Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Mice
2.2. Endothelial Cells, Macrophages, and Aortic Smooth Muscle Cell Isolations and Quantitative PCR
2.3. shRNA Infection
2.4. Hindlimb Ischemia Model
2.5. Micro-CT Imaging
2.6. Western Blot
2.7. Immunofluorescent Staining
2.8. xCELLigence Real-Time Cell Analysis (RTCA)
2.9. Endothelial Migration
2.10. Statistical Analyses
3. Results
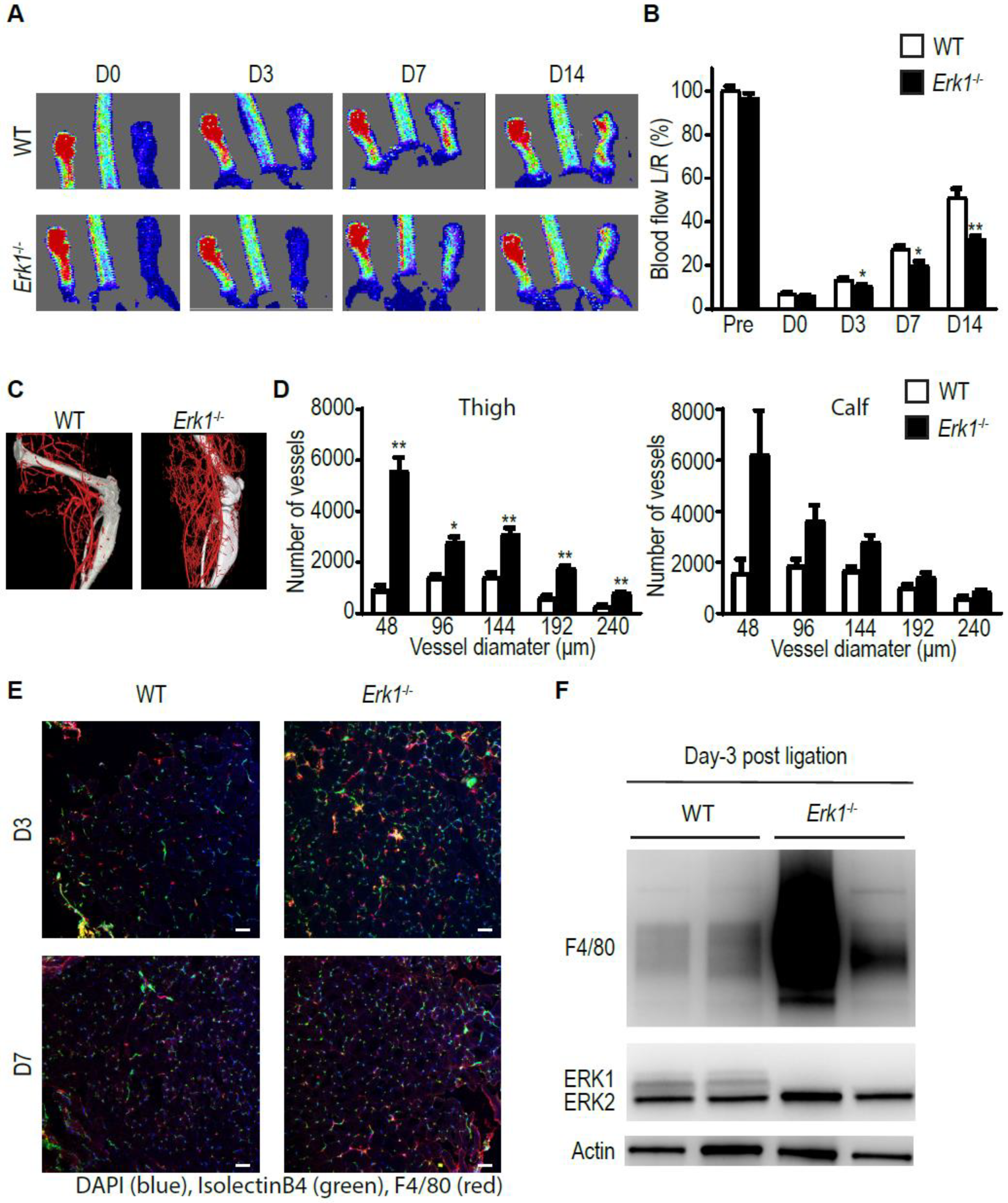
3.1. Erk1 KO Mice Exhibit Excessive but Poorly Functional Arteriogenesis
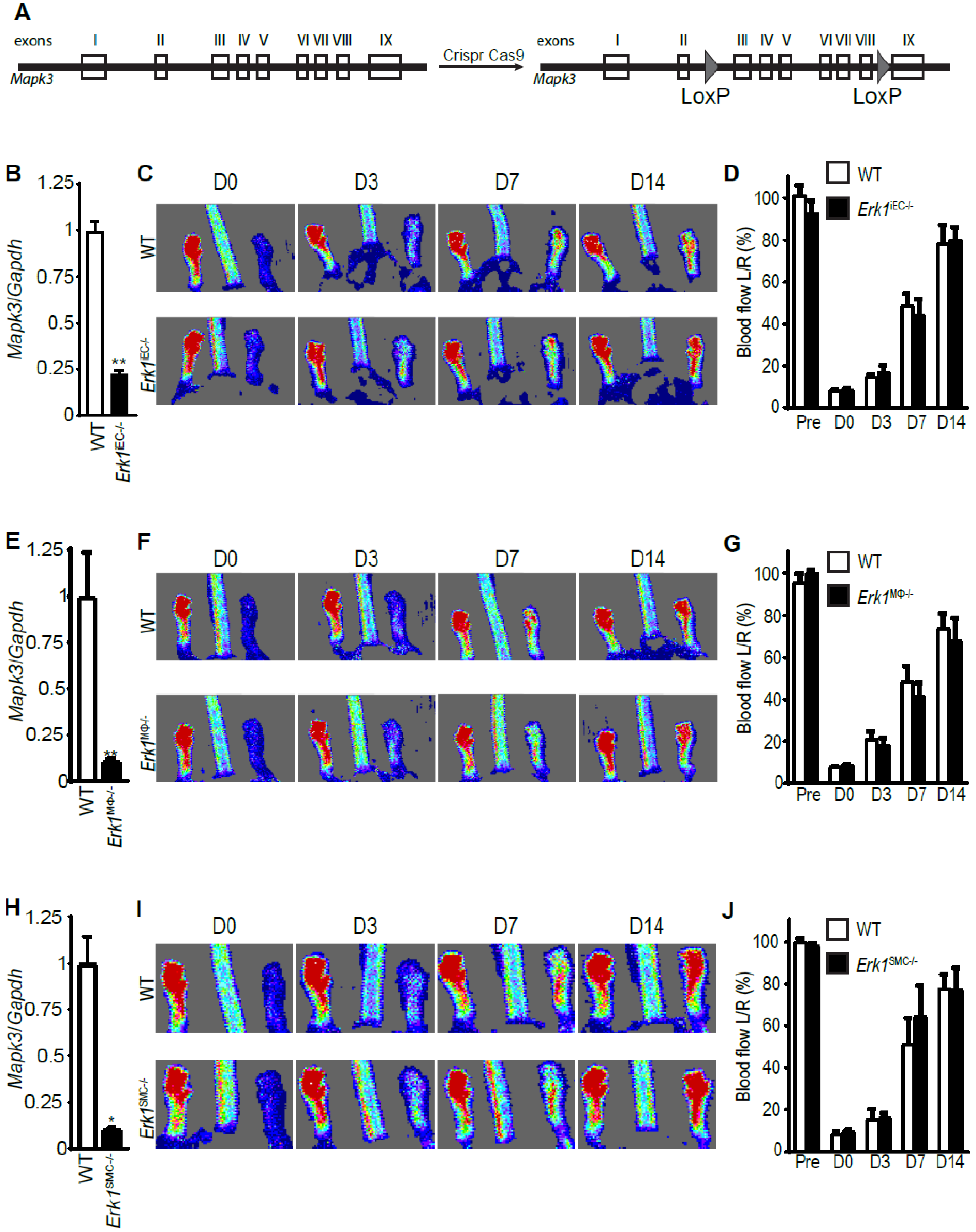
3.2. Erk1 Deletions in Endothelial Cells, Macrophages, or Smooth Muscle Cells Do Not Affect Arteriogenesis
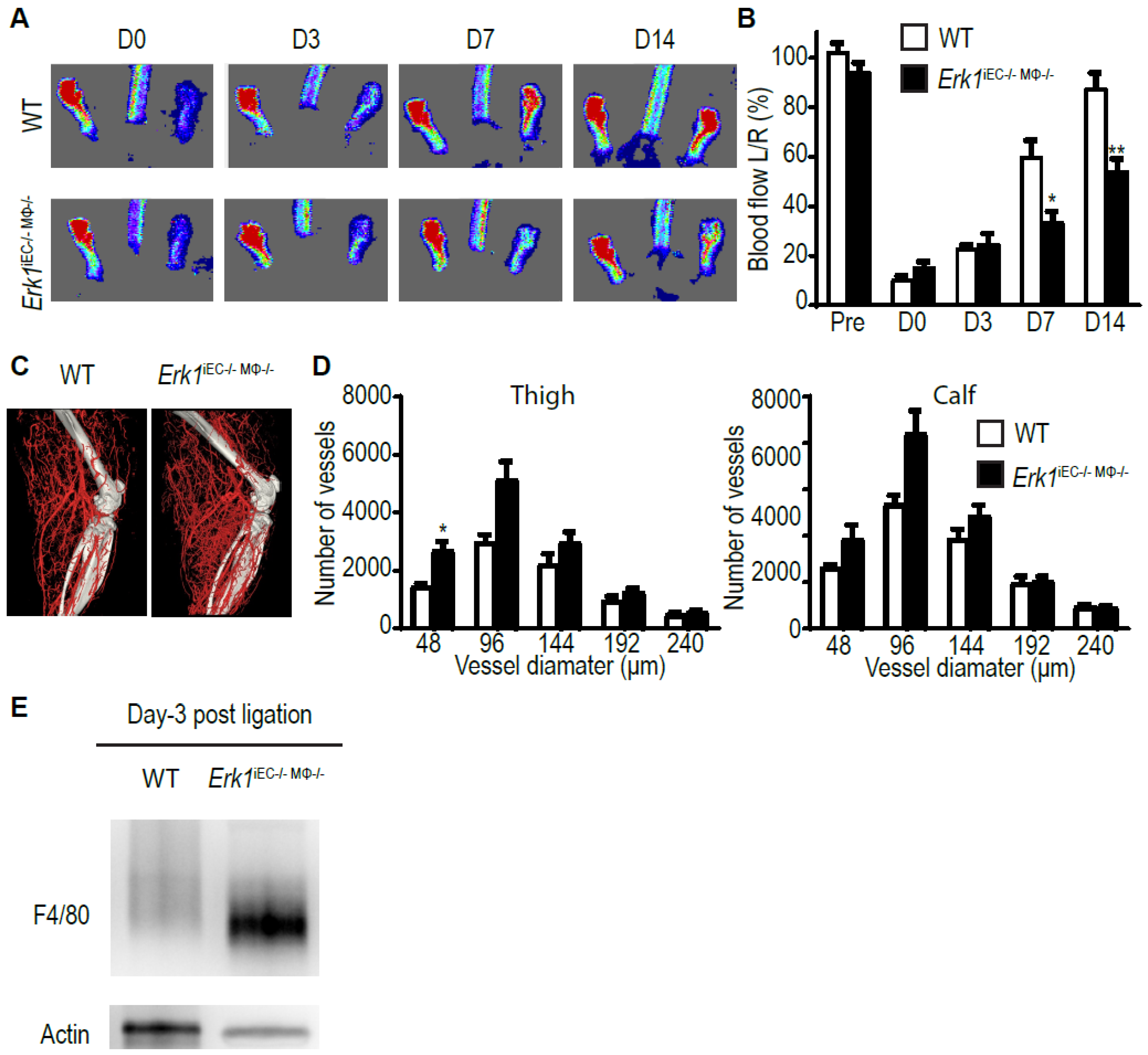
3.3. Erk1 Deletions in Endothelial Cells and Macrophages Leads to an Excessive but Poorly Functional Arteriogenesis
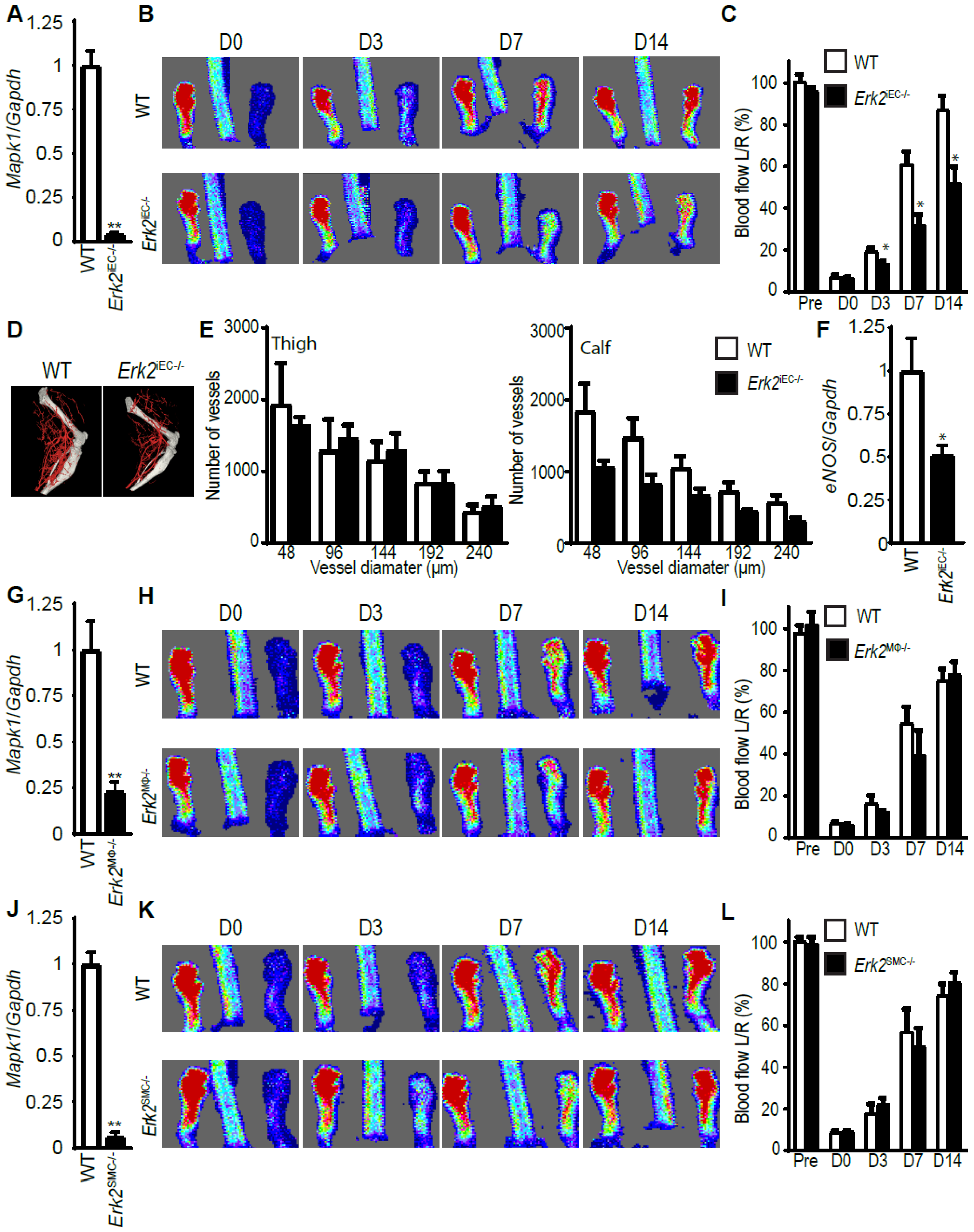
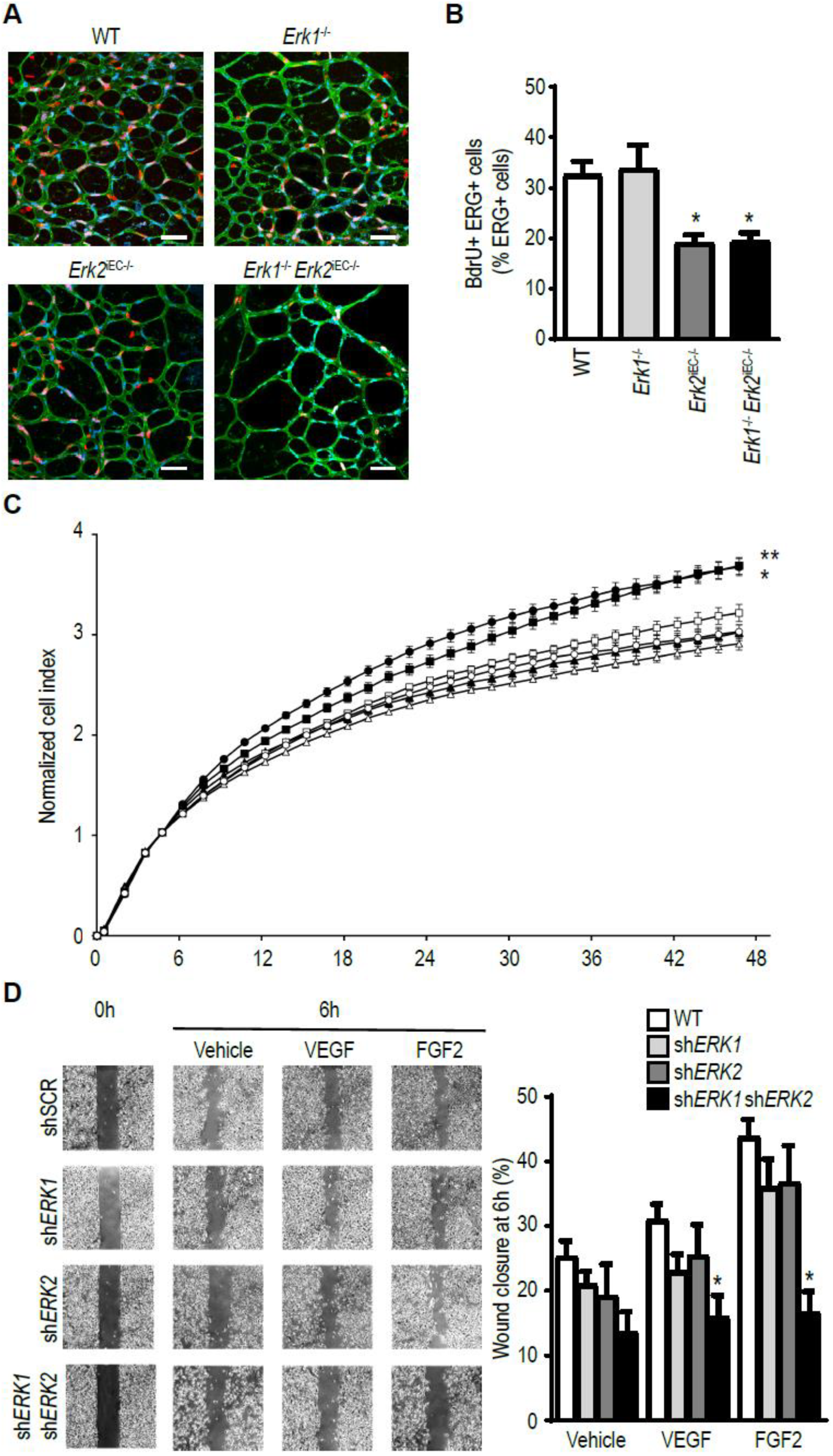
3.4. Erk2 Deletions in Endothelial, but Not Other Cell Types, Decreases Arteriogenesis
3.5. ERK Isoform Effect on Endothelial Cell Proliferation and Migration
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Rizzi, A.; Benagiano, V.; Ribatti, D. Angiogenesis versus arteriogenesis. Rom. J. Morphol. Embryol. 2017, 58, 15–19. [Google Scholar] [PubMed]
- Faber, J.E.; Chilian, W.M.; Deindl, E.; Van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Stabile, E.; Kinnaird, T.; Shou, M.; Devaney, J.M.; Epstein, S.E.; Burnett, M.S. Temporal patterns of gene expression after acute hindlimb ischemia in mice: Insights into the genomic program for collateral vessel development. J. Am. Coll. Cardiol. 