Profile of Matrix-Remodeling Proteinases in Osteoarthritis: Impact of Fibronectin

 ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
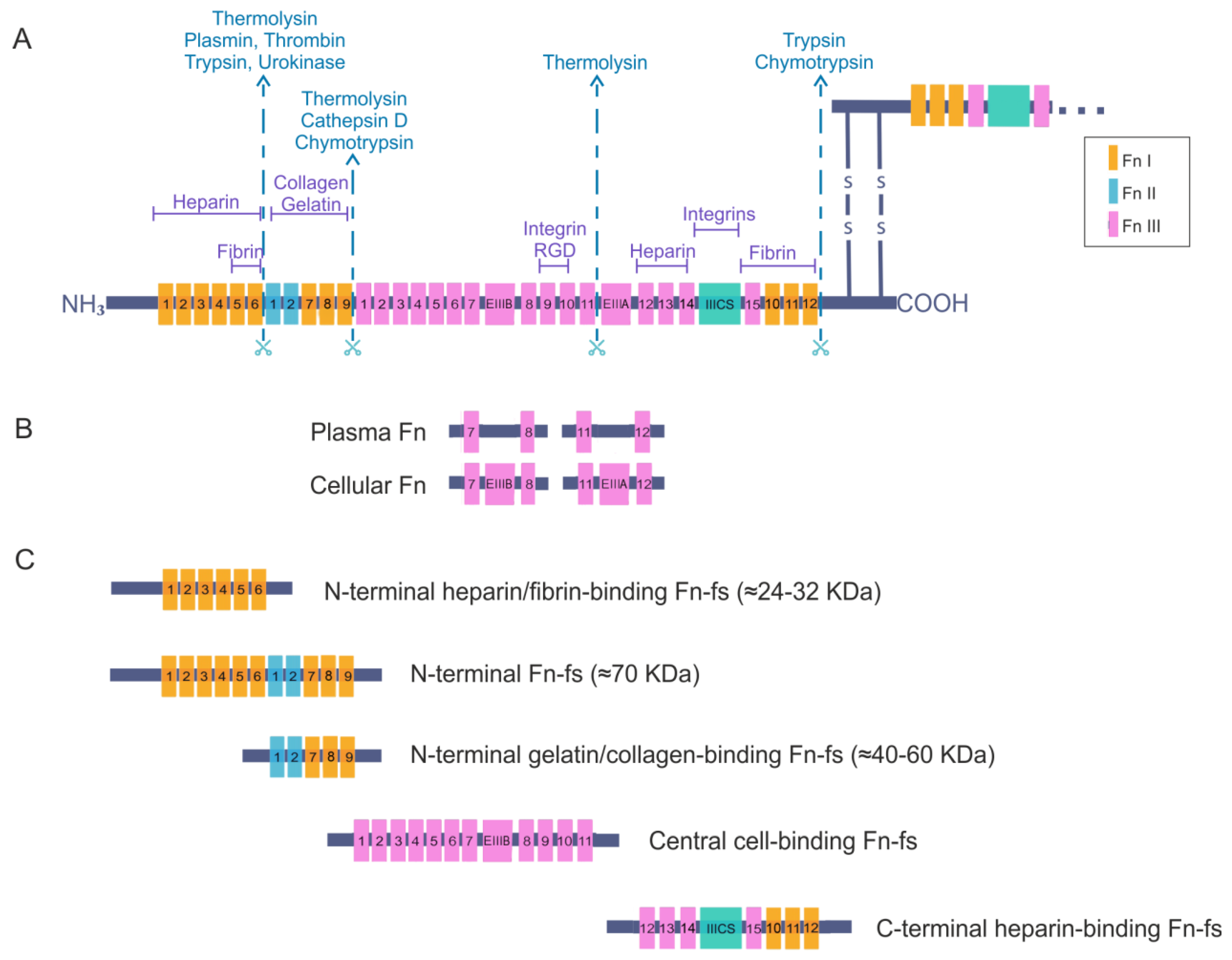
2. Fibronectin
Fibronectin Fragments
3. Osteoarthritis
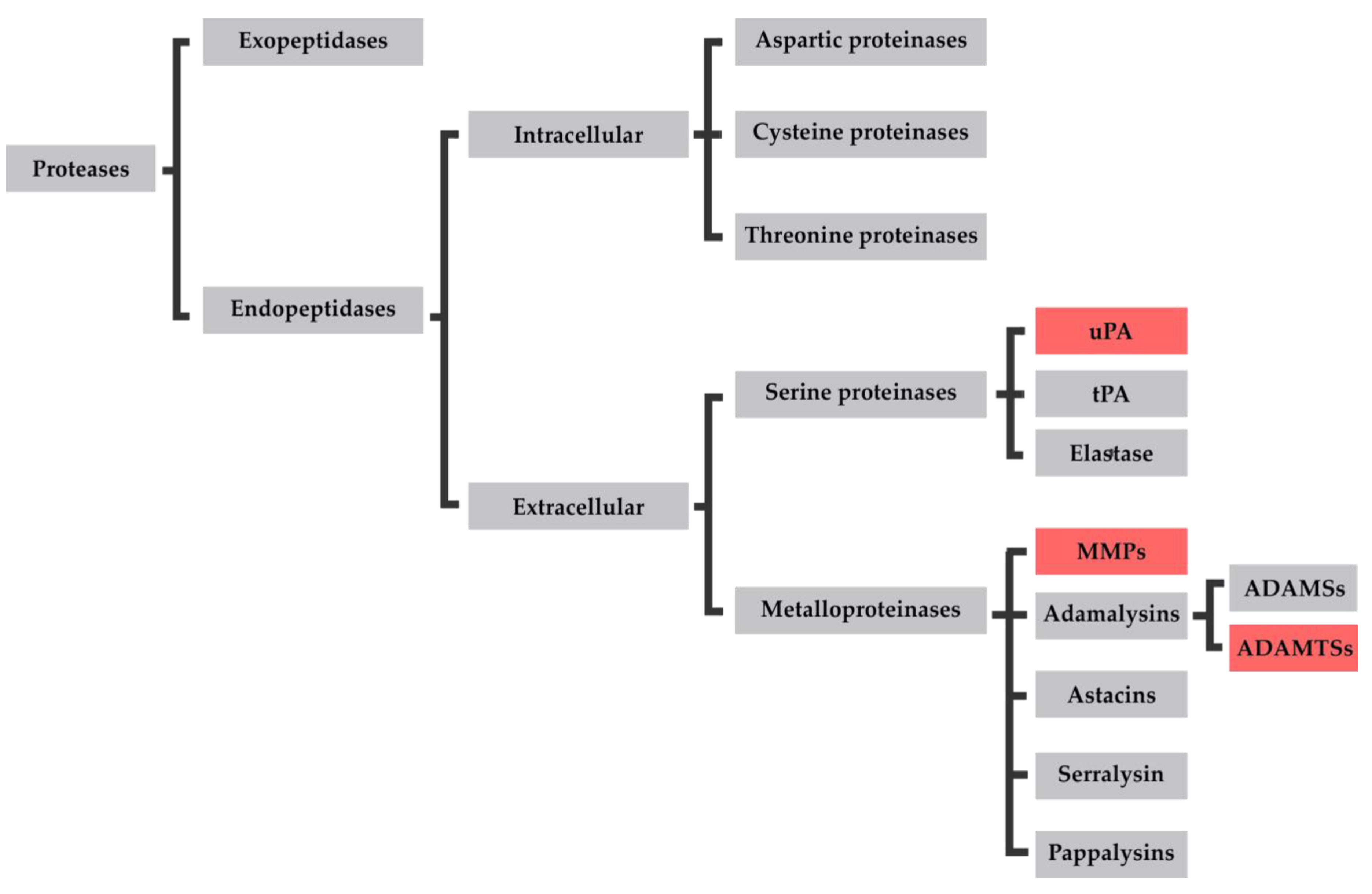
3.1. Proteases in Osteoarthritis
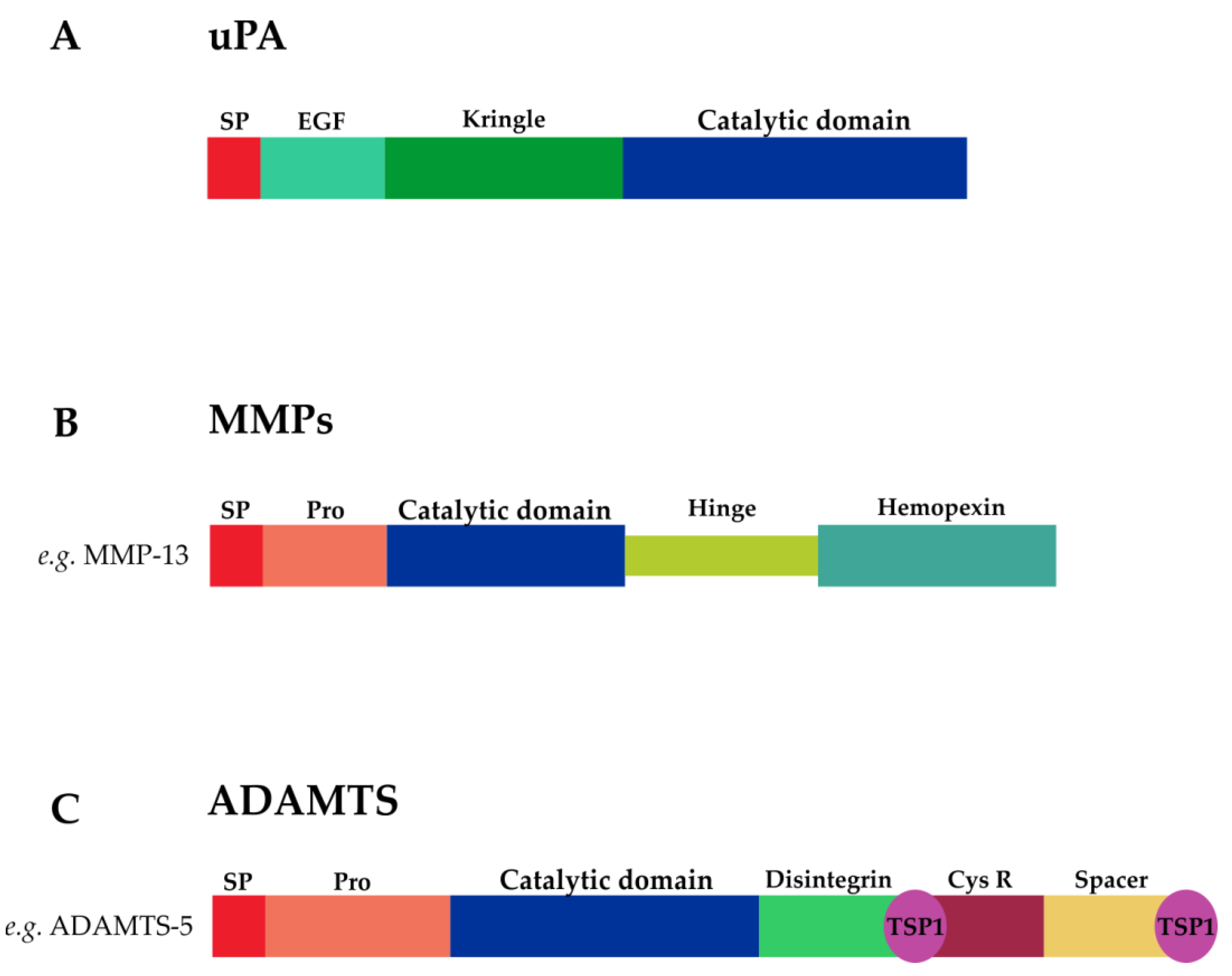
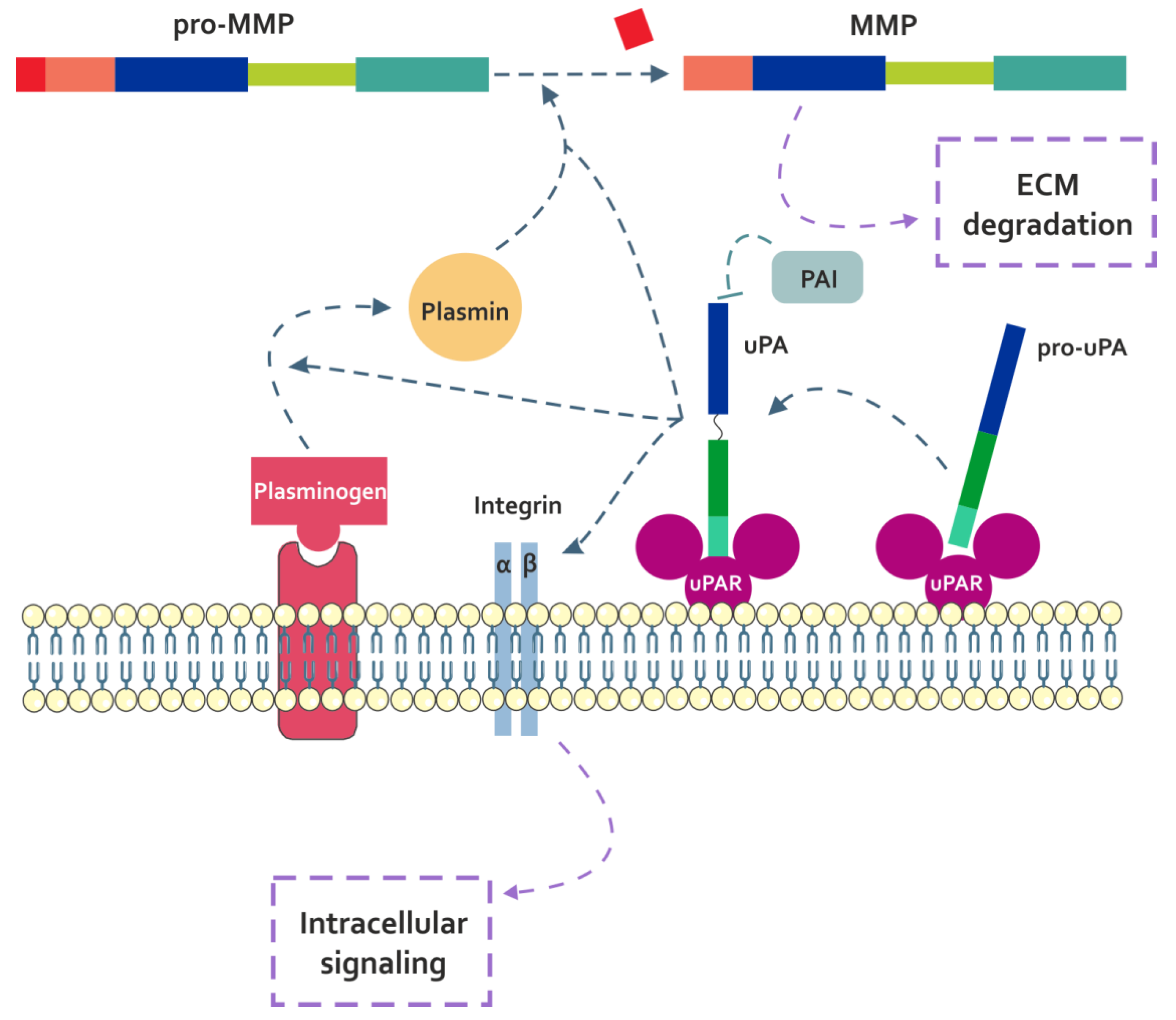
3.1.1. uPA
3.1.2. MMPs
Collagenases
Gelatinases
Other MMPs
3.1.3. ADAMTSs
Aggrecanases and Proteoglycanases
COMP Proteinases
Procollagen N-Propeptidases
von-Willebrand Factor Proteinase
Other ADAMTSs
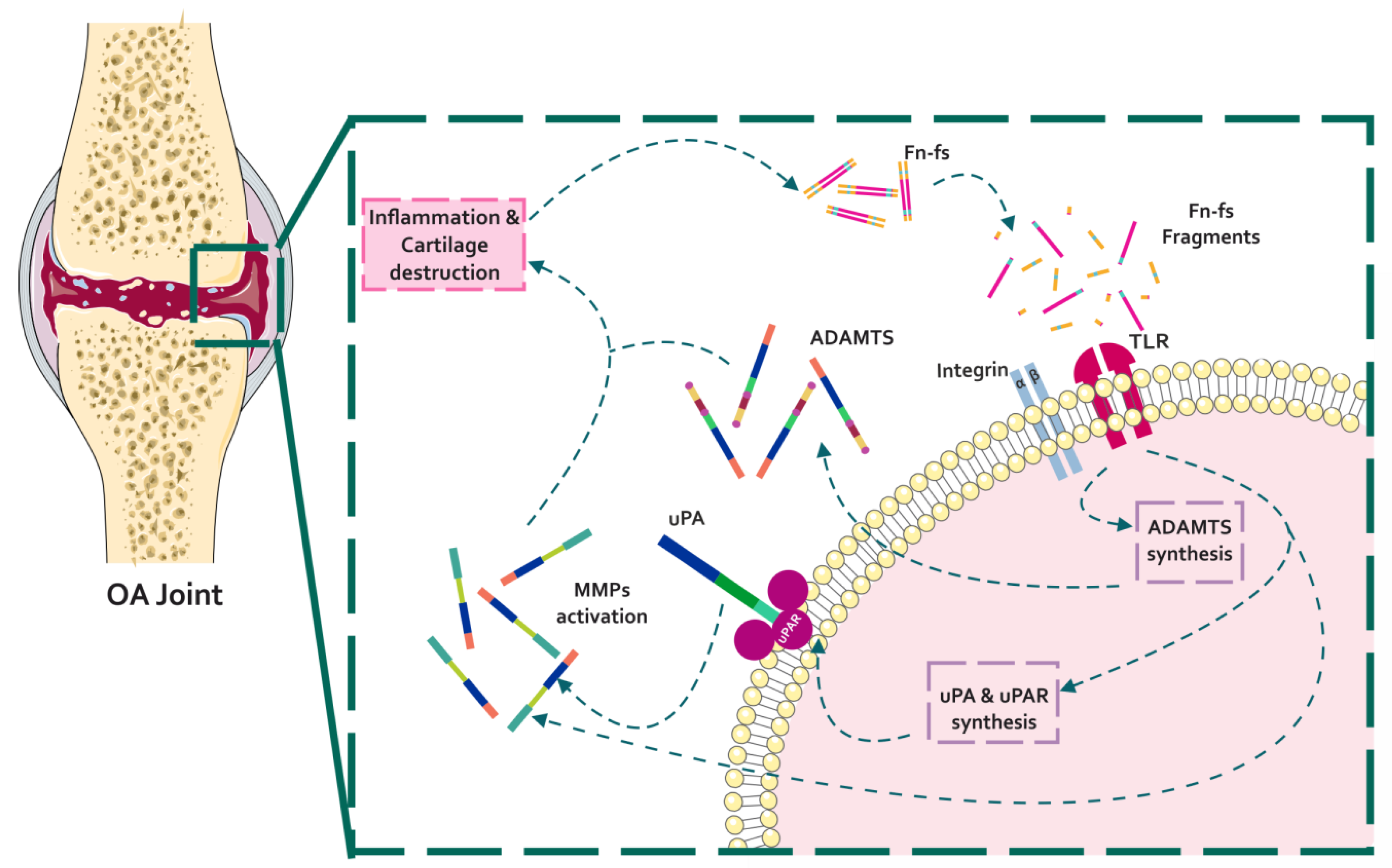
4. Fibronectin and Fibronectin Fragments in Osteoarthritis
4.1. Effect of Fibronectin Fragments in the Profile of OA Matrix-Remodeling Proteinases
4.1.1. Fn-fs Modulate the uPA System and Induce MMP Expression
4.1.2. Fn-fs Modifies ADAMTS Expression and Signaling
5. Conclusions
- Fibronectin is a component of the ECM essential to its assembly, which also regulates some cellular functions. However, cleavage of fibronectin in pathological conditions releases fibronectin fragments with pro-inflammatory and degradative properties.
- Fibronectin fragments mainly released from cartilage ECM during osteoarthritis induce the expression of different proteinases, such as uPA, MMPs, and ADAMTSs. These proteinases degrade several ECM components, including the type II collagen and the aggrecan.
- Overexpressed proteinases in the osteoarthritis joint microenvironment cleave fibronectin, therefore releasing new fibronectin fragments and promoting a feed-back loop of degradation and inflammation in the pathology.
