Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies


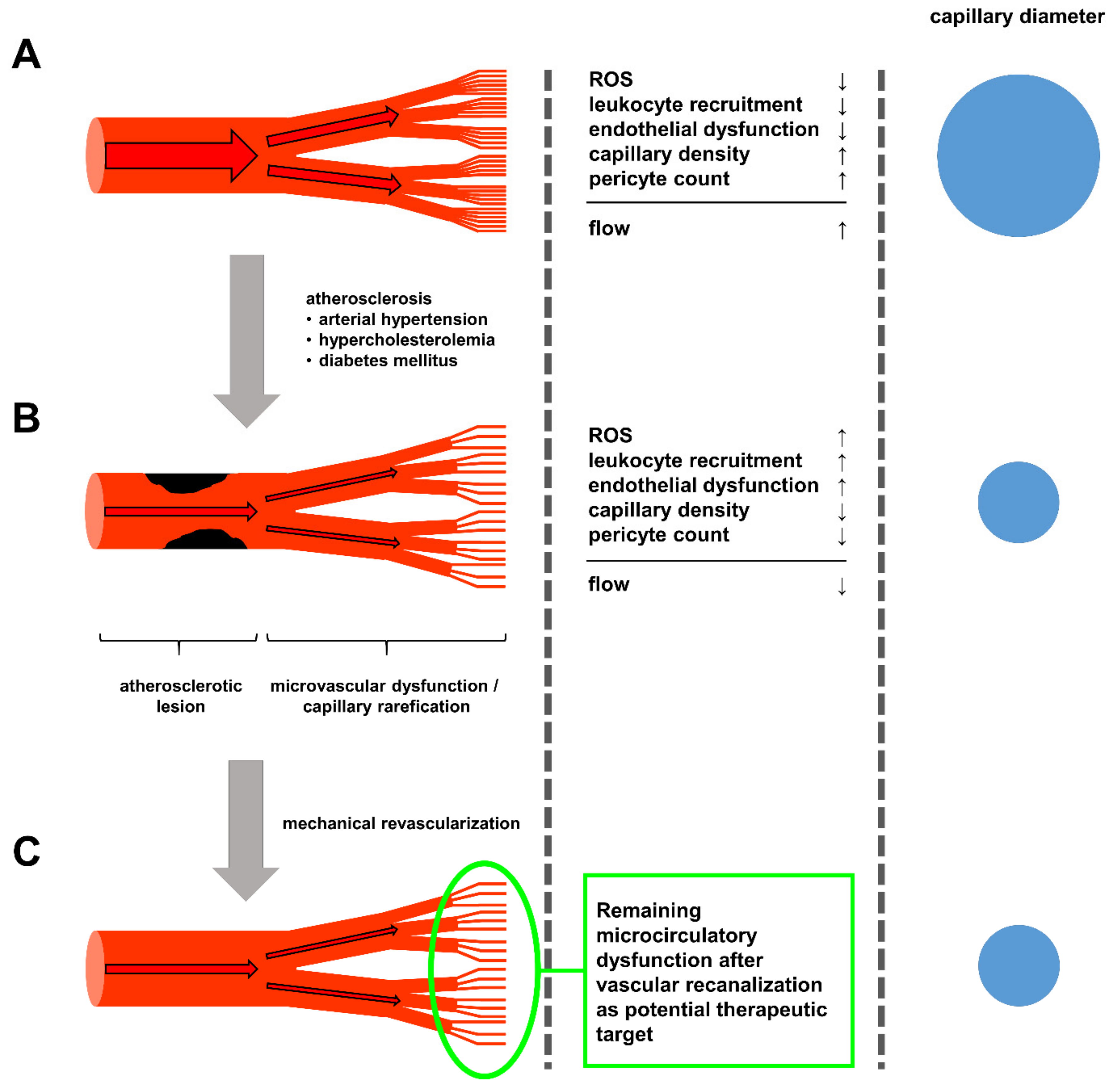{kind=link}
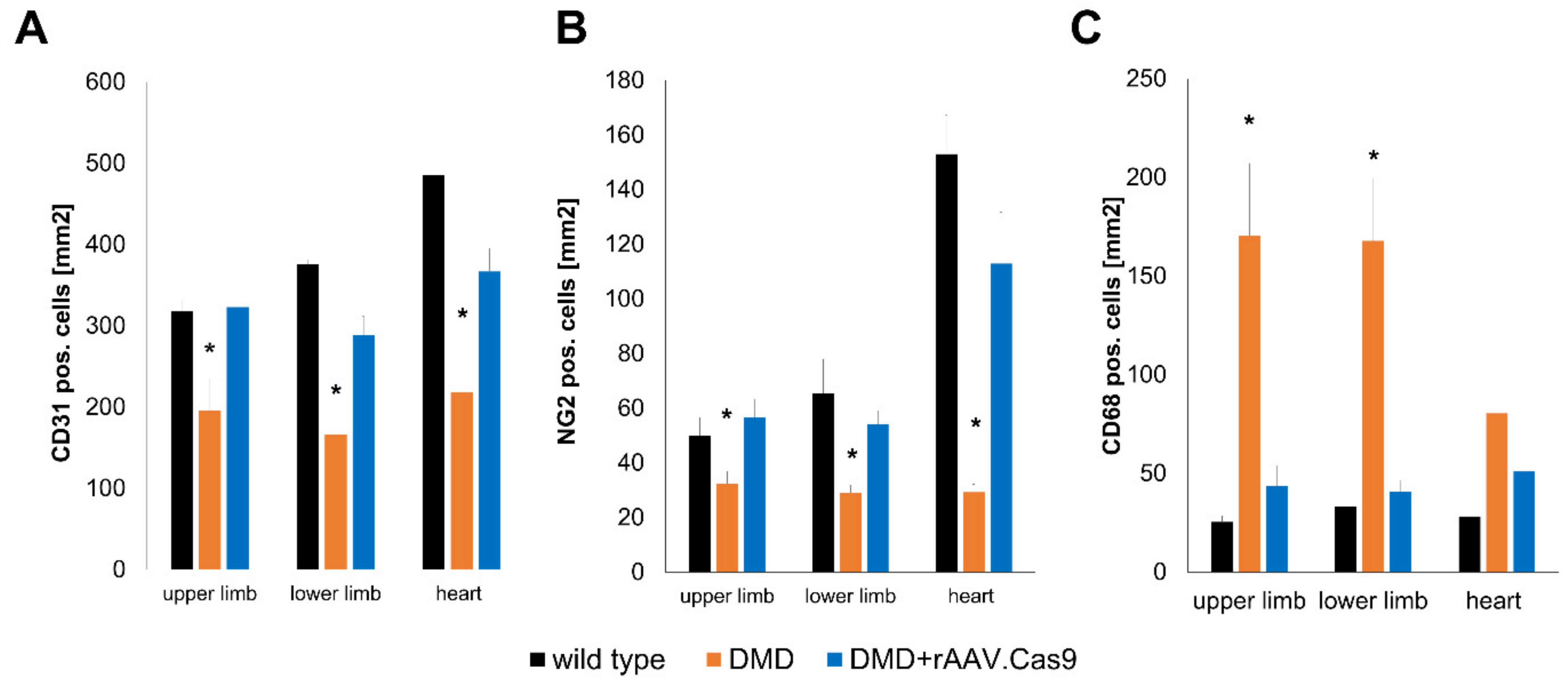{kind=link}
Abstract
1. Introduction
2. Cardiovascular Risk Factors Contributing to Atherosclerosis: The Macro
3. Hypertension, Hypercholesterolemia and Diabetes Mellitus in Capillaries: The Micro
4. Current and Future Treatment Strategies
5. Conclusions
Funding
Conflicts of Interest
References
- World Health Organization. Cardiovascular Disease (CVDs); World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Nakashima, Y.; Chen, Y.X.; Kinukawa, N.; Sueishi, K. Distributions of diffuse intimal thickening in human arteries: Preferential expression in atherosclerosis-prone arteries from an early age. Virchows Arch. 2002, 441, 279–288. [Google Scholar] [CrossRef]
- Guyton, J.R.; Bocan, T.M.; Schifani, T.A. Quantitative ultrastructural analysis of perifibrous lipid and its association with elastin in nonatherosclerotic human aorta. Arteriosclerosis 1985, 5, 644–652. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef]
- Lee, R.T.; Yamamoto, C.; Feng, Y.; Potter-Perigo, S.; Briggs, W.H.; Landschulz, K.T.; Turi, T.G.; Thompson, J.F.; Libby, P.; Wight, T.N. Mechanical strain induces specific changes in the synthesis and organization of proteoglycans by vascular smooth muscle cells. J. Biol. Chem. 2001, 276, 13847–13851. [Google Scholar] [CrossRef]
- Little, P.J.; Tannock, L.; Olin, K.L.; Chait, A.; Wight, T.N. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 55–60. [Google Scholar] [CrossRef]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef]
- Hurt-Camejo, E.; Camejo, G.; Rosengren, B.; Lopez, F.; Ahlstrom, C.; Fager, G.; Bondjers, G. Effect of arterial proteoglycans and glycosaminoglycans on low density lipoprotein oxidation and its uptake by human macrophages and arterial smooth muscle cells. Arterioscler. Thromb. 1992, 12, 569–583. [Google Scholar] [CrossRef]
- Karakikes, I.; Chaanine, A.H.; Kang, S.; Mukete, B.N.; Jeong, D.; Zhang, S.; Hajjar, R.J.; Lebeche, D. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J. Am. Heart Assoc. 2013, 2, e000078. [Google Scholar] [CrossRef]
- Kockx, M.M.; De Meyer, G.R.; Muhring, J.; Jacob, W.; Bult, H.; Herman, A.G. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation 1998, 97, 2307–2315. [Google Scholar] [CrossRef]
- Merrilees, M.J.; Beaumont, B. Structural heterogeneity of the diffuse intimal thickening and correlation with distribution of TGF-beta 1. J. Vasc. Res. 1993, 30, 293–302. [Google Scholar] [CrossRef]
- Murry, C.E.; Gipaya, C.T.; Bartosek, T.; Benditt, E.P.; Schwartz, S.M. Monoclonality of smooth muscle cells in human atherosclerosis. Am. J. Pathol. 1997, 151, 697–705. [Google Scholar]
- Nakata, A.; Miyagawa, J.; Yamashita, S.; Nishida, M.; Tamura, R.; Yamamori, K.; Nakamura, T.; Nozaki, S.; Kameda-Takemura, K.; Kawata, S.; et al. Localization of heparin-binding epidermal growth factor-like growth factor in human coronary arteries. Possible roles of HB-EGF in the formation of coronary atherosclerosis. Circulation 1996, 94, 2778–2786. [Google Scholar] [CrossRef]