2004, 43, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Scholz, D.; Ziegelhoeffer, T.; Helisch, A.; Wagner, S.; Friedrich, C.; Podzuweit, T.; Schaper, W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J. Mol. Cell. Cardiol. 2002, 34, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Eichmann, A. Molecular controls of arterial morphogenesis. Circ. Res. 2015, 116, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Moraes, F.; Paye, J.; Mac Gabhann, F.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Simons, M. Endothelial cell-dependent regulation of arteriogenesis. Circ. Res. 2013, 113, 1076–1086. [Google Scholar] [CrossRef]
- Lanahan, A.A.; Lech, D.; Dubrac, A.; Zhang, J.; Zhuang, Z.W.; Eichmann, A.; Simons, M. Ptp1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 2014, 130, 902–909. [Google Scholar] [CrossRef]
- Stabile, E.; Burnett, M.S.; Watkins, C.; Kinnaird, T.; Bachis, A.; La Sala, A.; Miller, J.M.; Shou, M.; Epstein, S.E.; Fuchs, S. Impaired arteriogenic response to acute hindlimb ischemia in cd4-knockout mice. Circulation 2003, 108, 205–210. [Google Scholar] [CrossRef]
- Stabile, E.; Kinnaird, T.; La Sala, A.; Hanson, S.K.; Watkins, C.; Campia, U.; Shou, M.; Zbinden, S.; Fuchs, S.; Kornfeld, H.; et al. Cd8+ t lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting cd4+ mononuclear cells through the expression of interleukin-16. Circulation 2006, 113, 118–124. [Google Scholar] [CrossRef]
- Van Weel, V.; Toes, R.E.; Seghers, L.; Deckers, M.M.; De Vries, M.R.; Eilers, P.H.; Sipkens, J.; Schepers, A.; Eefting, D.; Van Hinsbergh, V.W.; et al. Natural killer cells and cd4+ t-cells modulate collateral artery development. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2310–2318. [Google Scholar] [CrossRef]