- The study of fibronectin and proteinases in pathological conditions is important for the design of new strategies using them as potential therapeutic targets for the treatment of osteoarthritis, to control not only the symptoms but also the progression of the disease.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAMTSLs | ADAMTS-like proteins |
| ADAMTSs | A disintegrin and metalloproteinase with thrombospondin motifs |
| DAMPs | Damage-associated molecular patterns |
| dEDS | Dermatosparaxis Ehlers-Danlos syndrome |
| ECM | Extracellular matrix |
| EGF | Epidermal growth factor |
| Fn | Fibronectin |
| Fn-fs | Fibronectin fragments |
| GAGs | Glycosaminoglycans |
| GEP | Granulin-epithelin |
| GPCRs | G protein-coupled receptors |
| HD | Healthy donors |
| IVD | Intervertebral disc degeneration |
| LU | Ly-6 and uPAR domain |
| Ly-6 | Lymphocyte antigen 6 |
| MMPs | Matrix metalloproteinases |
| MT-MMPs | Membrane-type matrix metalloproteinases |
| NO | Nitric oxide |
| OA | Osteoarthritis |
| PAI | Plasminogen activator inhibitor |
| PAs | Plasminogen activators |
| PLAC | Protease and lacunin |
| RA | Rheumatoid arthritis |
| SF | Synovial fibroblasts |
| TIMP | Tissue inhibitors of metalloproteinases |
| TLR2 | Toll-like receptor 2 |
| tPA | Tissue-type plasminogen activator |
| TSP-1 | Thrombospondin type 1 |
| uPA | Urokinase-type plasminogen activator |
| uPAR | Urokinase-type plasminogen activator receptor |
| WMS | Weill–Marchesani syndrome |
| α2M | Alpha-2-macroglobulin |
References
- Bijlsma, J.W.; Berenbaum, F.; Lafeber, F.P. Osteoarthritis: An update with relevance for clinical practice. Lancet 2011, 377, 2115–2126. [Google Scholar] [CrossRef]
- Kraus, V.B.; Blanco, F.J.; Englund, M.; Karsdal, M.A.; Lohmander, L.S. Call for standardized definitions of osteoarthritis and risk stratification for clinical trials and clinical use. Osteoarthr. Cartil. 2015, 23, 1233–1241. [Google Scholar] [CrossRef]
- Monfort, J. Artrosis: Fisiopatología, Diagnóstico y Tratamiento; Médica Panamericana: Madrid, Spain, 2010. [Google Scholar]
- Raman, S.; FitzGerald, U.; Murphy, J.M. Interplay of Inflammatory Mediators with Epigenetics and Cartilage Modifications in Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 22. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Geurts, J.; Juric, D.; Muller, M.; Scharen, S.; Netzer, C. Novel Ex Vivo Human Osteochondral Explant Model of Knee and Spine Osteoarthritis Enables Assessment of Inflammatory and Drug Treatment Responses. Int. J. Mol. Sci. 2018, 19, 1314. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T. Cartilage destruction by matrix degradation products. Mod. Rheumatol. 2006, 16, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Structure and biology of proteoglycans. Annu. Rev. Cell Biol. 1988, 4, 229–255. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, G.; Baugh, L.; Vogel, V. Coexisting conformations of fibronectin in cell culture imaged using fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2001, 98, 14464–14468. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.P.; Carrell, N.; McDonagh, J. Fibronectin molecule visualized in electron microscopy: A long, thin, flexible strand. J. Cell Biol. 1981, 91, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Maurer, L.M.; Ma, W.; Mosher, D.F. Dynamic structure of plasma fibronectin. Crit. Rev. Biochem. Mol. Biol. 2015, 51, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- White, E.S.; Muro, A.F. Fibronectin splice variants: Understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life 2011, 63, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Barilla, M.L.; Carsons, S.E. Fibronectin fragments and their role in inflammatory arthritis. Semin. Arthritis Rheum. 2000, 29, 252–265. [Google Scholar] [CrossRef]
- Castellanos, M.; Leira, R.; Serena, J.; Blanco, M.; Pedraza, S.; Castillo, J.; Davalos, A. Plasma cellular-fibronectin concentration predicts hemorrhagic transformation after thrombolytic therapy in acute ischemic stroke. Stroke 2004, 35, 1671–1676. [Google Scholar] [CrossRef]
- Peters, J.H.; Loredo, G.A.; Chen, G.; Maunder, R.; Hahn, T.J.; Willits, N.H.; Hynes, R.O. Plasma levels of fibronectin bearing the alternatively spliced EIIIB segment are increased after major trauma. J. Lab. Clin. Med. 2003, 141, 401–410. [Google Scholar] [CrossRef]
- Song, K.S.; Kim, H.K.; Shim, W.; Jee, S.H. Plasma fibronectin levels in ischemic heart disease. Atherosclerosis 2001, 154, 449–453. [Google Scholar] [CrossRef]
- Claudepierre, P.; Allanore, Y.; Belec, L.; Larget-Piet, B.; Zardi, L.; Chevalier, X. Increased Ed-B fibronectin plasma levels in spondyloarthropathies: Comparison with rheumatoid arthritis patients and a healthy population. Rheumatology 1999, 38, 1099–1103. [Google Scholar] [CrossRef]
- Shiozawa, K.; Hino, K.; Shiozawa, S. Alternatively spliced EDA-containing fibronectin in synovial fluid as a predictor of rheumatoid joint destruction. Rheumatology 2001, 40, 739–742. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scanzello, C.R.; Markova, D.Z.; Chee, A.; Xiu, Y.; Adams, S.L.; Anderson, G.; Zgonis, M.; Qin, L.; An, H.S.; Zhang, Y. Fibronectin splice variation in human knee cartilage, meniscus and synovial membrane: Observations in osteoarthritic knee. J. Orthop. Res. 2015, 33, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Pickford, A.R.; Campbell, I.D. NMR studies of modular protein structures and their interactions. Chem. Rev. 2004, 104, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Hui, F.; Homandberg, G.A. Fibronectin fragments alter matrix protein synthesis in cartilage tissue cultured in vitro. Arch. Biochem. Biophys. 1993, 307, 110–118. [Google Scholar] [CrossRef]
- Yamada, K.M. Adhesive recognition sequences. J. Biol. Chem. 1991, 266, 12809–12812. [Google Scholar]
- Homandberg, G.A.; Costa, V.; Wen, C. Fibronectin fragments active in chondrocytic chondrolysis can be chemically cross-linked to the alpha5 integrin receptor subunit. Osteoarthr. Cartil. 2002, 10, 938–949. [Google Scholar] [CrossRef][Green Version]
- Hwang, H.S.; Park, S.J.; Cheon, E.J.; Lee, M.H.; Kim, H.A. Fibronectin fragment-induced expression of matrix metalloproteinases is mediated by MyD88-dependent TLR-2 signaling pathway in human chondrocytes. Arthritis Res. Ther. 2015, 17, 320. [Google Scholar] [CrossRef]
- Ding, L.; Guo, D.; Homandberg, G.A. The cartilage chondrolytic mechanism of fibronectin fragments involves MAP kinases: Comparison of three fragments and native fibronectin. Osteoarthr. Cartil. 2008, 16, 1253–1262. [Google Scholar] [CrossRef]
- Yasuda, T. Activation of Akt leading to NF-kappaB up-regulation in chondrocytes stimulated with fibronectin fragment. Biomed. Res. 2011, 32, 209–215. [Google Scholar] [CrossRef]
- You, R.; Zheng, M.; McKeown-Longo, P.J. The first type III repeat in fibronectin activates an inflammatory pathway in dermal fibroblasts. J. Biol. Chem. 2010, 285, 36255–36259. [Google Scholar] [CrossRef]
- Ou, J.; Deng, J.; Wei, X.; Xie, G.; Zhou, R.; Yu, L.; Liang, H. Fibronectin extra domain A (EDA) sustains CD133(+)/CD44(+) subpopulation of colorectal cancer cells. Stem Cell Res. 2013, 11, 820–833. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, S.; Gutiérrez-Cañas, I.; Seoane, I.V.; Fernández, J.; Mellado, M.; Leceta, J.; Tío, L.; Villanueva-Romero, R.; Juarranz, Y.; Gomariz, R.P. Healthy and Osteoarthritic Synovial Fibroblasts Produce a Disintegrin and Metalloproteinase with Thrombospondin Motifs 4, 5, 7, and 12: Induction by IL-1beta and Fibronectin and Contribution to Cartilage Damage. Am. J. Pathol. 2016, 186, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Batlle-Gualda, E.; Benito-Ruiz, P.; Blanco, F.J.; Martín, E. Manual SER de la Artrosis; IM&C: Madrid, Spain, 2002. [Google Scholar]
- Haseeb, A.; Haqqi, T.M. Immunopathogenesis of osteoarthritis. Clin. Immunol. 2013, 146, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Lepus, C.M. Role of inflammation in the pathogenesis of osteoarthritis: Latest findings and interpretations. Ther. Adv. Musculoskelet. Dis. 2013, 5, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Mimpen, J.Y.; Snelling, S.J.B. Chondroprotective Factors in Osteoarthritis: A Joint Affair. Curr. Rheumatol. Rep. 2019, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Pelletier, J.P. Is osteoarthritis a disease involving only cartilage or other articular tissues? Eklem Hast. Cerrahisi 2010, 21, 2–14. [Google Scholar]
- Puig-Junoy, J.; Ruiz Zamora, A. Socio-economic costs of osteoarthritis: A systematic review of cost-of-illness studies. Semin. Arthritis Rheum. 2015, 44, 531–541. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Wildi, L.M.; Pelletier, J.P. Future therapeutics for osteoarthritis. Bone 2012, 51, 297–311. [Google Scholar] [CrossRef]
