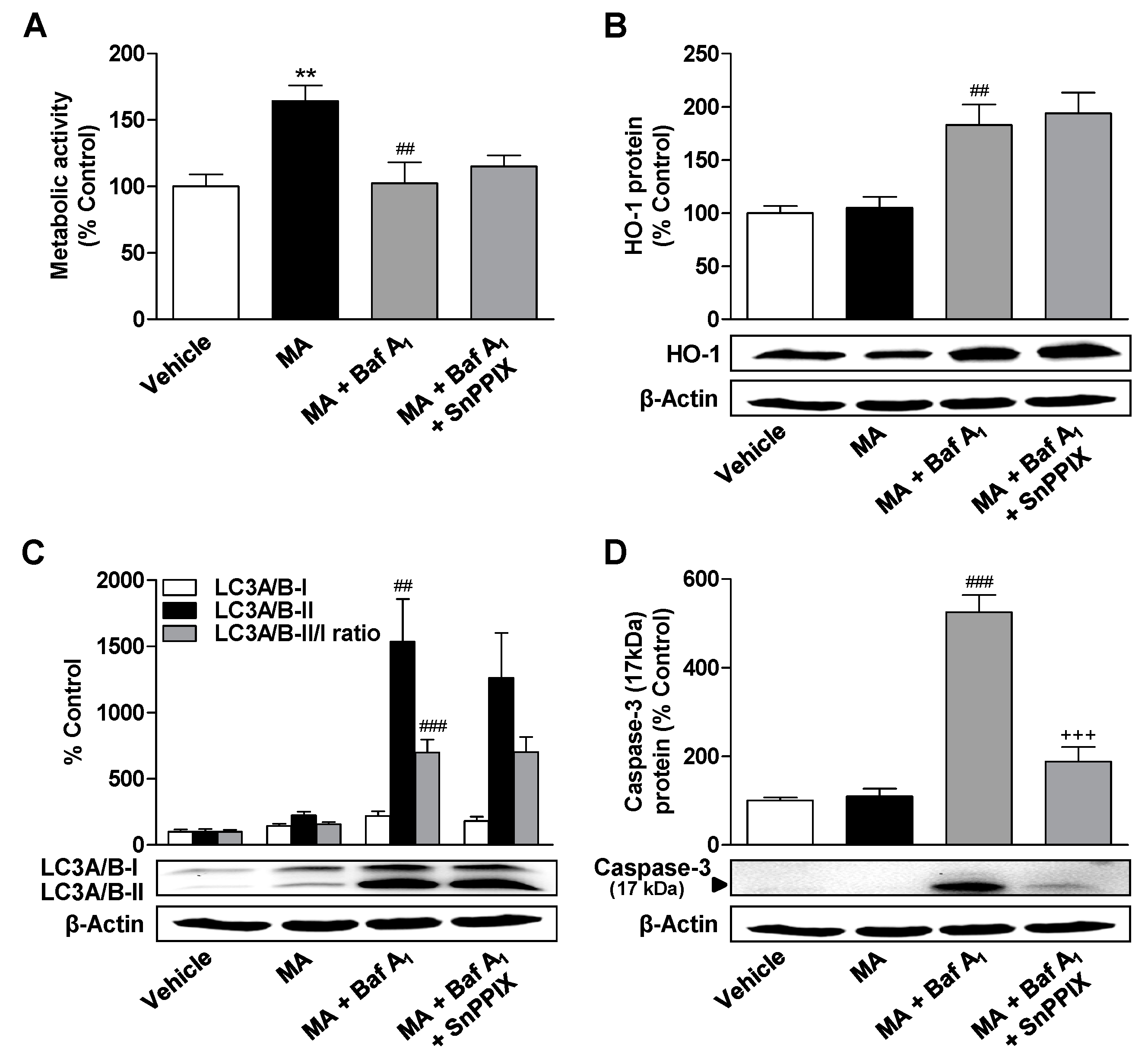
3.6. Inhibition of HO-1 Significantly Suppresses Apoptosis of ADMSCs Treated with Cannabinoids and the Autophagy Inhibitor Bafilomycin A1
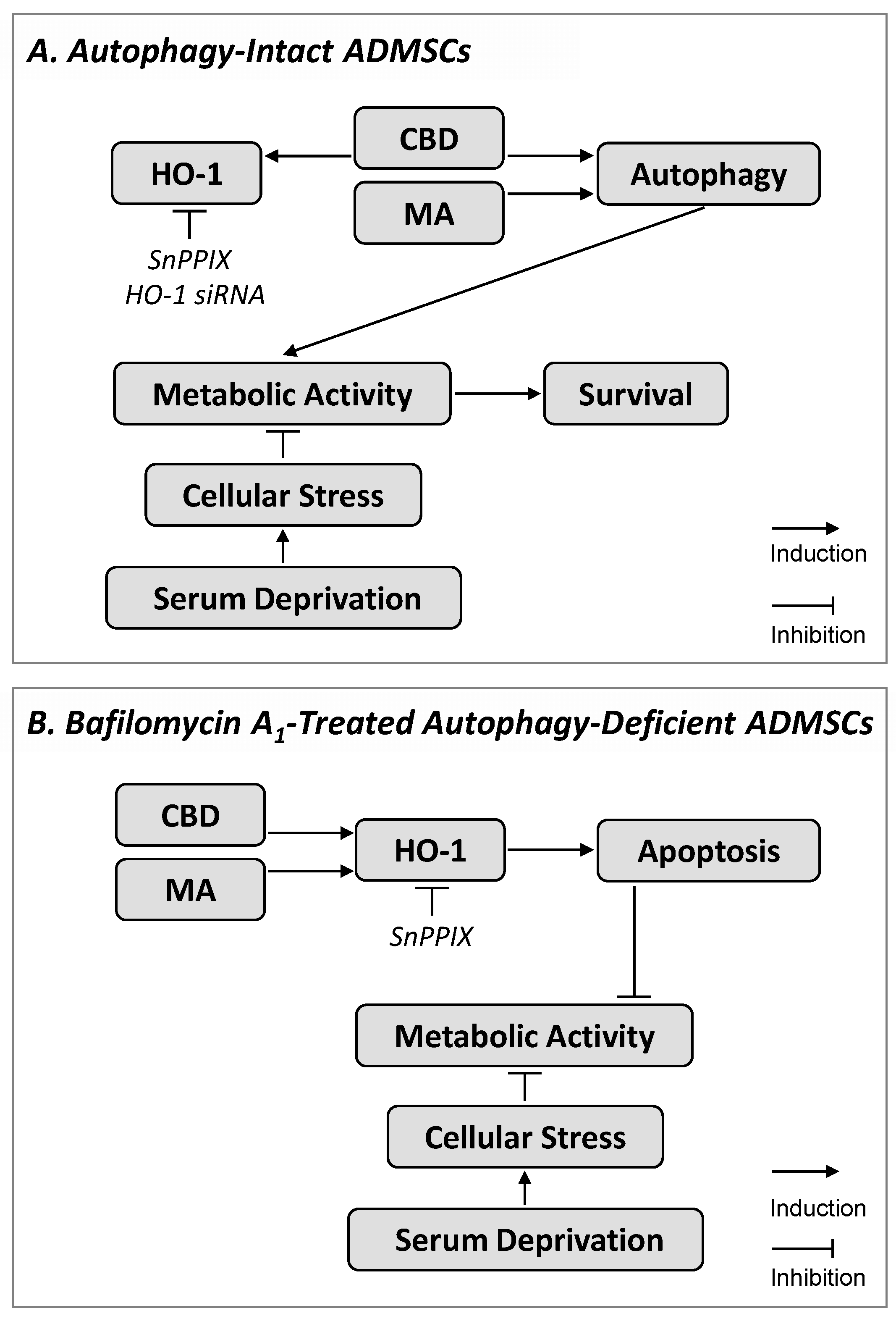
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef]
- Rehman, J.; Traktuev, D.; Li, J.; Merfeld-Clauss, S.; Temm-Grove, C.J.; Bovenkerk, J.E.; Pell, C.L.; Johnstone, B.H.; Considine, R.V.; March, K.L. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation 2004, 109, 1292–1298. [Google Scholar] [CrossRef]
- Deveza, L.; Choi, J.; Imanbayev, G.; Yang, F. Paracrine release from nonviral engineered adipose-derived stem cells promotes endothelial cell survival and migration in vitro. Stem Cells Dev. 2013, 22, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Ahn, A.; Frishman, W.H.; Gutwein, A.; Passeri, J.; Nelson, M. Therapeutic angiogenesis: A new treatment approach for ischemic heart disease–part I. Cardiol. Rev. 2008, 16, 163–171. [Google Scholar] [CrossRef]
- Boodhwani, M.; Sodha, N.R.; Mieno, S.; Xu, S.H.; Feng, J.; Ramlawi, B.; Clements, R.T.; Sellke, F.W. Functional, cellular, and molecular characterization of the angiogenic response to chronic myocardial ischemia in diabetes. Circulation 2007, 116, I31–I37. [Google Scholar] [CrossRef][Green Version]
- Tse, K.H.; Kingham, P.J.; Novikov, L.N.; Wiberg, M. Adipose tissue and bone marrow-derived stem cells react similarly in an ischaemia-like microenvironment. J. Tissue Eng. Regen. Med. 2012, 6, 473–485. [Google Scholar] [CrossRef]
- Pertwee, R.G. Elevating endocannabinoid levels: Pharmacological strategies and potential therapeutic applications. Proc. Nutr. Soc. 2014, 73, 96–105. [Google Scholar] [CrossRef]
- Hinz, B.; Ramer, R. Anti-tumour actions of cannabinoids. Br. J. Pharmacol. 2019, 176, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Durst, R.; Danenberg, H.; Gallily, R.; Mechoulam, R.; Meir, K.; Grad, E.; Beeri, R.; Pugatsch, T.; Tarsish, E.; Lotan, C. Cannabidiol, a nonpsychoactive Cannabis constituent, protects against myocardial ischemic reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3602–H3607. [Google Scholar] [CrossRef]
- Durst, R.; Lotan, C. The potential for clinical use of cannabinoids in treatment of cardiovascular diseases. Cardiovasc. Ther. 2011, 29, 17–22. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Chiurchiù, V.; Díaz-Alonso, J.; Bari, M.; Guzmán, M.; Maccarrone, M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lipid Res. 2013, 52, 633–650. [Google Scholar] [CrossRef]