- Krishnasamy, K.; Limbourg, A.; Kapanadze, T.; Gamrekelashvili, J.; Beger, C.; Hager, C.; Lozanovski, V.J.; Falk, C.S.; Napp, L.C.; Bauersachs, J.; et al. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun. 2017, 8, 952. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. A new role of mast cells in arteriogenesis. Microvasc. Res. 2018, 118, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hampton, T.; Morgan, J.P.; Simons, M. Stretch-induced vegf expression in the heart. J. Clin. Investig. 1997, 100, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Pipp, F.; Heil, M.; Issbrucker, K.; Ziegelhoeffer, T.; Martin, S.; Van Den Heuvel, J.; Weich, H.; Fernandez, B.; Golomb, G.; Carmeliet, P.; et al. Vegfr-1-selective vegf homologue plgf is arteriogenic: Evidence for a monocyte-mediated mechanism. Circ. Res. 2003, 92, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.R.; Yarovinsky, T.O.; Young, B.D.; Moraes, F.; Ross, T.D.; Ceneri, N.; Zhang, J.; Zhuang, Z.W.; Sinusas, A.J.; Pardi, R.; et al. Chemokine-coupled beta2 integrin-induced macrophage rac2-myosin iia interaction regulates vegf-a mrna stability and arteriogenesis. J. Exp. Med. 2014, 211, 1957–1968. [Google Scholar] [CrossRef] [PubMed]
- Ahn, G.O.; Seita, J.; Hong, B.J.; Kim, Y.E.; Bok, S.; Lee, C.J.; Kim, K.S.; Lee, J.C.; Leeper, N.J.; Cooke, J.P.; et al. Transcriptional activation of hypoxia-inducible factor-1 (hif-1) in myeloid cells promotes angiogenesis through vegf and s100a8. Proc. Natl. Acad. Sci. USA 2014, 111, 2698–2703. [Google Scholar] [CrossRef] [PubMed]
