An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
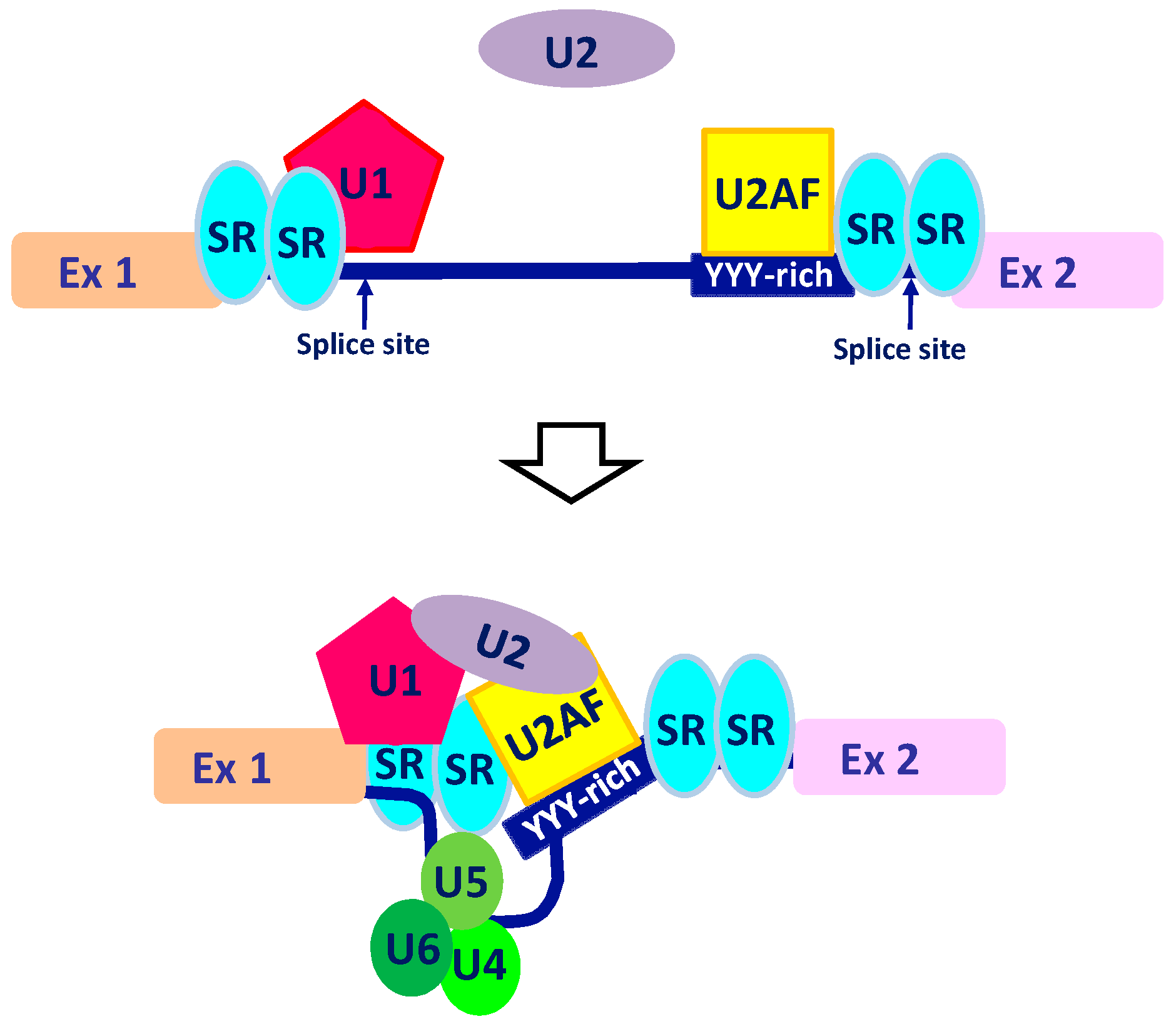
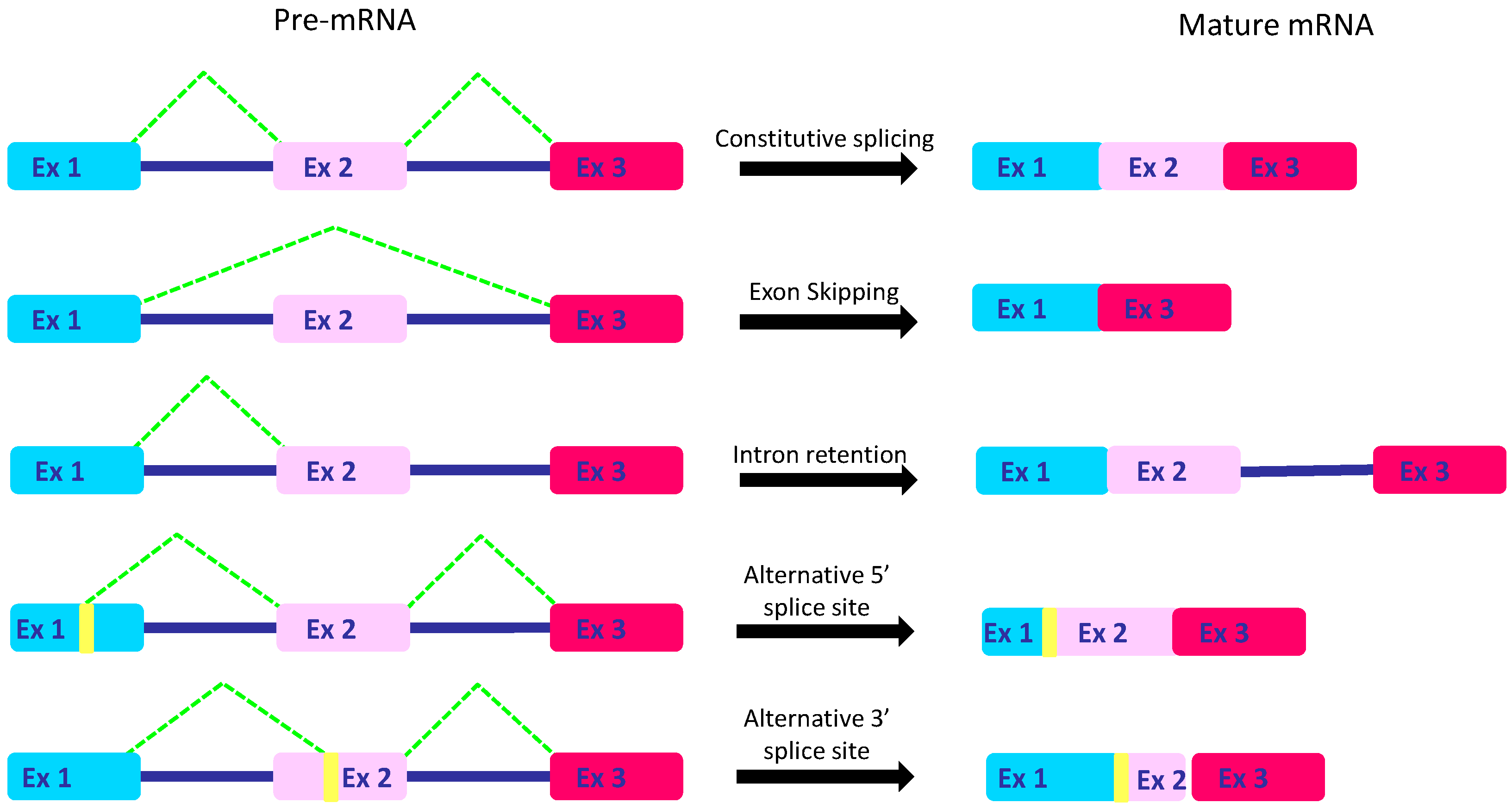