- Schmuhl, E.; Ramer, R.; Salamon, A.; Peters, K.; Hinz, B. Increase of mesenchymal stem cell migration by cannabidiol via activation of p42/44 MAPK. Biochem. Pharmacol. 2014, 87, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Wollank, Y.; Ramer, R.; Ivanov, I.; Salamon, A.; Peters, K.; Hinz, B. Inhibition of FAAH confers increased stem cell migration via PPARα. J. Lipid Res. 2015, 56, 1947–1960. [Google Scholar] [CrossRef]
- Lüder, E.; Ramer, R.; Peters, K.; Hinz, B. Decisive role of P42/44 mitogen-activated protein kinase in Δ(9)-tetrahydrocannabinol-induced migration of human mesenchymal stem cells. Oncotarget 2017, 8, 105984–105994. [Google Scholar] [CrossRef]
- Agarwal, A.; Bolisetty, S. Adaptive responses to tissue injury: Role of heme oxygenase-1. Trans. Am. Clin. Climatol. Assoc. 2013, 124, 111–122. [Google Scholar]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef]
- Yang, J.J.; Yang, X.; Liu, Z.Q.; Hu, S.Y.; Du, Z.Y.; Feng, L.L.; Liu, J.F.; Chen, Y.D. Transplantation of adipose tissue-derived stem cells overexpressing heme oxygenase-1 improves functions and remodeling of infarcted myocardium in rabbits. Tohoku J. Exp. Med. 2012, 226, 231–241. [Google Scholar] [CrossRef]
- Chen, K.D.; Hsu, L.W.; Goto, S.; Huang, K.T.; Nakano, T.; Wenig, W.T.; Lai, C.Y.; Kuo, Y.R.; Chiu, K.W.; Wang, C.C.; et al. Regulation of heme oxygenase 1 expression by miR-27b with stem cell therapy for liver regeneration in rats. Transplant. Proc. 2014, 46, 1198–1200. [Google Scholar] [CrossRef]
- Cremers, N.A.; Lundvig, D.M.; van Dalen, S.C.; Schelbergen, R.F.; van Lent, P.L.; Szarek, W.A.; Regan, R.F.; Carels, C.E.; Wagener, F.A. Curcumin-induced heme oxygenase-1 expression prevents H2O2-induced cell death in wild type and heme oxygenase-2 knockout adipose-derived mesenchymal stem cells. Int. J. Mol. Sci. 2014, 15, 17974–17999. [Google Scholar] [CrossRef]
- Zeng, B.; Ren, X.; Lin, G.; Zhu, C.; Chen, H.; Yin, J.; Jiang, H.; Yang, B.; Ding, D. Paracrine action of HO-1-modified mesenchymal stem cells mediates cardiac protection and functional improvement. Cell. Biol. Int. 2008, 32, 1256–1264. [Google Scholar] [CrossRef]