- Heil, M.; Ziegelhoeffer, T.; Pipp, F.; Kostin, S.; Martin, S.; Clauss, M.; Schaper, W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2411–H2419. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial vegf receptor signalling. Nat. Rev. Mol. Cell. Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Hatano, N.; Mori, Y.; Oh-hora, M.; Kosugi, A.; Fujikawa, T.; Nakai, N.; Niwa, H.; Miyazaki, J.; Hamaoka, T.; Ogata, M. Essential role for erk2 mitogen-activated protein kinase in placental development. Genes. Cells 2003, 8, 847–856. [Google Scholar] [CrossRef]
- Pages, G.; Guerin, S.; Grall, D.; Bonino, F.; Smith, A.; Anjuere, F.; Auberger, P.; Pouyssegur, J. Defective thymocyte maturation in p44 map kinase (erk 1) knockout mice. Science 1999, 286, 1374–1377. [Google Scholar]
- Srinivasan, R.; Zabuawala, T.; Huang, H.; Zhang, J.; Gulati, P.; Fernandez, S.; Karlo, J.C.; Landreth, G.E.; Leone, G.; Ostrowski, M.C. Erk1 and erk2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 2009, 4, e8283. [Google Scholar] [CrossRef] [PubMed]
- Ricard, N.; Scott, R.P.; Booth, C.J.; Velazquez, H.; Cilfone, N.A.; Baylon, J.L.; Gulcher, J.R.; Quaggin, S.E.; Chittenden, T.W.; Simons, M. Endothelial erk1/2 signaling maintains integrity of the quiescent endothelium. J. Exp. Med. 2019, 216, 1874–1890. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Dittel, B.N. Isolation of mouse peritoneal cavity cells. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Ziegelhoeffer, T.; Fernandez, B.; Kostin, S.; Heil, M.; Voswinckel, R.; Helisch, A.; Schaper, W. Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ. Res. 2004, 94, 230–238. [Google Scholar] [CrossRef]