- Alshehri, O.M.; Hughes, C.E.; Montague, S.; Watson, S.K.; Frampton, J.; Bender, M.; Watson, S.P. Fibrin activates GPVI in human and mouse platelets. Blood 2015, 126, 1601–1608. [Google Scholar] [CrossRef]
- Naimushin, Y.A.; Mazurov, A.V. Von Willebrand factor can support platelet aggregation via interaction with activated GPIIb-IIIa and GPIb. Platelets 2004, 15, 419–425. [Google Scholar] [CrossRef]
- Sarratt, K.L.; Chen, H.; Zutter, M.M.; Santoro, S.A.; Hammer, D.A.; Kahn, M.L. GPVI and alpha2beta1 play independent critical roles during platelet adhesion and aggregate formation to collagen under flow. Blood 2005, 106, 1268–1277. [Google Scholar] [CrossRef]
- Cassese, S.; Byrne, R.A.; Tada, T.; Pinieck, S.; Joner, M.; Ibrahim, T.; King, L.A.; Fusaro, M.; Laugwitz, K.L.; Kastrati, A. Incidence and predictors of restenosis after coronary stenting in 10 004 patients with surveillance angiography. Heart 2014, 100, 153–159. [Google Scholar] [CrossRef]
- Gray, W.A.; Keirse, K.; Soga, Y.; Benko, A.; Babaev, A.; Yokoi, Y.; Schroeder, H.; Prem, J.T.; Holden, A.; Popma, J.; et al. A polymer-coated, paclitaxel-eluting stent (Eluvia) versus a polymer-free, paclitaxel-coated stent (Zilver PTX) for endovascular femoropopliteal intervention (IMPERIAL): A randomised, non-inferiority trial. Lancet 2018, 392, 1541–1551. [Google Scholar] [CrossRef]
- Sander, M.; Chavoshan, B.; Victor, R.G. A large blood pressure-raising effect of nitric oxide synthase inhibition in humans. Hypertension 1999, 33, 937–942. [Google Scholar] [CrossRef]
- Bush, E.; Maeda, N.; Kuziel, W.A.; Dawson, T.C.; Wilcox, J.N.; DeLeon, H.; Taylor, W.R. CC chemokine receptor 2 is required for macrophage infiltration and vascular hypertrophy in angiotensin II-induced hypertension. Hypertension 2000, 36, 360–363. [Google Scholar] [CrossRef]
- Ryan, M.J.; Didion, S.P.; Mathur, S.; Faraci, F.M.; Sigmund, C.D. Angiotensin II-induced vascular dysfunction is mediated by the AT1A receptor in mice. Hypertension 2004, 43, 1074–1079. [Google Scholar] [CrossRef]
- Wang, H.D.; Xu, S.; Johns, D.G.; Du, Y.; Quinn, M.T.; Cayatte, A.J.; Cohen, R.A. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ. Res. 2001, 88, 947–953. [Google Scholar] [CrossRef]
- World Health Organization. Global Health Observatory; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Landsberg, L.; Aronne, L.J.; Beilin, L.J.; Burke, V.; Igel, L.I.; Lloyd-Jones, D.; Sowers, J. Obesity-related hypertension: Pathogenesis, cardiovascular risk, and treatment—A position paper of the The Obesity Society and The American Society of Hypertension. Obesity 2013, 21, 8–24. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K. Lifestyle effects on hematopoiesis and atherosclerosis. Circ. Res. 2015, 116, 884–894. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 2013, 22, 9–15. [Google Scholar] [CrossRef]