- Carchman, E.H.; Rao, J.; Loughran, P.A.; Rosengart, M.R.; Zuckerbraun, B.S. Heme oxygenase-1-mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology 2011, 53, 2053–2062. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, L.; Qiao, Y.; Zhou, X.; Wu, G.; Wang, L.; Peng, Y.; Dong, X.; Huang, H.; Si, L.; et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS ONE 2013, 8, e75927. [Google Scholar] [CrossRef]
- Lin, T.K.; Chen, S.D.; Chuang, Y.C.; Lin, H.Y.; Huang, C.R.; Chuang, J.H.; Wang, P.W.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol partially prevents rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through induction of heme oxygenase-1 dependent autophagy. Int. J. Mol. Sci. 2014, 15, 1625–1646. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Salemi, S.; Yousefi, S.; Constantinescu, M.A.; Fey, M.F.; Simon, H.U. Autophagy is required for self-renewal and differentiation of adult human stem cells. Cell. Res. 2012, 22, 432–435. [Google Scholar] [CrossRef]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef]
- Tang, A.H.; Rando, T.A. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef]
- Yang, C.M.; Huang, Y.J.; Hsu, S.H. Enhanced Autophagy of Adipose-Derived Stem Cells Grown on Chitosan Substrates. Biores. Open Access 2015, 4, 89–96. [Google Scholar] [CrossRef]
- Tao, J.; Wang, H.; Zhai, Y.; Park, H.; Wang, J.; Ji, F.; Zhang, Z. Downregulation of Nrf2 promotes autophagy-dependent osteoblastic differentiation of adipose-derived mesenchymal stem cells. Exp. Cell. Res. 2016, 349, 221–229. [Google Scholar] [CrossRef]
- Jacobsson, S.O.; Wallin, T.; Fowler, C.J. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther. 2001, 299, 951–959. [Google Scholar]
- Mukherjee, S.; Adams, M.; Whiteaker, K.; Daza, A.; Kage, K.; Cassar, S.; Meyer, M.; Yao, B.B. Species comparison and pharmacological characterization of rat and human CB2 cannabinoid receptors. Eur. J. Pharmacol. 2004, 505, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Hinz, B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J. Natl. Cancer Inst. 2008, 100, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, M.; Li Volti, G.; Barbagallo, I.A.; Vanella, L. Therapeutic Efficacy of Stem Cells Transplantation in Diabetes: Role of Heme Oxygenase. Front. Cell. Dev. Biol. 2016, 4, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Cai, N.; Li, M.; Liu, G.H.; Izpisua Belmonte, J.C. Autophagic control of cell ‘stemness’. EMBO Mol. Med. 2013, 5, 327–331. [Google Scholar] [CrossRef]