- Sorensen, I.; Adams, R.H.; Gossler, A. Dll1-mediated notch activation regulates endothelial identity in mouse fetal arteries. Blood 2009, 113, 5680–5688. [Google Scholar] [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Forster, I. Conditional gene targeting in macrophages and granulocytes using lysmcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef]
- Wirth, A.; Benyo, Z.; Lukasova, M.; Leutgeb, B.; Wettschureck, N.; Gorbey, S.; Orsy, P.; Horvath, B.; Maser-Gluth, C.; Greiner, E.; et al. G12-g13-larg-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat. Med. 2008, 14, 64–68. [Google Scholar] [CrossRef]
- Lee, M.Y.; Gamez-Mendez, A.; Zhang, J.; Zhuang, Z.; Vinyard, D.J.; Kraehling, J.; Velazquez, H.; Brudvig, G.W.; Kyriakides, T.R.; Simons, M.; et al. Endothelial cell autonomous role of akt1: Regulation of vascular tone and ischemia-induced arteriogenesis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 870–879. [Google Scholar] [CrossRef]
- Mac Gabhann, F.; Peirce, S.M. Collateral capillary arterialization following arteriolar ligation in murine skeletal muscle. Microcirculation 2010, 17, 333–347. [Google Scholar] [CrossRef][Green Version]
- Heil, M.; Schaper, W. Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis). Circ. Res. 2004, 95, 449–458. [Google Scholar] [CrossRef]
- Shin, M.; Beane, T.J.; Quillien, A.; Male, I.; Zhu, L.J.; Lawson, N.D. Vegfa signals through erk to promote angiogenesis, but not artery differentiation. Development 2016, 143, 3796–3805. [Google Scholar] [CrossRef] [PubMed]