- Fogelstrand, P.; Boren, J. Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 1–7. [Google Scholar] [CrossRef]
- Iverius, P.H. The interaction between human plasma lipoproteins and connective tissue glycosaminoglycans. J. Biol. Chem. 1972, 247, 2607–2613. [Google Scholar]
- Flood, C.; Gustafsson, M.; Pitas, R.E.; Arnaboldi, L.; Walzem, R.L.; Boren, J. Molecular mechanism for changes in proteoglycan binding on compositional changes of the core and the surface of low-density lipoprotein-containing human apolipoprotein B100. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 564–570. [Google Scholar] [CrossRef]
- Ungvari, Z.; Wolin, M.S.; Csiszar, A. Mechanosensitive production of reactive oxygen species in endothelial and smooth muscle cells: Role in microvascular remodeling? Antioxid. Redox Signal. 2006, 8, 1121–1129. [Google Scholar] [CrossRef]
- Krieger, M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J. Clin. Investig. 2001, 108, 793–797. [Google Scholar] [CrossRef]
- Lara-Guzman, O.J.; Gil-Izquierdo, A.; Medina, S.; Osorio, E.; Alvarez-Quintero, R.; Zuluaga, N.; Oger, C.; Galano, J.M.; Durand, T.; Munoz-Durango, K. Oxidized LDL triggers changes in oxidative stress and inflammatory biomarkers in human macrophages. Redox Biol. 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Khan, B.V.; Parthasarathy, S.S.; Alexander, R.W.; Medford, R.M. Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J. Clin. Investig. 1995, 95, 1262–1270. [Google Scholar] [CrossRef]
- Vora, D.K.; Fang, Z.T.; Liva, S.M.; Tyner, T.R.; Parhami, F.; Watson, A.D.; Drake, T.A.; Territo, M.C.; Berliner, J.A. Induction of P-selectin by oxidized lipoproteins. Separate effects on synthesis and surface expression. Circ. Res. 1997, 80, 810–818. [Google Scholar] [CrossRef]
- Berliner, J.A.; Schwartz, D.S.; Territo, M.C.; Andalibi, A.; Almada, L.; Lusis, A.J.; Quismorio, D.; Fang, Z.P.; Fogelman, A.M. Induction of chemotactic cytokines by minimally oxidized LDL. Adv. Exp. Med. Biol. 1993, 351, 13–18. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Basta, G.; Schmidt, A.M.; De Caterina, R. Advanced glycation end products and vascular inflammation: Implications for accelerated atherosclerosis in diabetes. Cardiovasc. Res. 2004, 63, 582–592. [Google Scholar] [CrossRef]
- Neumann, A.; Schinzel, R.; Palm, D.; Riederer, P.; Munch, G. High molecular weight hyaluronic acid inhibits advanced glycation endproduct-induced NF-kappaB activation and cytokine expression. FEBS Lett. 1999, 453, 283–287. [Google Scholar] [CrossRef]
- Iwashima, Y.; Eto, M.; Hata, A.; Kaku, K.; Horiuchi, S.; Ushikubi, F.; Sano, H. Advanced glycation end products-induced gene expression of scavenger receptors in cultured human monocyte-derived macrophages. Biochem. Biophys. Res. Commun. 2000, 277, 368–380. [Google Scholar] [CrossRef]
- Ziegler, T.; Horstkotte, M.; Lange, P.; Ng, J.; Bongiovanni, D.; Hinkel, R.; Laugwitz, K.L.; Sperandio, M.; Horstkotte, J.; Kupatt, C. Endothelial RAGE exacerbates acute postischaemic cardiac inflammation. Thromb. Haemost. 2016, 116, 300–308. [Google Scholar] [CrossRef]