- Oliver, L.; Hue, E.; Priault, M.; Vallette, F.M. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012, 21, 2779–2788. [Google Scholar] [CrossRef]
- Schwartz, M.; Böckmann, S.; Hinz, B. Up-regulation of heme oxygenase-1 expression and inhibition of disease-associated features by cannabidiol in vascular smooth muscle cells. Oncotarget 2018, 9, 34595–34616. [Google Scholar] [CrossRef]
- Böckmann, S.; Hinz, B. Cannabidiol Promotes Endothelial Cell Survival by Heme Oxygenase-1-Mediated Autophagy. Cells 2020, 9, 1703. [Google Scholar] [CrossRef]
- Oh, H.A.; Kwon, S.; Choi, S.; Shin, H.; Yoon, K.H.; Kim, W.J.; Lim, H.J. Uncovering a role for endocannabinoid signaling in autophagy in preimplantation mouse embryos. Mol. Human Repr. 2013, 19, 93–101. [Google Scholar] [CrossRef]
- Dodson, M.; Redmann, M.; Rajasekaran, N.S.; Darley-Usmar, V.; Zhang, J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem. J. 2015, 469, 347–355. [Google Scholar] [CrossRef]
- Waltz, P.; Carchman, E.H.; Young, A.C.; Rao, J.; Rosengart, M.R.; Kaczorowski, D.; Zuckerbraun, B.S. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 2011, 7, 315–320. [Google Scholar] [CrossRef]
- Chang, C.Y.; Chen, P.H.; Lu, S.C.; Hsieh, M.C.; Lin, C.W.; Lee, H.M.; Jawan, B.; Kao, Y.H. Propofol-enhanced autophagy increases motility and angiogenic capacity of cultured human umbilical vascular endothelial cells. Life Sci. 2015, 142, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Surolia, R.; Karkim, S.; Kim, H.; Yu, Z.; Kulkarni, T.; Mirov, S.B.; Carter, A.B.; Rowe, S.M.; Matalon, S.; Thannickal, V.J.; et al. Heme oxygenase-1-mediated autophagy protects against pulmonary endothelial cell death and development of emphysema in cadmium-treated mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L280–L292. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, X.; Jiang, L.; Liu, X.; Chen, M.; Yao, X.; Sun, Q.; Yang, G. 6-Gingerol induces autophagy to protect HUVECs survival from apoptosis. Chem. Biol. Interact. 2016, 256, 249–256. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Shen, C.; Zhang, X.; Sun, S.; Cho, M.; Sun, C.; Song, Z. tert-Butylhydroquinone (tBHQ) protects hepatocytes against lipotoxicity via inducing autophagy independently of Nrf2 activation. Biochim. Biophys. Acta. 2014, 1841, 22–33. [Google Scholar] [CrossRef]