- Kofler, N.M.; Simons, M. Angiogenesis versus arteriogenesis: Neuropilin 1 modulation of vegf signaling. F1000Prime Rep. 2015, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Lucitti, J.L.; Mackey, J.K.; Morrison, J.C.; Haigh, J.J.; Adams, R.H.; Faber, J.E. Formation of the collateral circulation is regulated by vascular endothelial growth factor-a and a disintegrin and metalloprotease family members 10 and 17. Circ. Res. 2012, 111, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.C.; Peterson, Q.P.; Hong, J.Y.; Peterson, R.T. Artery/vein specification is governed by opposing phosphatidylinositol-3 kinase and map kinase/erk signaling. Curr. Biol. 2006, 16, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Deng, Y.; Mukhopadhyay, A.; Lanahan, A.A.; Zhuang, Z.W.; Moodie, K.L.; Mulligan-Kehoe, M.J.; Byzova, T.V.; Peterson, R.T.; Simons, M. Erk1/2-akt1 crosstalk regulates arteriogenesis in mice and zebrafish. J. Clin. Investig. 2010, 120, 1217–1228. [Google Scholar] [CrossRef]
- Lefloch, R.; Pouyssegur, J.; Lenormand, P. Single and combined silencing of erk1 and erk2 reveals their positive contribution to growth signaling depending on their expression levels. Mol. Cell. Biol. 2008, 28, 511–527. [Google Scholar] [CrossRef]
- Takeda, Y.; Costa, S.; Delamarre, E.; Roncal, C.; De Oliveira, R.L.; Squadrito, M.L.; Finisguerra, V.; Deschoemaeker, S.; Bruyere, F.; Wenes, M.; et al. Macrophage skewing by phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 2011, 479, 122–126. [Google Scholar] [CrossRef]
- Troidl, C.; Jung, G.; Troidl, K.; Hoffmann, J.; Mollmann, H.; Nef, H.; Schaper, W.; Hamm, C.W.; Schmitz-Rixen, T. The temporal and spatial distribution of macrophage subpopulations during arteriogenesis. Curr. Vasc. Pharmacol. 2013, 11, 5–12. [Google Scholar] [CrossRef]