- Natarajan, R.; Gerrity, R.G.; Gu, J.L.; Lanting, L.; Thomas, L.; Nadler, J.L. Role of 12-lipoxygenase and oxidant stress in hyperglycaemia-induced acceleration of atherosclerosis in a diabetic pig model. Diabetologia 2002, 45, 125–133. [Google Scholar] [CrossRef]
- Wu, L.; Vikramadithyan, R.; Yu, S.; Pau, C.; Hu, Y.; Goldberg, I.J.; Dansky, H.M. Addition of dietary fat to cholesterol in the diets of LDL receptor knockout mice: Effects on plasma insulin, lipoproteins, and atherosclerosis. J. Lipid Res. 2006, 47, 2215–2222. [Google Scholar] [CrossRef]
- Ho, K.L. Ultrastructure of cerebellar capillary hemangioblastoma. IV. Pericytes and their relationship to endothelial cells. Acta Neuropathol. 1985, 67, 254–264. [Google Scholar] [CrossRef]
- Larson, D.M.; Carson, M.P.; Haudenschild, C.C. Junctional transfer of small molecules in cultured bovine brain microvascular endothelial cells and pericytes. Microvasc. Res. 1987, 34, 184–199. [Google Scholar] [CrossRef]
- Rucker, H.K.; Wynder, H.J.; Thomas, W.E. Cellular mechanisms of CNS pericytes. Brain Res. Bull. 2000, 51, 363–369. [Google Scholar] [CrossRef]
- Davis, S.; Aldrich, T.H.; Jones, P.F.; Acheson, A.; Compton, D.L.; Jain, V.; Ryan, T.E.; Bruno, J.; Radziejewski, C.; Maisonpierre, P.C.; et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 1996, 87, 1161–1169. [Google Scholar] [CrossRef]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Fulton, D.; Mahboubi, K.; Kalb, R.G.; O’Connor, D.S.; Li, F.; Altieri, D.C.; Sessa, W.C. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J. Biol. Chem. 2000, 275, 9102–9105. [Google Scholar] [CrossRef]
- Kobayashi, H.; DeBusk, L.M.; Babichev, Y.O.; Dumont, D.J.; Lin, P.C. Hepatocyte growth factor mediates angiopoietin-induced smooth muscle cell recruitment. Blood 2006, 108, 1260–1266. [Google Scholar] [CrossRef]
- Iivanainen, E.; Nelimarkka, L.; Elenius, V.; Heikkinen, S.M.; Junttila, T.T.; Sihombing, L.; Sundvall, M.; Maatta, J.A.; Laine, V.J.; Yla-Herttuala, S.; et al. Angiopoietin-regulated recruitment of vascular smooth muscle cells by endothelial-derived heparin binding EGF-like growth factor. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1609–1621. [Google Scholar] [CrossRef]
- Orlidge, A.; D’Amore, P.A. Inhibition of capillary endothelial cell growth by pericytes and smooth muscle cells. J. Cell Biol. 1987, 105, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Bonewald, L.F.; Park-Snyder, S.; Park, I.S.; Woodruff, K.A.; Abboud, H.E. Characterization and regulation of the latent transforming growth factor-beta complex secreted by vascular pericytes. J. Cell. Physiol. 1996, 166, 537–546. [Google Scholar] [CrossRef]
- Watanabe, S.; Morisaki, N.; Tezuka, M.; Fukuda, K.; Ueda, S.; Koyama, N.; Yokote, K.; Kanzaki, T.; Yoshida, S.; Saito, Y. Cultured retinal pericytes stimulate in vitro angiogenesis of endothelial cells through secretion of a fibroblast growth factor-like molecule. Atherosclerosis 1997, 130, 101–107. [Google Scholar] [CrossRef]