- So, K.Y.; Oh, S.H. Heme oxygenase-1-mediated apoptosis under cadmium-induced oxidative stress is regulated by autophagy, which is sensitized by tumor suppressor p53. Biochem. Biophys. Res. Commun. 2016, 479, 80–85. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Y.J.; Wang, H.; Dong, Q.T.; Wang, T.J.; Qian, H.Y.; Xu, H. Autophagy activation: A novel mechanism of atorvastatin to protect mesenchymal stem cells from hypoxia and serum deprivation via AMP-activated protein kinase/mammalian target of rapamycin pathway. Stem Cells Dev. 2012, 21, 1321–1332. [Google Scholar] [CrossRef]
- Wagers, A.J. How stem cells get “turned on”. EMBO J. 2014, 33, 2743–2744. [Google Scholar] [CrossRef]
- Li, Q.; Yin, Y.; Zheng, Y.; Chen, F.; Jin, P. Inhibition of autophagy promoted high glucose/ROS-mediated apoptosis in ADSCs. Stem Cell Res. Ther. 2018, 9, 289–300. [Google Scholar] [CrossRef]
- Sardana, M.K.; Kappas, A. Dual control mechanism for heme oxygenase: Tin(IV)-protoporphyrin potently inhibits enzyme activity while markedly increasing content of enzyme protein in liver. Proc. Natl. Acad. Sci. USA. 1987, 84, 2464–2468. [Google Scholar] [CrossRef]
- Chang, T.; Wu, L.; Wang, R. Inhibition of vascular smooth muscle cell proliferation by chronic hemin treatment. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H999–H1007. [Google Scholar] [CrossRef]
- Hsu, F.F.; Yeh, C.T.; Sun, Y.J.; Chiang, M.T.; Lan, W.M.; Li, F.A.; Lee, W.H.; Chau, L.Y. Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene 2015, 34, 2360–2370. [Google Scholar] [CrossRef]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; Di Raimondo, F.; Li Volti, G. Heme oxygenase-1 nuclear translocation regulates bortezomib-induced cytotoxicity and mediates genomic instability in myeloma cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Shah, N.; Muthu, M.; La, P.; Fernando, A.P.; Sengupta, S.; Yang, G.; Dennery, P.A. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J. Biol. Chem. 2014, 289, 26882–26894. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell. Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell. Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhao, J.; Xue, J.; Zhao, X.; Liu, P. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am. J. Cancer Res. 2016, 6, 2162–2177. [Google Scholar]