- Ghouri, A.; Conaghan, P.G. Update on novel pharmacological therapies for osteoarthritis. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1759720X19864492. [Google Scholar] [CrossRef]
- Schulze-Tanzil, G. Intraarticular Ligament Degeneration Is Interrelated with Cartilage and Bone Destruction in Osteoarthritis. Cells 2019, 8, 990. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, J.; Blom, A.B.; Wainwright, S.; Hughes, C.; Caterson, B.; van den Berg, W.B. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 2010, 62, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M.; Tsuchimochi, K.; Ijiri, K.; Li, Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann. Rheum. Dis. 2008, 67 (Suppl. 3), 75–82. [Google Scholar] [CrossRef]
- Kato, T.; Miyaki, S.; Ishitobi, H.; Nakamura, Y.; Nakasa, T.; Lotz, M.K.; Ochi, M. Exosomes from IL-1beta stimulated synovial fibroblasts induce osteoarthritic changes in articular chondrocytes. Arthritis Res. Ther. 2014, 16, R163. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Lin, S.N.; Kurz, B.; Cole, A.A.; Kumar, S.; Grodzinsky, A.J. Potent inhibition of cartilage biosynthesis by coincubation with joint capsule through an IL-1-independent pathway. Scand. J. Med. Sci. Sports 2009, 19, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Attur, M.; Samuels, J.; Krasnokutsky, S.; Abramson, S.B. Targeting the synovial tissue for treating osteoarthritis (OA): Where is the evidence? Best Pract. Res. Clin. Rheumatol. 2010, 24, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Pérez-García, S.; Carrión, M.; Villanueva-Romero, R.; Hermida-Gomez, T.; Fernández-Moreno, M.; Mellado, M.; Blanco, F.J.; Juarranz, Y.; Gomariz, R.P. Wnt and RUNX2 mediate cartilage breakdown by osteoarthritis synovial fibroblast-derived ADAMTS-7 and -12. J. Cell Mol. Med. 2019, 23, 3974–3983. [Google Scholar] [CrossRef]
- Funck-Brentano, T.; Cohen-Solal, M. Crosstalk between cartilage and bone: When bone cytokines matter. Cytokine Growth Factor Rev. 2011, 22, 91–97. [Google Scholar] [CrossRef]
- Maldonado, M.; Nam, J. The role of changes in extracellular matrix of cartilage in the presence of inflammation on the pathology of osteoarthritis. Biomed. Res. Int. 2013, 2013, 284873. [Google Scholar] [CrossRef]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Rengel, Y.; Ospelt, C.; Gay, S. Proteinases in the joint: Clinical relevance of proteinases in joint destruction. Arthritis Res. Ther. 2007, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, S. El Sinoviocito Fibroblástico en la Artrosis. Efecto Protector de VIP y CRF; UCM, Faculty of Biology: Madrid, Spain, 2016. [Google Scholar]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Lee, M.H. What are the roles of metalloproteinases in cartilage and bone damage? Ann. Rheum. Dis. 2005, 64 (Suppl. 4), iv44–iv47. [Google Scholar] [CrossRef]
- Mehana, E.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Kiani, C.; Chen, L.; Wu, Y.J.; Yee, A.J.; Yang, B.B. Structure and function of aggrecan. Cell Res. 2002, 12, 19–32. [Google Scholar] [CrossRef]
- Chana-Munoz, A.; Jendroszek, A.; Sonnichsen, M.; Wang, T.; Ploug, M.; Jensen, J.K.; Andreasen, P.A.; Bendixen, C.; Panitz, F. Origin and diversification of the plasminogen activation system among chordates. BMC Evol. Biol. 2019, 19, 27. [Google Scholar] [CrossRef]
- Madunic, J. The Urokinase Plasminogen Activator System in Human Cancers: An Overview of Its Prognostic and Predictive Role. Thromb. Haemost. 2018, 118, 2020–2036. [Google Scholar] [CrossRef]
- Buckley, B.J.; Ali, U.; Kelso, M.J.; Ranson, M. The Urokinase Plasminogen Activation System in Rheumatoid Arthritis: Pathophysiological Roles and Prospective Therapeutic Targets. Curr. Drug Targets 2019, 20, 970–981. [Google Scholar] [CrossRef]
- Blasi, F.; Sidenius, N. The urokinase receptor: Focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010, 584, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Del Rosso, M.; Fibbi, G.; Pucci, M.; Margheri, F.; Serrati, S. The plasminogen activation system in inflammation. Front. Biosci. A J. Virtual Libr. 2008, 13, 4667–4686. [Google Scholar] [CrossRef] [PubMed]
- Huai, Q.; Mazar, A.P.; Kuo, A.; Parry, G.C.; Shaw, D.E.; Callahan, J.; Li, Y.; Yuan, C.; Bian, C.; Chen, L.; et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science 2006, 311, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Blouse, G.E.; Botkjaer, K.A.; Deryugina, E.; Byszuk, A.A.; Jensen, J.M.; Mortensen, K.K.; Quigley, J.P.; Andreasen, P.A. A novel mode of intervention with serine protease activity: Targeting zymogen activation. J. Biol. Chem. 2009, 284, 4647–4657. [Google Scholar] [CrossRef]
- Liu, G.; Yang, Y.; Yang, S.; Banerjee, S.; De Freitas, A.; Friggeri, A.; Davis, K.I.; Abraham, E. The receptor for urokinase regulates TLR2 mediated inflammatory responses in neutrophils. PLoS ONE 2011, 6, e25843. [Google Scholar] [CrossRef]
- Jin, T.; Tarkowski, A.; Carmeliet, P.; Bokarewa, M. Urokinase, a constitutive component of the inflamed synovial fluid, induces arthritis. Arthritis Res. Ther. 2003, 5, R9–R17. [Google Scholar] [CrossRef]
- Gyetko, M.R.; Shollenberger, S.B.; Sitrin, R.G. Urokinase expression in mononuclear phagocytes: Cytokine-specific modulation by interferon-gamma and tumor necrosis factor-alpha. J. Leukoc. Biol. 1992, 51, 256–263. [Google Scholar] [CrossRef][Green Version]
- Hamilton, J.A.; Piccoli, D.S.; Leizer, T.; Butler, D.M.; Croatto, M.; Royston, A.K. Transforming growth factor beta stimulates urokinase-type plasminogen activator and DNA synthesis, but not prostaglandin E2 production, in human synovial fibroblasts. Proc. Natl. Acad. Sci. USA 1991, 88, 7180–7184. [Google Scholar] [CrossRef]
- Ito, A.; Itoh, Y.; Sasaguri, Y.; Morimatsu, M.; Mori, Y. Effects of interleukin-6 on the metabolism of connective tissue components in rheumatoid synovial fibroblasts. Arthritis Rheum. 1992, 35, 1197–1201. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Faure, M.P.; McCollum, R.; Mineau, F.; Cloutier, J.M.; Pelletier, J.P. Plasmin, plasminogen activators and inhibitor in human osteoarthritic cartilage. J. Rheumatol. 1991, 18, 1863–1871. [Google Scholar]
- Medcalf, R.L.; Hamilton, J.A. Human synovial fibroblasts produce urokinase-type plasminogen activator. Arthritis Rheum. 1986, 29, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Schwab, W.; Schulze-Tanzil, G.; Mobasheri, A.; Dressler, J.; Kotzsch, M.; Shakibaei, M. Interleukin-1beta-induced expression of the urokinase-type plasminogen activator receptor and its co-localization with MMPs in human articular chondrocytes. Histol. Histopathol. 2004, 19, 105–112. [Google Scholar] [PubMed]
- Hamilton, J.A.; Campbell, I.K.; Wojta, J.; Cheung, D. Plasminogen activators and their inhibitors in arthritic disease. Ann. N. Y. Acad. Sci. 1992, 667, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Vaughan, D.E. PAI-1 in tissue fibrosis. J. Cell. Physiol. 2012, 227, 493–507. [Google Scholar] [CrossRef]
- Li, J.; Ny, A.; Leonardsson, G.; Nandakumar, K.S.; Holmdahl, R.; Ny, T. The plasminogen activator/plasmin system is essential for development of the joint inflammatory phase of collagen type II-induced arthritis. Am. J. Pathol. 2005, 166, 783–792. [Google Scholar] [CrossRef]
- Del Rosso, M.; Fibbi, G.; Matucci Cerinic, M. The urokinase-type plasminogen activator system and inflammatory joint diseases. Clin. Exp. Rheumatol. 1999, 17, 485–498. [Google Scholar]
- Pérez-García, S.; Carrión, M.; Jimeno, R.; Ortiz, A.M.; González-Álvaro, I.; Fernández, J.; Gomariz, R.P.; Juarranz, Y. Urokinase plasminogen activator system in synovial fibroblasts from osteoarthritis patients: Modulation by inflammatory mediators and neuropeptides. J. Mol. Neurosci. 2014, 52, 18–27. [Google Scholar] [CrossRef]
- Cerinic, M.M.; Generini, S.; Partsch, G.; Pignone, A.; Dini, G.; Konttinen, Y.T.; Del Rosso, M. Synoviocytes from osteoarthritis and rheumatoid arthritis produce plasminogen activators and plasminogen activator inhibitor-1 and display u-PA receptors on their surface. Life Sci. 1998, 63, 441–453. [Google Scholar] [CrossRef]
- Ronday, H.K.; Smits, H.H.; Van Muijen, G.N.; Pruszczynski, M.S.; Dolhain, R.J.; Van Langelaan, E.J.; Breedveld, F.C.; Verheijen, J.H. Difference in expression of the plasminogen activation system in synovial tissue of patients with rheumatoid arthritis and osteoarthritis. Br. J. Rheumatol. 1996, 35, 416–423. [Google Scholar] [CrossRef][Green Version]
- Belcher, C.; Fawthrop, F.; Bunning, R.; Doherty, M. Plasminogen activators and their inhibitors in synovial fluids from normal, osteoarthritis, and rheumatoid arthritis knees. Ann. Rheum. Dis. 1996, 55, 230–236. [Google Scholar] [CrossRef]