- Takagi, H.; King, G.L.; Robinson, G.S.; Ferrara, N.; Aiello, L.P. Adenosine mediates hypoxic induction of vascular endothelial growth factor in retinal pericytes and endothelial cells. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2165–2176. [Google Scholar]
- Austin, K.M.; Nguyen, N.; Javid, G.; Covic, L.; Kuliopulos, A. Noncanonical matrix metalloprotease-1-protease-activated receptor-1 signaling triggers vascular smooth muscle cell dedifferentiation and arterial stenosis. J. Biol. Chem. 2013, 288, 23105–23115. [Google Scholar] [CrossRef] [PubMed]
- Pfister, F.; Feng, Y.; vom Hagen, F.; Hoffmann, S.; Molema, G.; Hillebrands, J.L.; Shani, M.; Deutsch, U.; Hammes, H.P. Pericyte migration: A novel mechanism of pericyte loss in experimental diabetic retinopathy. Diabetes 2008, 57, 2495–2502. [Google Scholar] [CrossRef]
- Zehendner, C.M.; Wedler, H.E.; Luhmann, H.J. A novel in vitro model to study pericytes in the neurovascular unit of the developing cortex. PLoS ONE 2013, 8, e81637. [Google Scholar] [CrossRef]
- Ziegler, T.; Horstkotte, J.; Schwab, C.; Pfetsch, V.; Weinmann, K.; Dietzel, S.; Rohwedder, I.; Hinkel, R.; Gross, L.; Lee, S.; et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J. Clin. Investig. 2013, 123, 3436–3445. [Google Scholar] [CrossRef]
- Herman, I.M.; Jacobson, S. In situ analysis of microvascular pericytes in hypertensive rat brains. Tissue Cell 1988, 20, 1–12. [Google Scholar] [CrossRef]
- Wallow, I.H.; Bindley, C.D.; Reboussin, D.M.; Gange, S.J.; Fisher, M.R. Systemic hypertension produces pericyte changes in retinal capillaries. Investig. Ophthalmol. Vis. Sci. 1993, 34, 420–430. [Google Scholar]
- Ricard, N.; Tu, L.; Le Hiress, M.; Huertas, A.; Phan, C.; Thuillet, R.; Sattler, C.; Fadel, E.; Seferian, A.; Montani, D.; et al. Increased pericyte coverage mediated by endothelial-derived fibroblast growth factor-2 and interleukin-6 is a source of smooth muscle-like cells in pulmonary hypertension. Circulation 2014, 129, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.I.; Prewitt, R.L.; Dowell, R.F. Microvascular rarefaction in spontaneously hypertensive rat cremaster muscle. Am. J. Physiol. 1981, 241, H306–H310. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Daskalakis, C.; Falkner, B. Capillary rarefaction in treated and untreated hypertensive subjects. Ther. Adv. Cardiovasc. Dis. 2008, 2, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Wolfle, S.E.; de Wit, C. Intact endothelium-dependent dilation and conducted responses in resistance vessels of hypercholesterolemic mice in vivo. J. Vasc. Res. 2005, 42, 475–482. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Bayazeed, B.; Hook, G.; Johnson, A.; Cronin, J.; Baron, A.D. Endothelial dysfunction is associated with cholesterol levels in the high normal range in humans. Circulation 1997, 96, 3287–3293. [Google Scholar] [CrossRef]