- Harada, S.; Nakagawa, T.; Yokoe, S.; Edogawa, S.; Takeuchi, T.; Inoue, T.; Higuchi, K.; Asahi, M. Autophagy Deficiency Diminishes Indomethacin-Induced Intestinal Epithelial Cell Damage through Activation of the ERK/Nrf2/HO-1 Pathway. J. Pharmacol. Exp. Ther. 2015, 355, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Por, E.D.; Kwon, Y.G.; Kim, Y.M. Regulation of ROS production and vascular function by carbon monoxide. Oxid. Med. Cell. Longev. 2012, 2012, 794237–794254. [Google Scholar] [CrossRef]
- Bansal, S.; Biswas, G.; Avadhani, N.G. Mitochondria-targeted heme oxygenase-1 induces oxidative stress and mitochondrial dysfunction in macrophages, kidney fibroblasts and in chronic alcohol hepatotoxicity. Redox Biol. 2013, 2, 273–283. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Varadharaj, S.; Khanderao, G.D.; Davidson, C.J.; Kannan, S.; Firpo, M.A.; Zweier, J.L.; Benjamin, I.J. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid. Redox Signal. 2011, 14, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260–2280. [Google Scholar] [CrossRef] [PubMed]
- Pan, A.; Weintraub, N.L.; Tang, Y. Enhancing stem cell survival in an ischemic heart by CRISPR-dCas9-based gene regulation. Med. Hypotheses 2014, 83, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. Biotechnol. Annu. Rev. 2005, 11, 127–152. [Google Scholar]
- Kimura, T.; Takahashi, A.; Takabatake, Y.; Namba, T.; Yamamoto, T.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; Niimura, F.; Matsusaka, T.; et al. Autophagy protects kidney proximal tubule epithelial cells from mitochondrial metabolic stress. Autophagy 2013, 9, 1876–1886. [Google Scholar] [CrossRef]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef]
- Sun, S.; Hu, F.; Wu, J.; Zhang, S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox Biol. 2017, 11, 577–585. [Google Scholar] [CrossRef]
- Hao, E.; Mukhopadhyay, P.; Cao, Z.; Erdélyi, K.; Holovac, E.; Liaudet, L.; Lee, W.S.; Haskó, G.; Mechoulam, R.; Pacher, P. Cannabidiol Protects against Doxorubicin-Induced Cardiomyopathy by Modulating Mitochondrial Function and Biogenesis. Mol. Med. 2015, 21, 38–45. [Google Scholar] [CrossRef]
- Tan, Q.; Wang, H.; Hu, Y.; Hu, M.; Li, X.; Aodengqimuge; Ma, Y.; Wei, C.; Song, L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015, 106, 1023–1032. [Google Scholar] [CrossRef]