- Hart, P.H.; Vitti, G.F.; Burgess, D.R.; Whitty, G.A.; Royston, K.; Hamilton, J.A. Activation of human monocytes by granulocyte-macrophage colony-stimulating factor: Increased urokinase-type plasminogen activator activity. Blood 1991, 77, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Wu, L.D. Aggrecanase and aggrecan degradation in osteoarthritis: A review. J. Int. Med. Res. 2008, 36, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, M.; Tortorella, M.; Nagase, H.; Brew, K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J. Biol. Chem. 2001, 276, 12501–12504. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Asp. Med. 2008, 29, 290–308. [Google Scholar] [CrossRef] [PubMed]
- Kevorkian, L.; Young, D.A.; Darrah, C.; Donell, S.T.; Shepstone, L.; Porter, S.; Brockbank, S.M.; Edwards, D.R.; Parker, A.E.; Clark, I.M. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004, 50, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Davidson, R.K.; Waters, J.G.; Kevorkian, L.; Darrah, C.; Cooper, A.; Donell, S.T.; Clark, I.M. Expression profiling of metalloproteinases and their inhibitors in synovium and cartilage. Arthritis Res. Ther. 2006, 8, R124. [Google Scholar] [CrossRef]
- Vu, T.H.; Werb, Z. Matrix metalloproteinases: Effectors of development and normal physiology. Genes Dev. 2000, 14, 2123–2133. [Google Scholar] [CrossRef]
- Tokuhara, C.K.; Santesso, M.R.; Oliveira, G.S.N.; Ventura, T.; Doyama, J.T.; Zambuzzi, W.F.; Oliveira, R.C. Updating the role of matrix metalloproteinases in mineralized tissue and related diseases. J. Appl. Oral Sci. 2019, 27, e20180596. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Welsch, D.J.; Pelletier, J.P. Metalloproteases and inhibitors in arthritic diseases. Best Pract. Res. Clin. Rheumatol. 2001, 15, 805–829. [Google Scholar] [CrossRef]
- Clutterbuck, A.L.; Asplin, K.E.; Harris, P.; Allaway, D.; Mobasheri, A. Targeting matrix metalloproteinases in inflammatory conditions. Curr. Drug Targets 2009, 10, 1245–1254. [Google Scholar] [CrossRef]
- Moura-da-Silva, A.M.; Theakston, R.D.; Crampton, J.M. Evolution of disintegrin cysteine-rich and mammalian matrix-degrading metalloproteinases: Gene duplication and divergence of a common ancestor rather than convergent evolution. J. Mol. Evol. 1996, 43, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Pennati, R.; Ficetola, G.F.; Brunetti, R.; Caicci, F.; Gasparini, F.; Griggio, F.; Sato, A.; Stach, T.; Kaul-Strehlow, S.; Gissi, C.; et al. Morphological Differences between Larvae of the Ciona intestinalis Species Complex: Hints for a Valid Taxonomic Definition of Distinct Species. PLoS ONE 2015, 10, e0122879. [Google Scholar] [CrossRef] [PubMed]
- Cancemi, P.; Di Falco, F.; Feo, S.; Arizza, V.; Vizzini, A. The gelatinase MMP-9like is involved in regulation of LPS inflammatory response in Ciona robusta. Fish Shellfish Immunol 2019, 86, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Nagase, H. Activation mechanisms of matrix metalloproteinases. Biol. Chem. 1997, 378, 151–160. [Google Scholar] [PubMed]
- Buisson-Legendre, N.; Smith, S.; March, L.; Jackson, C. Elevation of activated protein C in synovial joints in rheumatoid arthritis and its correlation with matrix metalloproteinase 2. Arthritis Rheum. 2004, 50, 2151–2156. [Google Scholar] [CrossRef]
- Murphy, G.; Knauper, V.; Atkinson, S.; Butler, G.; English, W.; Hutton, M.; Stracke, J.; Clark, I. Matrix metalloproteinases in arthritic disease. Arthritis Res. 2002, 4 (Suppl. 3), S39–S49. [Google Scholar] [CrossRef]
- Jackson, M.T.; Moradi, B.; Smith, M.M.; Jackson, C.J.; Little, C.B. Activation of matrix metalloproteinases 2, 9, and 13 by activated protein C in human osteoarthritic cartilage chondrocytes. Arthritis Rheumatol. 2014, 66, 1525–1536. [Google Scholar] [CrossRef]
- Fernandes, J.C.; Martel-Pelletier, J.; Lascau-Coman, V.; Moldovan, F.; Jovanovic, D.; Raynauld, J.P.; Pelletier, J.P. Collagenase-1 and collagenase-3 synthesis in normal and early experimental osteoarthritic canine cartilage: An immunohistochemical study. J. Rheumatol. 1998, 25, 1585–1594. [Google Scholar]
- Shiomi, T.; Lemaitre, V.; D’Armiento, J.; Okada, Y. Matrix metalloproteinases, a disintegrin and metalloproteinases, and a disintegrin and metalloproteinases with thrombospondin motifs in non-neoplastic diseases. Pathol. Int. 2010, 60, 477–496. [Google Scholar] [CrossRef]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 2001, 107, 35–44. [Google Scholar] [CrossRef]
- Wang, M.; Sampson, E.R.; Jin, H.; Li, J.; Ke, Q.H.; Im, H.J.; Chen, D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 2013, 15, R5. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I.; Yamada, N.; Cheung, K.S.; Tilley, S.; Clarke, N.M.; Oreffo, R.O.; Kokubun, S.; Bronner, F. Association between the abnormal expression of matrix-degrading enzymes by human osteoarthritic chondrocytes and demethylation of specific CpG sites in the promoter regions. Arthritis Rheum. 2005, 52, 3110–3124. [Google Scholar] [CrossRef] [PubMed]
- Piecha, D.; Weik, J.; Kheil, H.; Becher, G.; Timmermann, A.; Jaworski, A.; Burger, M.; Hofmann, M.W. Novel selective MMP-13 inhibitors reduce collagen degradation in bovine articular and human osteoarthritis cartilage explants. Inflamm. Res. 2010, 59, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Lee, Y.A.; Choi, H.M.; Yoo, M.C.; Yang, H.I. Implication of MMP-9 and urokinase plasminogen activator (uPA) in the activation of pro-matrix metalloproteinase (MMP)-13. Rheumatol. Int. 2012, 32, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Skwara, A.; Bloch, M.; Dankbar, B. Differential induction and regulation of matrix metalloproteinases in osteoarthritic tissue and fluid synovial fibroblasts. Osteoarthr. Cartil. 2004, 12, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Wernicke, D.; Seyfert, C.; Hinzmann, B.; Gromnica-Ihle, E. Cloning of collagenase 3 from the synovial membrane and its expression in rheumatoid arthritis and osteoarthritis. J. Rheumatol. 1996, 23, 590–595. [Google Scholar] [PubMed]
- Yoshihara, Y.; Nakamura, H.; Obata, K.; Yamada, H.; Hayakawa, T.; Fujikawa, K.; Okada, Y. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in synovial fluids from patients with rheumatoid arthritis or osteoarthritis. Ann. Rheum. Dis. 2000, 59, 455–461. [Google Scholar] [CrossRef]
- Su, J.; Yu, J.; Ren, T.; Zhang, W.; Zhang, Y.; Liu, X.; Sun, T.; Lu, H.; Miyazawa, K.; Yao, L. Discoidin domain receptor 2 is associated with the increased expression of matrix metalloproteinase-13 in synovial fibroblasts of rheumatoid arthritis. Mol. Cell. Biochem. 2009, 330, 141–152. [Google Scholar] [CrossRef]
- Vandooren, J.; Van den Steen, P.E.; Opdenakker, G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): The next decade. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 222–272. [Google Scholar] [CrossRef]
- Bau, B.; Gebhard, P.M.; Haag, J.; Knorr, T.; Bartnik, E.; Aigner, T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002, 46, 2648–2657. [Google Scholar] [CrossRef]
- Xie, D.L.; Hui, F.; Meyers, R.; Homandberg, G.A. Cartilage chondrolysis by fibronectin fragments is associated with release of several proteinases: Stromelysin plays a major role in chondrolysis. Arch. Biochem. Biophys. 1994, 311, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Ohta, S.; Imai, K.; Yamashita, K.; Matsumoto, T.; Azumano, I.; Okada, Y. Expression of matrix metalloproteinase 7 (matrilysin) in human osteoarthritic cartilage. Lab. Investig. 1998, 78, 79–87. [Google Scholar] [PubMed]
- Yuan, Q.; Kan, W.B.; Song, P.F.; Zhao, J.; Yu, W.G.; Wang, Y.J. Influence of Bushen Huoxue decoction on beta-catenin, MMP-7 of synoviocytes in rats with knee osteoarthritis. Zhongguo Gu Shang 2012, 25, 761–765. [Google Scholar] [PubMed]
- Pap, T.; Shigeyama, Y.; Kuchen, S.; Fernihough, J.K.; Simmen, B.; Gay, R.E.; Billingham, M.; Gay, S. Differential expression pattern of membrane-type matrix metalloproteinases in rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1226–1232. [Google Scholar] [CrossRef]
- Hubmacher, D.; Apte, S.S. Genetic and functional linkage between ADAMTS superfamily proteins and fibrillin-1: A novel mechanism influencing microfibril assembly and function. Cell Mol. Life Sci. 2011, 68, 3137–3148. [Google Scholar] [CrossRef]
- Hubmacher, D.; Apte, S.S. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015, 47, 34–43. [Google Scholar] [CrossRef]
- Apte, S.S. ADAMTS Proteases: Methods and Protocols; Springer Nature. Humana: New York, NY, USA, 2019; Volume 2043. [Google Scholar]
- Mead, T.J.; Apte, S.S. ADAMTS proteins in human disorders. Matrix Biol. 71–72, 225–239.