- Ha, J.M.; Jin, S.Y.; Lee, H.S.; Shin, H.K.; Lee, D.H.; Song, S.H.; Kim, C.D.; Bae, S.S. Regulation of retinal angiogenesis by endothelial nitric oxide synthase signaling pathway. Korean J. Physiol. Pharmacol. 2016, 20, 533–538. [Google Scholar] [CrossRef]
- Garcia-Quintans, N.; Sanchez-Ramos, C.; Prieto, I.; Tierrez, A.; Arza, E.; Alfranca, A.; Redondo, J.M.; Monsalve, M. Oxidative stress induces loss of pericyte coverage and vascular instability in PGC-1alpha-deficient mice. Angiogenesis 2016, 19, 217–228. [Google Scholar] [CrossRef]
- Radice, G.L. N-cadherin-mediated adhesion and signaling from development to disease: Lessons from mice. Prog. Mol. Biol. Transl. Sci. 2013, 116, 263–289. [Google Scholar] [CrossRef]
- Zechariah, A.; ElAli, A.; Hagemann, N.; Jin, F.; Doeppner, T.R.; Helfrich, I.; Mies, G.; Hermann, D.M. Hyperlipidemia attenuates vascular endothelial growth factor-induced angiogenesis, impairs cerebral blood flow, and disturbs stroke recovery via decreased pericyte coverage of brain endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1561–1567. [Google Scholar] [CrossRef]
- Beltramo, E.; Porta, M. Pericyte loss in diabetic retinopathy: Mechanisms and consequences. Curr. Med. Chem. 2013, 20, 3218–3225. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, S.Y.; Park, J.; Kim, Y.S.; Hwang, E.M.; Park, J.Y.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; et al. Reduction of experimental diabetic vascular leakage and pericyte apoptosis in mice by delivery of alphaA-crystallin with a recombinant adenovirus. Diabetologia 2012, 55, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Hinkel, R.; Howe, A.; Renner, S.; Ng, J.; Lee, S.; Klett, K.; Kaczmarek, V.; Moretti, A.; Laugwitz, K.L.; Skroblin, P.; et al. Diabetes Mellitus-Induced Microvascular Destabilization in the Myocardium. J. Am. Coll. Cardiol. 2017, 69, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Takeuchi, M.; Matsui, T.; Nakamura, K.; Imaizumi, T.; Inoue, H. Angiotensin II augments advanced glycation end product-induced pericyte apoptosis through RAGE overexpression. FEBS Lett. 2005, 579, 4265–4270. [Google Scholar] [CrossRef] [PubMed]
- Fiordaliso, F.; Cuccovillo, I.; Bianchi, R.; Bai, A.; Doni, M.; Salio, M.; De Angelis, N.; Ghezzi, P.; Latini, R.; Masson, S. Cardiovascular oxidative stress is reduced by an ACE inhibitor in a rat model of streptozotocin-induced diabetes. Life Sci. 2006, 79, 121–129. [Google Scholar] [CrossRef]
- Lupo, E.; Locher, R.; Weisser, B.; Vetter, W. In vitro antioxidant activity of calcium antagonists against LDL oxidation compared with alpha-tocopherol. Biochem. Biophys. Res. Commun. 1994, 203, 1803–1808. [Google Scholar] [CrossRef]
- Algire, C.; Moiseeva, O.; Deschenes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef]
- Sabatine, M.S.; Leiter, L.A.; Wiviott, S.D.; Giugliano, R.P.; Deedwania, P.; De Ferrari, G.M.; Murphy, S.A.; Kuder, J.F.; Gouni-Berthold, I.; Lewis, B.S.; et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: A prespecified analysis of the FOURIER randomised controlled trial. Lancet Diabetes Endocrinol. 2017, 5, 941–950. [Google Scholar] [CrossRef]