- Pei, L.; Kong, Y.; Shao, C.; Yue, X.; Wang, Z.; Zhang, N. Heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to pharmorubicin by promoting autophagy via PI3K/Akt pathway. J. Cell. Mol. Med. 2018, 22, 5311–5321. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Groups | CB Corresponds to CBD (3 µM) | CB Corresponds to MA (3 µM) | ||
|---|---|---|---|---|
| Metabolic Activity (%) | HO-1 Protein (%) | HO-2 Protein (%) | Metabolic Activity (%) | |
| Vehicle | 100 ± 35 | 100 ± 9 | 100 ± 4 | 100 ± 8 |
| CB | 202 ± 50 | 515 ± 30 * | 109 ± 8 | 205 ± 23 * |
| CB + AM-630 | 205 ± 40 | 624 ± 108 | 102 ± 3 | 214 ± 26 |
| CB + AM-251 | 242 ± 83 | 488 ± 75 | 89 ± 9 | 237 ± 29 |
| CB + AM-630 + AM-251 | 220 ± 83 | 499 ± 91 | 89 ± 10 | 217 ± 23 |
| CB + Capsa | 211 ± 59 | 615 ± 122 | 89 ± 8 | 242 ± 18 |
| Vehicle | 100 ± 35 | 100 ± 18 | 100 ± 6 | 100 ± 26 |
| AM-630 | 88 ± 6 | 127 ± 19 | 125 ± 9 | 87 ± 4 |
| AM-251 | 125 ± 15 | 216 ± 75 | 123 ± 6 | 110 ± 9 |
| AM-630 + AM-251 | 144 ± 19 | 218 ± 88 | 126 ± 19 | 120 ± 3 |
| Capsa | 93 ± 3 | 124 ± 13 | 122 ± 18 | 108 ± 5 |
| Treatment Groups | Metabolic Activity (%) | HO-1 Protein (%) |
|---|---|---|
| Vehicle | 100 ± 29 | 100 ± 16 |
| 3 µM CBD | 174 ± 32 | 277 ± 64 |
| CBD + O-1602 | 188 ± 33 | 255 ± 61 |
| O-1602 | 106 ± 10 | 103 ± 21 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bublitz, K.; Böckmann, S.; Peters, K.; Hinz, B. Cannabinoid-Induced Autophagy and Heme Oxygenase-1 Determine the Fate of Adipose Tissue-Derived Mesenchymal Stem Cells under Stressful Conditions. Cells 2020, 9, 2298. https://doi.org/10.3390/cells9102298
Bublitz K, Böckmann S, Peters K, Hinz B. Cannabinoid-Induced Autophagy and Heme Oxygenase-1 Determine the Fate of Adipose Tissue-Derived Mesenchymal Stem Cells under Stressful Conditions. Cells. 2020; 9(10):2298. https://doi.org/10.3390/cells9102298
Chicago/Turabian StyleBublitz, Katharina, Sabine Böckmann, Kirsten Peters, and Burkhard Hinz. 2020. "Cannabinoid-Induced Autophagy and Heme Oxygenase-1 Determine the Fate of Adipose Tissue-Derived Mesenchymal Stem Cells under Stressful Conditions" Cells 9, no. 10: 2298. https://doi.org/10.3390/cells9102298
APA StyleBublitz, K., Böckmann, S., Peters, K., & Hinz, B. (2020). Cannabinoid-Induced Autophagy and Heme Oxygenase-1 Determine the Fate of Adipose Tissue-Derived Mesenchymal Stem Cells under Stressful Conditions. Cells, 9(10), 2298. https://doi.org/10.3390/cells9102298