- Kuno, K.; Iizasa, H.; Ohno, S.; Matsushima, K. The exon/intron organization and chromosomal mapping of the mouse ADAMTS-1 gene encoding an ADAM family protein with TSP motifs. Genomics 1997, 46, 466–471. [Google Scholar] [CrossRef]
- Kelwick, R.; Desanlis, I.; Wheeler, G.N.; Edwards, D.R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015, 16, 113. [Google Scholar] [CrossRef]
- Porter, S.; Clark, I.M.; Kevorkian, L.; Edwards, D.R. The ADAMTS metalloproteinases. Biochem. J. 2005, 386, 15–27. [Google Scholar] [CrossRef]
- Huxley-Jones, J.; Apte, S.S.; Robertson, D.L.; Boot-Handford, R.P. The characterisation of six ADAMTS proteases in the basal chordate Ciona intestinalis provides new insights into the vertebrate ADAMTS family. Int. J. Biochem. Cell Biol. 2005, 37, 1838–1845. [Google Scholar] [CrossRef]
- Somerville, R.P.; Longpre, J.M.; Jungers, K.A.; Engle, J.M.; Ross, M.; Evanko, S.; Wight, T.N.; Leduc, R.; Apte, S.S. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J. Biol. Chem. 2003, 278, 9503–9513. [Google Scholar] [CrossRef] [PubMed]
- Brunet, F.G.; Fraser, F.W.; Binder, M.J.; Smith, A.D.; Kintakas, C.; Dancevic, C.M.; Ward, A.C.; McCulloch, D.R. The evolutionary conservation of the A Disintegrin-like and Metalloproteinase domain with Thrombospondin-1 motif metzincins across vertebrate species and their expression in teleost zebrafish. BMC Evol. Biol. 2015, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.S. A disintegrin-like and metalloprotease (reprolysin-type) with thrombospondin type 1 motif (ADAMTS) superfamily: Functions and mechanisms. J. Biol. Chem. 2009, 284, 31493–31497. [Google Scholar] [CrossRef] [PubMed]
- Apte, S.S. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: The ADAMTS family. Int. J. Biochem Cell Biol. 2004, 36, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.A.; Liu, C.J. The role of ADAMTSs in arthritis. Protein Cell 2010, 1, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Fosang, A.J.; Rogerson, F.M.; East, C.J.; Stanton, H. ADAMTS-5: The story so far. Eur. Cell Mater. 2008, 15, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.C.; Riley, G.P. ADAMTS proteinases: A multi-domain, multi-functional family with roles in extracellular matrix turnover and arthritis. Arthritis Res. Ther. 2005, 7, 160–169. [Google Scholar] [CrossRef][Green Version]
- Longpre, J.M.; McCulloch, D.R.; Koo, B.H.; Alexander, J.P.; Apte, S.S.; Leduc, R. Characterization of proADAMTS5 processing by proprotein convertases. Int. J. Biochem. Cell Biol. 2009, 41, 1116–1126. [Google Scholar] [CrossRef]
- Stanton, H.; Melrose, J.; Little, C.B.; Fosang, A.J. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim Biophys Acta 2011, 1812, 1616–1629. [Google Scholar] [CrossRef]
- Arner, E.C.; Pratta, M.A.; Trzaskos, J.M.; Decicco, C.P.; Tortorella, M.D. Generation and characterization of aggrecanase. A soluble, cartilage-derived aggrecan-degrading activity. J. Biol. Chem. 1999, 274, 6594–6601. [Google Scholar] [CrossRef]
- Lohmander, L.S.; Neame, P.J.; Sandy, J.D. The structure of aggrecan fragments in human synovial fluid. Evidence that aggrecanase mediates cartilage degradation in inflammatory joint disease, joint injury, and osteoarthritis. Arthritis Rheum. 1993, 36, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Caterson, B.; Fosang, A.J.; Roughley, P.J.; Mort, J.S. Monoclonal antibodies that specifically recognize neoepitope sequences generated by ‘aggrecanase’ and matrix metalloproteinase cleavage of aggrecan: Application to catabolism in situ and in vitro. Biochem. J. 1995, 305, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Lark, M.W.; Bayne, E.K.; Flanagan, J.; Harper, C.F.; Hoerrner, L.A.; Hutchinson, N.I.; Singer, I.I.; Donatelli, S.A.; Weidner, J.R.; Williams, H.R.; et al. Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J. Clin. Investig. 1997, 100, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, M.D.; Vincent, T.L.; Driscoll, C.; Burleigh, A.; Bou-Gharios, G.; Saklatvala, J.; Nagase, H.; Chanalaris, A. Transcriptional analysis of micro-dissected articular cartilage in post-traumatic murine osteoarthritis. Osteoarthr. Cartil. 2015, 23, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bu, Y.; Zhu, B.; Zhao, Q.; Lv, Z.; Li, B.; Liu, J. Global transcriptome analysis to identify critical genes involved in the pathology of osteoarthritis. Bone Jt. Res. 2018, 7, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Yan, X.; Zhang, M.; Chang, X.; Bai, Z.; He, Y.; Yuan, Z. Aggrecanases in the human synovial fluid at different stages of osteoarthritis. Clin. Rheumatol. 2013, 32, 797–803. [Google Scholar] [CrossRef]
- Fosang, A.J.; Rogerson, F.M. Identifying the human aggrecanase. Osteoarthr. Cartil. 2010, 18, 1109–1116. [Google Scholar] [CrossRef]
- Kataoka, Y.; Ariyoshi, W.; Okinaga, T.; Kaneuji, T.; Mitsugi, S.; Takahashi, T.; Nishihara, T. Mechanisms involved in suppression of ADAMTS4 expression in synoviocytes by high molecular weight hyaluronic acid. Biochem. Biophys. Res. Commun. 2013, 432, 580–585. [Google Scholar] [CrossRef]
- Sylvester, J.; El Mabrouk, M.; Ahmad, R.; Chaudry, A.; Zafarullah, M. Interleukin-1 induction of aggrecanase gene expression in human articular chondrocytes is mediated by mitogen-activated protein kinases. Cell. Physiol. Biochem. 2012, 30, 563–574. [Google Scholar] [CrossRef]
- Imada, K.; Oka, H.; Kawasaki, D.; Miura, N.; Sato, T.; Ito, A. Anti-arthritic action mechanisms of natural chondroitin sulfate in human articular chondrocytes and synovial fibroblasts. Biol. Pharm. Bull. 2010, 33, 410–414. [Google Scholar] [CrossRef]
- Kobayashi, H.; Hirata, M.; Saito, T.; Itoh, S.; Chung, U.I.; Kawaguchi, H. Transcriptional induction of ADAMTS5 by an NF-kappaB family member RelA/p65 in chondrocytes during osteoarthritis development. J. Biol. Chem. 2013, 288, 28620–28629. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Chanalaris, A.; Troeberg, L. ADAMTS and ADAM metalloproteinases in osteoarthritis—looking beyond the ‘usual suspects’. Osteoarthr. Cartil. 2017, 25, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Somerville, R.P.; Longpre, J.M.; Apel, E.D.; Lewis, R.M.; Wang, L.W.; Sanes, J.R.; Leduc, R.; Apte, S.S. ADAMTS7B, the full-length product of the ADAMTS7 gene, is a chondroitin sulfate proteoglycan containing a mucin domain. J. Biol. Chem. 2004, 279, 35159–35175. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.A.; Liu, C.J. The emerging roles of ADAMTS-7 and ADAMTS-12 matrix metalloproteinases. Rheumatol. Res. Rev. 2009, 1, 121–131. [Google Scholar]
- Di Cesare, P.E.; Chen, F.S.; Moergelin, M.; Carlson, C.S.; Leslie, M.P.; Perris, R.; Fang, C. Matrix-matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix Biol. 2002, 21, 461–470. [Google Scholar] [CrossRef]
- Rosenberg, K.; Olsson, H.; Morgelin, M.; Heinegard, D. Cartilage oligomeric matrix protein shows high affinity zinc-dependent interaction with triple helical collagen. J. Biol. Chem. 1998, 273, 20397–20403. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, M.; Hauser, N.; Paulsson, M.; DiCesare, P.E.; Michel, B.A.; Hauselmann, H.J. Small fragments of cartilage oligomeric matrix protein in synovial fluid and serum as markers for cartilage degradation. Br. J. Rheumatol. 1997, 36, 1151–1160. [Google Scholar] [CrossRef]
- Wei, J.; Richbourgh, B.; Jia, T.; Liu, C. ADAMTS-12: A multifaced metalloproteinase in arthritis and inflammation. Mediat. Inflamm. 2014, 2014, 649718. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, Y.; Ilalov, K.; Carlson, C.S.; Feng, J.Q.; Di Cesare, P.E.; Liu, C.J. Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J. Biol. Chem. 2007, 282, 11347–11355. [Google Scholar] [CrossRef]
- Luan, Y.; Kong, L.; Howell, D.R.; Ilalov, K.; Fajardo, M.; Bai, X.H.; Di Cesare, P.E.; Goldring, M.B.; Abramson, S.B.; Liu, C.J. Inhibition of ADAMTS-7 and ADAMTS-12 degradation of cartilage oligomeric matrix protein by alpha-2-macroglobulin. Osteoarthr. Cartil. 2008, 16, 1413–1420. [Google Scholar] [CrossRef]
- Liu, C.J.; Kong, W.; Xu, K.; Luan, Y.; Ilalov, K.; Sehgal, B.; Yu, S.; Howell, R.D.; Di Cesare, P.E. ADAMTS-12 associates with and degrades cartilage oligomeric matrix protein. J. Biol. Chem. 2006, 281, 15800–15808. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J.; Kong, W.; Ilalov, K.; Yu, S.; Xu, K.; Prazak, L.; Fajardo, M.; Sehgal, B.; Di Cesare, P.E. ADAMTS-7: A metalloproteinase that directly binds to and degrades cartilage oligomeric matrix protein. FASEB J. 2006, 20, 988–990. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Bai, X.; Zhao, Y.; Tian, Q.; Liu, B.; Lin, E.A.; Chen, Y.; Lee, B.; CT, G.A.; Beier, F.; et al. ADAMTS-7 forms a positive feedback loop with TNF-alpha in the pathogenesis of osteoarthritis. Ann. Rheum. Dis. 2014, 73, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Lapiere, C.M.; Lenaers, A.; Kohn, L.D. Procollagen peptidase: An enzyme excising the coordination peptides of procollagen. Proc. Natl. Acad. Sci. USA 1971, 68, 3054–3058. [Google Scholar] [CrossRef]