- Inoue, I.; Goto, S.; Mizotani, K.; Awata, T.; Mastunaga, T.; Kawai, S.; Nakajima, T.; Hokari, S.; Komoda, T.; Katayama, S. Lipophilic HMG-CoA reductase inhibitor has an anti-inflammatory effect: Reduction of MRNA levels for interleukin-1beta, interleukin-6, cyclooxygenase-2, and p22phox by regulation of peroxisome proliferator-activated receptor alpha (PPARalpha) in primary endothelial cells. Life Sci. 2000, 67, 863–876. [Google Scholar] [CrossRef]
- Bouitbir, J.; Charles, A.L.; Echaniz-Laguna, A.; Kindo, M.; Daussin, F.; Auwerx, J.; Piquard, F.; Geny, B.; Zoll, J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: A ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur. Heart J. 2012, 33, 1397–1407. [Google Scholar] [CrossRef]
- Raiteri, M.; Arnaboldi, L.; McGeady, P.; Gelb, M.H.; Verri, D.; Tagliabue, C.; Quarato, P.; Ferraboschi, P.; Santaniello, E.; Paoletti, R.; et al. Pharmacological control of the mevalonate pathway: Effect on arterial smooth muscle cell proliferation. J. Pharmcol. Exp. Ther. 1997, 281, 1144–1153. [Google Scholar]
- Tokuda, T.; Hirano, K.; Sakamoto, Y.; Takimura, H.; Kobayashi, N.; Araki, M.; Yamawaki, M.; Ito, Y. Incidence and clinical outcomes of the slow-flow phenomenon after infrapopliteal balloon angioplasty. J. Vasc. Surg. 2017, 65, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.W.; Aggarwal, A.; Ou, F.S.; Klein, L.W.; Rumsfeld, J.S.; Roe, M.T.; Wang, T.Y.; American College of Cardiology National Cardiovascular Data, Registry. Incidence and outcomes of no-reflow phenomenon during percutaneous coronary intervention among patients with acute myocardial infarction. Am. J. Cardiol. 2013, 111, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Hayashi, K. G-actin sequestering protein thymosin-beta4 regulates the activity of myocardin-related transcription factor. Biochem. Biophys. Res. Commun. 2013, 437, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Hinkel, R.; Trenkwalder, T.; Petersen, B.; Husada, W.; Gesenhues, F.; Lee, S.; Hannappel, E.; Bock-Marquette, I.; Theisen, D.; Leitner, L.; et al. MRTF-A controls vessel growth and maturation by increasing the expression of CCN1 and CCN2. Nat. Commun. 2014, 5, 3970. [Google Scholar] [CrossRef]
- Hall-Glenn, F.; De Young, R.A.; Huang, B.L.; van Handel, B.; Hofmann, J.J.; Chen, T.T.; Choi, A.; Ong, J.R.; Benya, P.D.; Mikkola, H.; et al. CCN2/connective tissue growth factor is essential for pericyte adhesion and endothelial basement membrane formation during angiogenesis. PLoS ONE 2012, 7, e30562. [Google Scholar] [CrossRef]
- Hanna, M.; Liu, H.; Amir, J.; Sun, Y.; Morris, S.W.; Siddiqui, M.A.; Lau, L.F.; Chaqour, B. Mechanical regulation of the proangiogenic factor CCN1/CYR61 gene requires the combined activities of MRTF-A and CREB-binding protein histone acetyltransferase. J. Biol. Chem. 2009, 284, 23125–23136. [Google Scholar] [CrossRef]