- Li, J.; Brown, L.F.; Hibberd, M.G.; Grossman, J.D.; Morgan, J.P.; Simons, M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am. J. Physiol. 1996, 270, H1803–H1811. [Google Scholar] [CrossRef]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; Van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of th2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef]
- Limbourg, A.; Von Felden, J.; Jagavelu, K.; Krishnasamy, K.; Napp, L.C.; Kapopara, P.R.; Gaestel, M.; Schieffer, B.; Bauersachs, J.; Limbourg, F.P.; et al. Map-kinase activated protein kinase 2 links endothelial activation and monocyte/macrophage recruitment in arteriogenesis. PLoS ONE 2015, 10, e0138542. [Google Scholar] [CrossRef] [PubMed]
- Tirziu, D.; Jaba, I.M.; Yu, P.; Larrivee, B.; Coon, B.G.; Cristofaro, B.; Zhuang, Z.W.; Lanahan, A.A.; Schwartz, M.A.; Eichmann, A.; et al. Endothelial nuclear factor-kappab-dependent regulation of arteriogenesis and branching. Circulation 2012, 126, 2589–2600. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, B.; Shi, Y.; Faria, M.; Suchting, S.; Leroyer, A.S.; Trindade, A.; Duarte, A.; Zovein, A.C.; Iruela-Arispe, M.L.; Nih, L.R.; et al. Dll4-notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development 2013, 140, 1720–1729. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Majmundar, A.J.; Krock, B.L.; Mesquita, R.C.; Mathew, L.K.; Quinn, Z.L.; Runge, A.; Liu, L.; Kim, M.N.; Liang, J.; et al. Endothelial hif-2alpha regulates murine pathological angiogenesis and revascularization processes. J. Clin. Investig. 2012, 122, 1427–1443. [Google Scholar] [CrossRef]
- Busca, R.; Pouyssegur, J.; Lenormand, P. Erk1 and erk2 map kinases: Specific roles or functional redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar] [CrossRef]