- Colige, A.; Nuytinck, L.; Hausser, I.; van Essen, A.J.; Thiry, M.; Herens, C.; Ades, L.C.; Malfait, F.; Paepe, A.D.; Franck, P.; et al. Novel types of mutation responsible for the dermatosparactic type of Ehlers-Danlos syndrome (Type VIIC) and common polymorphisms in the ADAMTS2 gene. J. Investig. Dermatol. 2004, 123, 656–663. [Google Scholar] [CrossRef]
- Colige, A.; Li, S.W.; Sieron, A.L.; Nusgens, B.V.; Prockop, D.J.; Lapiere, C.M. cDNA cloning and expression of bovine procollagen I N-proteinase: A new member of the superfamily of zinc-metalloproteinases with binding sites for cells and other matrix components. Proc. Natl. Acad. Sci. USA 1997, 94, 2374–2379. [Google Scholar] [CrossRef]
- Dubail, J.; Kesteloot, F.; Deroanne, C.; Motte, P.; Lambert, V.; Rakic, J.M.; Lapiere, C.; Nusgens, B.; Colige, A. ADAMTS-2 functions as anti-angiogenic and anti-tumoral molecule independently of its catalytic activity. Cell Mol. Life Sci. 2010, 67, 4213–4232. [Google Scholar] [CrossRef]
- Fernandes, R.J.; Hirohata, S.; Engle, J.M.; Colige, A.; Cohn, D.H.; Eyre, D.R.; Apte, S.S. Procollagen II amino propeptide processing by ADAMTS-3. Insights on dermatosparaxis. J. Biol. Chem. 2001, 276, 31502–31509. [Google Scholar] [CrossRef]
- Le Goff, C.; Somerville, R.P.; Kesteloot, F.; Powell, K.; Birk, D.E.; Colige, A.C.; Apte, S.S. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: Insights on collagen biosynthesis and dermatosparaxis. Development 2006, 133, 1587–1596. [Google Scholar] [CrossRef]
- Bekhouche, M.; Colige, A. The procollagen N-proteinases ADAMTS2, 3 and 14 in pathophysiology. Matrix Biol. 2015, 44–46, 46–53. [Google Scholar] [CrossRef]
- Loeser, R.F.; Olex, A.L.; McNulty, M.A.; Carlson, C.S.; Callahan, M.; Ferguson, C.; Fetrow, J.S. Disease progression and phasic changes in gene expression in a mouse model of osteoarthritis. PLoS ONE 2013, 8, e54633. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lopez, J.; Pombo-Suarez, M.; Loughlin, J.; Tsezou, A.; Blanco, F.J.; Meulenbelt, I.; Slagboom, P.E.; Valdes, A.M.; Spector, T.D.; Gomez-Reino, J.J.; et al. Association of a nsSNP in ADAMTS14 to some osteoarthritis phenotypes. Osteoarthr. Cartil. 2009, 17, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Poonpet, T.; Honsawek, S.; Tammachote, N.; Kanitnate, S.; Tammachote, R. ADAMTS14 gene polymorphism associated with knee osteoarthritis in Thai women. Genet. Mol Res. 2013, 12, 5301–5309. [Google Scholar] [CrossRef] [PubMed]
- Cain, S.A.; Mularczyk, E.J.; Singh, M.; Massam-Wu, T.; Kielty, C.M. ADAMTS-10 and -6 differentially regulate cell-cell junctions and focal adhesions. Sci. Rep. 2016, 6, 35956. [Google Scholar] [CrossRef]
- Brocker, C.N.; Vasiliou, V.; Nebert, D.W. Evolutionary divergence and functions of the ADAM and ADAMTS gene families. Hum. Genom. 2009, 4, 43–55. [Google Scholar] [CrossRef]
- Jorgenson, E.; Makki, N.; Shen, L.; Chen, D.C.; Tian, C.; Eckalbar, W.L.; Hinds, D.; Ahituv, N.; Avins, A. A genome-wide association study identifies four novel susceptibility loci underlying inguinal hernia. Nat. Commun. 2015, 6, 10130. [Google Scholar] [CrossRef]
- Morales, J.; Al-Sharif, L.; Khalil, D.S.; Shinwari, J.M.; Bavi, P.; Al-Mahrouqi, R.A.; Al-Rajhi, A.; Alkuraya, F.S.; Meyer, B.F.; Al Tassan, N. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am. J. Hum. Genet. 2009, 85, 558–568. [Google Scholar] [CrossRef]
- Dutu, A.; Vlaicu-Rus, V.; Bolosiu, H.D.; Parasca, I.; Cristea, A. Fibronectin in plasma and synovial fluid of patients with rheumatic diseases. Med. Interne 1986, 24, 61–68. [Google Scholar]
- Homandberg, G.A.; Meyers, R.; Xie, D.L. Fibronectin fragments cause chondrolysis of bovine articular cartilage slices in culture. J. Biol. Chem. 1992, 267, 3597–3604. [Google Scholar]
- Scott, D.L.; Wainwright, A.C.; Walton, K.W.; Williamson, N. Significance of fibronectin in rheumatoid arthritis and osteoarthrosis. Ann. Rheum. Dis. 1981, 40, 142–153. [Google Scholar] [CrossRef]
- Homandberg, G.A.; Wen, C. Exposure of cartilage to a fibronectin fragment amplifies catabolic processes while also enhancing anabolic processes to limit damage. J. Orthop. Res. 1998, 16, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Homandberg, G.A.; Wen, C.; Hui, F. Cartilage damaging activities of fibronectin fragments derived from cartilage and synovial fluid. Osteoarthr. Cartil. 1998, 6, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, K.; Shanthi, C. Matrikines for therapeutic and biomedical applications. Life Sci. 2018, 214, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Guo, D.; Homandberg, G.A.; Buckwalter, J.A.; Martin, J.A. A single blunt impact on cartilage promotes fibronectin fragmentation and upregulates cartilage degrading stromelysin-1/matrix metalloproteinase-3 in a bovine ex vivo model. J. Orthop. Res. 2014, 32, 811–818. [Google Scholar] [CrossRef]
- Hwang, H.S.; Choi, M.H.; Kim, H.A. 29-kDa FN-f inhibited autophagy through modulating localization of HMGB1 in human articular chondrocytes. BMB Rep. 2018, 51, 508–513. [Google Scholar] [CrossRef]
- Buchbinder, R.; van Tulder, M.; Oberg, B.; Costa, L.M.; Woolf, A.; Schoene, M.; Croft, P.; Lancet Low Back Pain Series Working Group. Low back pain: A call for action. Lancet 2018, 391, 2384–2388. [Google Scholar] [CrossRef]
- Scheele, J.; de Schepper, E.I.; van Meurs, J.B.; Hofman, A.; Koes, B.W.; Luijsterburg, P.A.; Bierma-Zeinstra, S.M. Association between spinal morning stiffness and lumbar disc degeneration: The Rotterdam Study. Osteoarthr. Cartil. 2012, 20, 982–987. [Google Scholar] [CrossRef]
- Rustenburg, C.M.E.; Emanuel, K.S.; Peeters, M.; Lems, W.F.; Vergroesen, P.A.; Smit, T.H. Osteoarthritis and intervertebral disc degeneration: Quite different, quite similar. Jor Spine 2018, 1, e1033. [Google Scholar] [CrossRef]
- Oegema, T.R., Jr.; Johnson, S.L.; Aguiar, D.J.; Ogilvie, J.W. Fibronectin and its fragments increase with degeneration in the human intervertebral disc. Spine 2000, 25, 2742–2747. [Google Scholar] [CrossRef]
- Ruel, N.; Markova, D.Z.; Adams, S.L.; Scanzello, C.; Cs-Szabo, G.; Gerard, D.; Shi, P.; Anderson, D.G.; Zack, M.; An, H.S.; et al. Fibronectin fragments and the cleaving enzyme ADAM-8 in the degenerative human intervertebral disc. Spine 2014, 39, 1274–1279. [Google Scholar] [CrossRef]
- Baker, M.; Brook, B.S.; Owen, M.R. Mathematical modelling of cytokines, MMPs and fibronectin fragments in osteoarthritic cartilage. J. Math. Biol. 2017, 75, 985–1024. [Google Scholar] [CrossRef] [PubMed]
- Werb, Z.; Tremble, P.M.; Behrendtsen, O.; Crowley, E.; Damsky, C.H. Signal transduction through the fibronectin receptor induces collagenase and stromelysin gene expression. J. Cell Biol. 1989, 109, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Stanton, H.; Ung, L.; Fosang, A.J. The 45 kDa collagen-binding fragment of fibronectin induces matrix metalloproteinase-13 synthesis by chondrocytes and aggrecan degradation by aggrecanases. Biochem. J. 2002, 364, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Homandberg, G.A.; Meyers, R.; Williams, J.M. Intraarticular injection of fibronectin fragments causes severe depletion of cartilage proteoglycans in vivo. J. Rheumatol. 1993, 20, 1378–1382. [Google Scholar] [PubMed]
- Homandberg, G.A.; Kang, Y.; Zhang, J.; Cole, A.A.; Williams, J.M. A single injection of fibronectin fragments into rabbit knee joints enhances catabolism in the articular cartilage followed by reparative responses but also induces systemic effects in the non-injected knee joints. Osteoarthr. Cartil. 2001, 9, 673–683. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Homandberg, G.A.; Costa, V.; Ummadi, V.; Pichika, R. Antisense oligonucleotides to the integrin receptor subunit alpha(5) decrease fibronectin fragment mediated cartilage chondrolysis. Osteoarthr. Cartil. 2002, 10, 381–393. [Google Scholar] [CrossRef]
- Arner, E.C.; Tortorella, M.D. Signal transduction through chondrocyte integrin receptors induces matrix metalloproteinase synthesis and synergizes with interleukin-1. Arthritis Rheum. 1995, 38, 1304–1314. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Pulai, J.; Loeser, R.F. Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002, 46, 2368–2376. [Google Scholar] [CrossRef]
- Long, D.L.; Willey, J.S.; Loeser, R.F. Rac1 is required for matrix metalloproteinase-13 production by chondrocytes in response to fibronectin fragments. Arthritis Rheum. 2013, 65, 1561–1568. [Google Scholar] [CrossRef]
- Tremble, P.; Damsky, C.H.; Werb, Z. Components of the nuclear signaling cascade that regulate collagenase gene expression in response to integrin-derived signals. J. Cell Biol. 1995, 129, 1707–1720. [Google Scholar] [CrossRef]