- Ziegler, T.; Bahr, A.; Howe, A.; Klett, K.; Husada, W.; Weber, C.; Laugwitz, K.L.; Kupatt, C.; Hinkel, R. Tbeta4 Increases Neovascularization and Cardiac Function in Chronic Myocardial Ischemia of Normo- and Hypercholesterolemic Pigs. Mol. Ther. 2018, 26, 1706–1714. [Google Scholar] [CrossRef]
- Chirmule, N.; Propert, K.; Magosin, S.; Qian, Y.; Qian, R.; Wilson, J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999, 6, 1574–1583. [Google Scholar] [CrossRef]
- Nowrouzi, A.; Penaud-Budloo, M.; Kaeppel, C.; Appelt, U.; Le Guiner, C.; Moullier, P.; von Kalle, C.; Snyder, R.O.; Schmidt, M. Integration frequency and intermolecular recombination of rAAV vectors in non-human primate skeletal muscle and liver. Mol. Ther. 2012, 20, 1177–1186. [Google Scholar] [CrossRef]
- Yla-Herttuala, S.; Rissanen, T.T.; Vajanto, I.; Hartikainen, J. Vascular endothelial growth factors: Biology and current status of clinical applications in cardiovascular medicine. J. Am. Coll. Cardiol. 2007, 49, 1015–1026. [Google Scholar] [CrossRef]
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar] [PubMed]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.M.; Lagerstrom-Fermer, M.; Carlsson, L.G.; Arfvidsson, C.; Egnell, A.C.; Rudvik, A.; Kjaer, M.; Collen, A.; Thompson, J.D.; Joyal, J.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- White, S.; Kalf, M.; Liu, Q.; Villerius, M.; Engelsma, D.; Kriek, M.; Vollebregt, E.; Bakker, B.; van Ommen, G.J.; Breuning, M.H.; et al. Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am. J. Hum. Genet. 2002, 71, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Motohashi, Y.; Asakura, A. Angiogenesis as a novel therapeutic strategy for Duchenne muscular dystrophy through decreased ischemia and increased satellite cells. Front. Physiol. 2014, 5, 50. [Google Scholar] [CrossRef][Green Version]
- Klymiuk, N.; Blutke, A.; Graf, A.; Krause, S.; Burkhardt, K.; Wuensch, A.; Krebs, S.; Kessler, B.; Zakhartchenko, V.; Kurome, M.; et al. Dystrophin-deficient pigs provide new insights into the hierarchy of physiological derangements of dystrophic muscle. Hum. Mol. Genet. 2013, 22, 4368–4382. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziegler, T.; Abdel Rahman, F.; Jurisch, V.; Kupatt, C. Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies. Cells 2020, 9, 50. https://doi.org/10.3390/cells9010050
Ziegler T, Abdel Rahman F, Jurisch V, Kupatt C. Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies. Cells. 2020; 9(1):50. https://doi.org/10.3390/cells9010050
Chicago/Turabian StyleZiegler, Tilman, Farah Abdel Rahman, Victoria Jurisch, and Christian Kupatt. 2020. "Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies" Cells 9, no. 1: 50. https://doi.org/10.3390/cells9010050
APA StyleZiegler, T., Abdel Rahman, F., Jurisch, V., & Kupatt, C. (2020). Atherosclerosis and the Capillary Network; Pathophysiology and Potential Therapeutic Strategies. Cells, 9(1), 50. https://doi.org/10.3390/cells9010050