- Busca, R.; Christen, R.; Lovern, M.; Clifford, A.M.; Yue, J.X.; Goss, G.G.; Pouyssegur, J.; Lenormand, P. Erk1 and erk2 present functional redundancy in tetrapods despite higher evolution rate of erk1. BMC Evol. Biol. 2015, 15, 179. [Google Scholar] [CrossRef]
- Fremin, C.; Saba-El-Leil, M.K.; Levesque, K.; Ang, S.L.; Meloche, S. Functional redundancy of erk1 and erk2 map kinases during development. Cell. Rep. 2015, 12, 913–921. [Google Scholar] [CrossRef]
- Blasco, R.B.; Francoz, S.; Santamaria, D.; Canamero, M.; Dubus, P.; Charron, J.; Baccarini, M.; Barbacid, M. C-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell 2011, 19, 652–663. [Google Scholar] [CrossRef]
- Lips, D.J.; Bueno, O.F.; Wilkins, B.J.; Purcell, N.H.; Kaiser, R.A.; Lorenz, J.N.; Voisin, L.; Saba-El-Leil, M.K.; Meloche, S.; Pouysségur, J.; et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation 2004, 109, 1938–1941. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ricard, N.; Zhang, J.; Zhuang, Z.W.; Simons, M. Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis. Cells 2020, 9, 38. https://doi.org/10.3390/cells9010038
Ricard N, Zhang J, Zhuang ZW, Simons M. Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis. Cells. 2020; 9(1):38. https://doi.org/10.3390/cells9010038
Chicago/Turabian StyleRicard, Nicolas, Jiasheng Zhang, Zhen W. Zhuang, and Michael Simons. 2020. "Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis" Cells 9, no. 1: 38. https://doi.org/10.3390/cells9010038
APA StyleRicard, N., Zhang, J., Zhuang, Z. W., & Simons, M. (2020). Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis. Cells, 9(1), 38. https://doi.org/10.3390/cells9010038