- Yasuda, T.; Poole, A.R. A fibronectin fragment induces type II collagen degradation by collagenase through an interleukin-1-mediated pathway. Arthritis Rheum. 2002, 46, 138–148. [Google Scholar] [CrossRef]
- Yasuda, T.; Poole, A.R.; Shimizu, M.; Nakagawa, T.; Julovi, S.M.; Tamamura, H.; Fujii, N.; Nakamura, T. Involvement of CD44 in induction of matrix metalloproteinases by a COOH-terminal heparin-binding fragment of fibronectin in human articular cartilage in culture. Arthritis Rheum. 2003, 48, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Shimizu, M.; Nakagawa, T.; Julovi, S.M.; Nakamura, T. Matrix metalloproteinase production by COOH-terminal heparin-binding fibronectin fragment in rheumatoid synovial cells. Lab. Investig. 2003, 83, 153–162. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hartney, J.M.; Gustafson, C.E.; Bowler, R.P.; Pelanda, R.; Torres, R.M. Thromboxane receptor signaling is required for fibronectin-induced matrix metalloproteinase 9 production by human and murine macrophages and is attenuated by the Arhgef1 molecule. J. Biol. Chem. 2011, 286, 44521–44531. [Google Scholar] [CrossRef]
- Moroz, A.; Delella, F.K.; Lacorte, L.M.; Deffune, E.; Felisbino, S.L. Fibronectin induces MMP2 expression in human prostate cancer cells. Biochem. Biophys. Res. Commun. 2013, 430, 1319–1321. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, C.T.; Bhargava, M.; Torzilli, P.A. A Comparative Study of Fibronectin Cleavage by MMP-1, -3, -13, and -14. Cartilage 2012, 3, 267–277. [Google Scholar] [CrossRef]
- Zhen, E.Y.; Brittain, I.J.; Laska, D.A.; Mitchell, P.G.; Sumer, E.U.; Karsdal, M.A.; Duffin, K.L. Characterization of metalloprotease cleavage products of human articular cartilage. Arthritis Rheum. 2008, 58, 2420–2431. [Google Scholar] [CrossRef]
- Ding, L.; Buckwalter, J.A.; Martin, J.A. DAMPs Synergize with Cytokines or Fibronectin Fragment on Inducing Chondrolysis but Lose Effect When Acting Alone. Mediat. Inflamm 2017, 2017, 2642549. [Google Scholar] [CrossRef]
- Guo, D.; Ding, L.; Homandberg, G.A. Telopeptides of type II collagen upregulate proteinases and damage cartilage but are less effective than highly active fibronectin fragments. Inflamm. Res. 2009, 58, 161–169. [Google Scholar] [CrossRef]
- Gendron, C.; Kashiwagi, M.; Lim, N.H.; Enghild, J.J.; Thogersen, I.B.; Hughes, C.; Caterson, B.; Nagase, H. Proteolytic activities of human ADAMTS-5: Comparative studies with ADAMTS-4. J. Biol. Chem. 2007, 282, 18294–18306. [Google Scholar] [CrossRef]
- Hashimoto, G.; Shimoda, M.; Okada, Y. ADAMTS4 (aggrecanase-1) interaction with the C-terminal domain of fibronectin inhibits proteolysis of aggrecan. J. Biol. Chem. 2004, 279, 32483–32491. [Google Scholar] [CrossRef] [PubMed]
- Schnellmann, R.; Sack, R.; Hess, D.; Annis, D.S.; Mosher, D.F.; Apte, S.S.; Chiquet-Ehrismann, R. A Selective Extracellular Matrix Proteomics Approach Identifies Fibronectin Proteolysis by A Disintegrin-like and Metalloprotease Domain with Thrombospondin Type 1 Motifs (ADAMTS16) and Its Impact on Spheroid Morphogenesis. Mol. Cell Proteom. 2018, 17, 1410–1425. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Nandadasa, S.; Annis, D.S.; Dubail, J.; Mosher, D.F.; Willard, B.B.; Apte, S.S. A disintegrin-like and metalloproteinase domain with thrombospondin type 1 motif 9 (ADAMTS9) regulates fibronectin fibrillogenesis and turnover. J. Biol. Chem. 2019, 294, 9924–9936. [Google Scholar] [CrossRef] [PubMed]
- De Langhe, S.P.; Sala, F.G.; Del Moral, P.M.; Fairbanks, T.J.; Yamada, K.M.; Warburton, D.; Burns, R.C.; Bellusci, S. Dickkopf-1 (DKK1) reveals that fibronectin is a major target of Wnt signaling in branching morphogenesis of the mouse embryonic lung. Dev. Biol. 2005, 277, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Sen, M.; Reifert, J.; Lauterbach, K.; Wolf, V.; Rubin, J.S.; Corr, M.; Carson, D.A. Regulation of fibronectin and metalloproteinase expression by Wnt signaling in rheumatoid arthritis synoviocytes. Arthritis Rheum. 2002, 46, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Bielefeld, K.A.; Amini-Nik, S.; Whetstone, H.; Poon, R.; Youn, A.; Wang, J.; Alman, B.A. Fibronectin and beta-catenin act in a regulatory loop in dermal fibroblasts to modulate cutaneous healing. J. Biol. Chem. 2011, 286, 27687–27697. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Enzyme | Main Substrates |
|---|---|---|
| Collagenases | MMP-1 (collagenase-1) | Collagen I, II, III, VII, VIII, X, and XI, gelatin, entactin, tenascin, aggrecan, fibronectin, vitronectin, myelin basic protein, ovostatin, casein, MMP-2, MMP-9, proMMPs |
| MMP-8 (collagenase-2) | Collagen I, II, and III, fibronectin, prostaglandins, aggrecan, ovostatin | |
| MMP-13 (collagenase-3) | Collagen I, II, III, IV, IX, X and XIV, gelatin, tenascin, plasminogen, osteonectin, fibronectin, aggrecan, casein | |
| MMP-18 | Collagen, gelatin | |
| Gelatinases | MMP-2 (gelatinase A) | Collagen I, III, IV, V, VII and X, gelatin, fibronectin, laminin, aggrecan, elastin, vitronectin, tenascin, myelin basic protein |
| MMP-9 (gelatinase B) | Collagen IV, V, XI, elastin, aggrecan, decorin, laminin, entactin, myelin basic protein, casein | |
| Stromelysins | MMP-3 (stromelysin-1) | Collagen III, IV, V, IX, X and XI, gelatin, aggrecan, elastin, fibronectin, vitronectin, laminin, entactin, tenascin, decorin, myelin basic protein, ovostatin, casein, osteonectin, proMMPs |
| MMP-10 (stromelysin-2) | Collagen III, IV and V, gelatin, elastin, fibronectin, aggrecan, casein | |
| MMP-11 (stromelysin-3) | Gelatin, fibronectin, collagen IV, laminin, elastin, casein, prostaglandins | |
| MMP-27 | Gelatin | |
| Matrilysins | MMP-7 | Collagen I and IV, gelatin, elastin fibronectin, vitronectin, laminin, entactin, tenascin, aggrecan, myelin, proMMP-1, proMMP-2, proMMP-9 transferrin, casein |
| MMP-26 | Collagen IV, gelatin, fibronectin, fibrinogen, pro-MMP9 | |
| MT-MMPs | MMP-14 (MT1-MMP) | Collagen I, II and III, gelatin, fibronectin, tenascin, vitronectin, laminin, entactin, aggrecan, vibronectin, pro-MMP2 |
| MMP-15 (MT2-MMP) | Fibronectin, tenascin, entactin, laminin, aggrecan, gelatin, vibronectin, pro-MMP2 | |
| MMP-16 (MT3-MMP) | Collagen III, gelatin, fibronectin, casein, laminin, pro MMP-2 | |
| MMP-17 (MT4-MMP) | Gelatin, fibrinogen, pro MMP-2 | |
| MMP-24 (MT5-MMP) | Fibronectin, gelatin, proteoglycans, pro-MMP2 | |
| MMP-25 (MT6-MMP) | Collagen IV, gelatin, fibronectin, proteoglycans, pro-MMP2 | |
| Other MMPs | MMP-12 | Collagen I, V and IV, gelatin, elastin, fibronectin, vitronectin, laminin, entactin, osteonectin, aggrecan, myelin, vitronectin, fibrinogen |
| MMP-19 | Collagen I and IV, gelatin, lamin, entactin, fibronectin, aggrecan | |
| MMP-20 | Amelogenin, aggrecan | |
| MMP-21 | Not defined | |
| MMP-23 | Gelatin | |
| MMP-28 | Caseine |
| Family | Enzyme | Main Substrates |
|---|---|---|
| Agrecanases and proteoglycanases | ADAMTS1 ADAMTS4 ADAMTS5 ADAMTS8 ADAMTS9 ADAMTS15 ADAMTS20 | Proteoglycans: aggrecan, versican, brevican, neurocan, fibromodulin, decorin, carboxylmethylated transferrin |
| COMP proteinases | ADAMTS7 ADAMTS12 | COMP |
| Procollagen N-peptidases | ADAMTS2 ADAMTS3 ADAMTS14 | Procollagen |
| von-Willebrand factor proteinases | ADAMTS13 | von-Willebrand coagulation factor |
| Other ADAMTS | ADAMTS6 ADAMTS10 ADAMTS16 ADAMTS17 ADAMTS18 ADAMTS19 | Not defined |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-García, S.; Carrión, M.; Gutiérrez-Cañas, I.; Villanueva-Romero, R.; Castro, D.; Martínez, C.; González-Álvaro, I.; Blanco, F.J.; Juarranz, Y.; Gomariz, R.P. Profile of Matrix-Remodeling Proteinases in Osteoarthritis: Impact of Fibronectin. Cells 2020, 9, 40. https://doi.org/10.3390/cells9010040
Pérez-García S, Carrión M, Gutiérrez-Cañas I, Villanueva-Romero R, Castro D, Martínez C, González-Álvaro I, Blanco FJ, Juarranz Y, Gomariz RP. Profile of Matrix-Remodeling Proteinases in Osteoarthritis: Impact of Fibronectin. Cells. 2020; 9(1):40. https://doi.org/10.3390/cells9010040
Chicago/Turabian StylePérez-García, Selene, Mar Carrión, Irene Gutiérrez-Cañas, Raúl Villanueva-Romero, David Castro, Carmen Martínez, Isidoro González-Álvaro, Francisco J. Blanco, Yasmina Juarranz, and Rosa P. Gomariz. 2020. "Profile of Matrix-Remodeling Proteinases in Osteoarthritis: Impact of Fibronectin" Cells 9, no. 1: 40. https://doi.org/10.3390/cells9010040
APA StylePérez-García, S., Carrión, M., Gutiérrez-Cañas, I., Villanueva-Romero, R., Castro, D., Martínez, C., González-Álvaro, I., Blanco, F. J., Juarranz, Y., & Gomariz, R. P. (2020). Profile of Matrix-Remodeling Proteinases in Osteoarthritis: Impact of Fibronectin. Cells, 9(1), 40. https://doi.org/10.3390/cells9010040