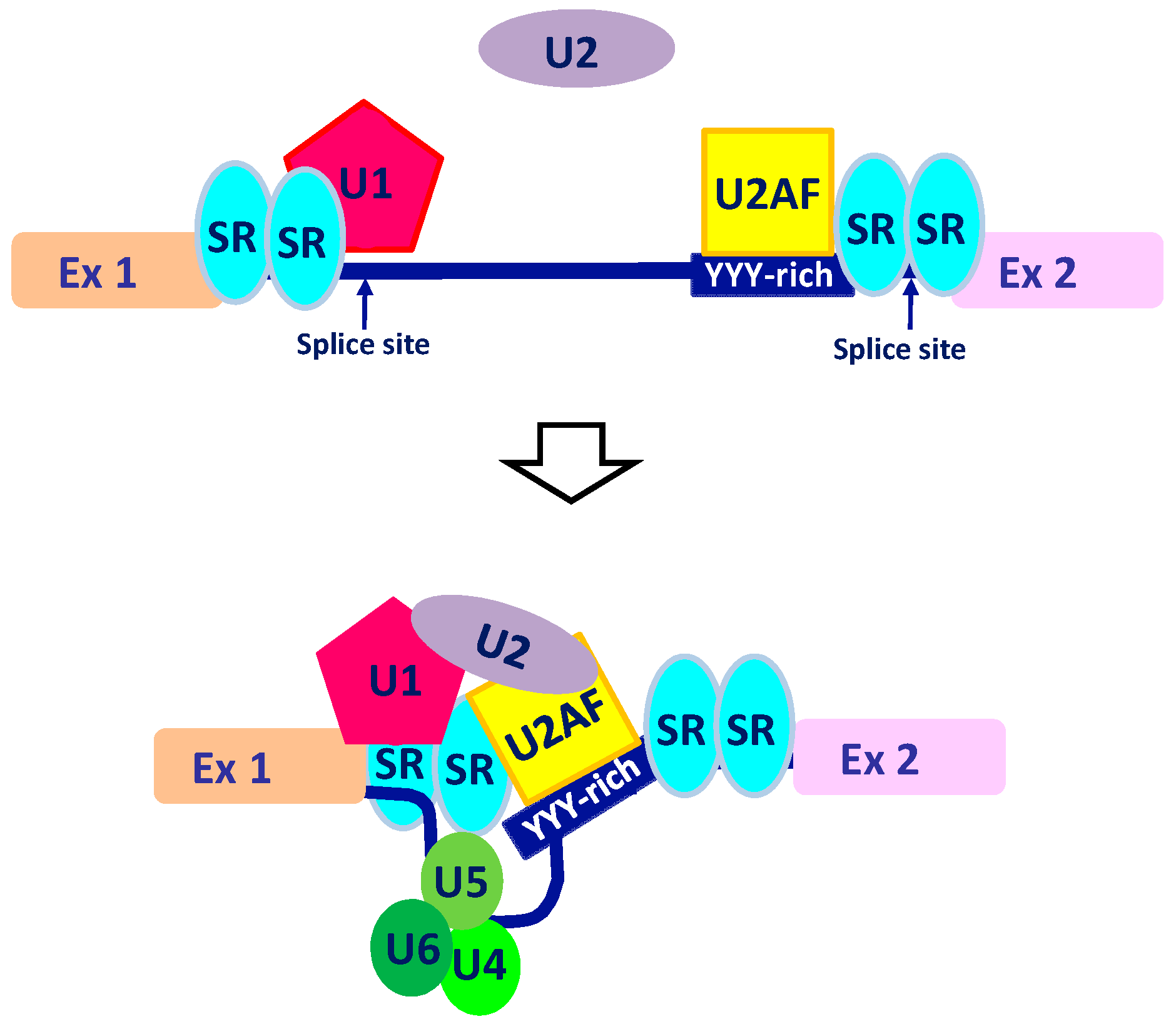
2. The Intricate Molecular Mechanisms Controlling RNA Maturation
3. AS in Development
3.1. A Finely Regulated AS Network Controls Human Embryonic Stem Cells (hESCs) Pluripotency during Embryogenesis
3.2. AS Isoforms Control Cell-Lineage Differentiation during Organogenesis
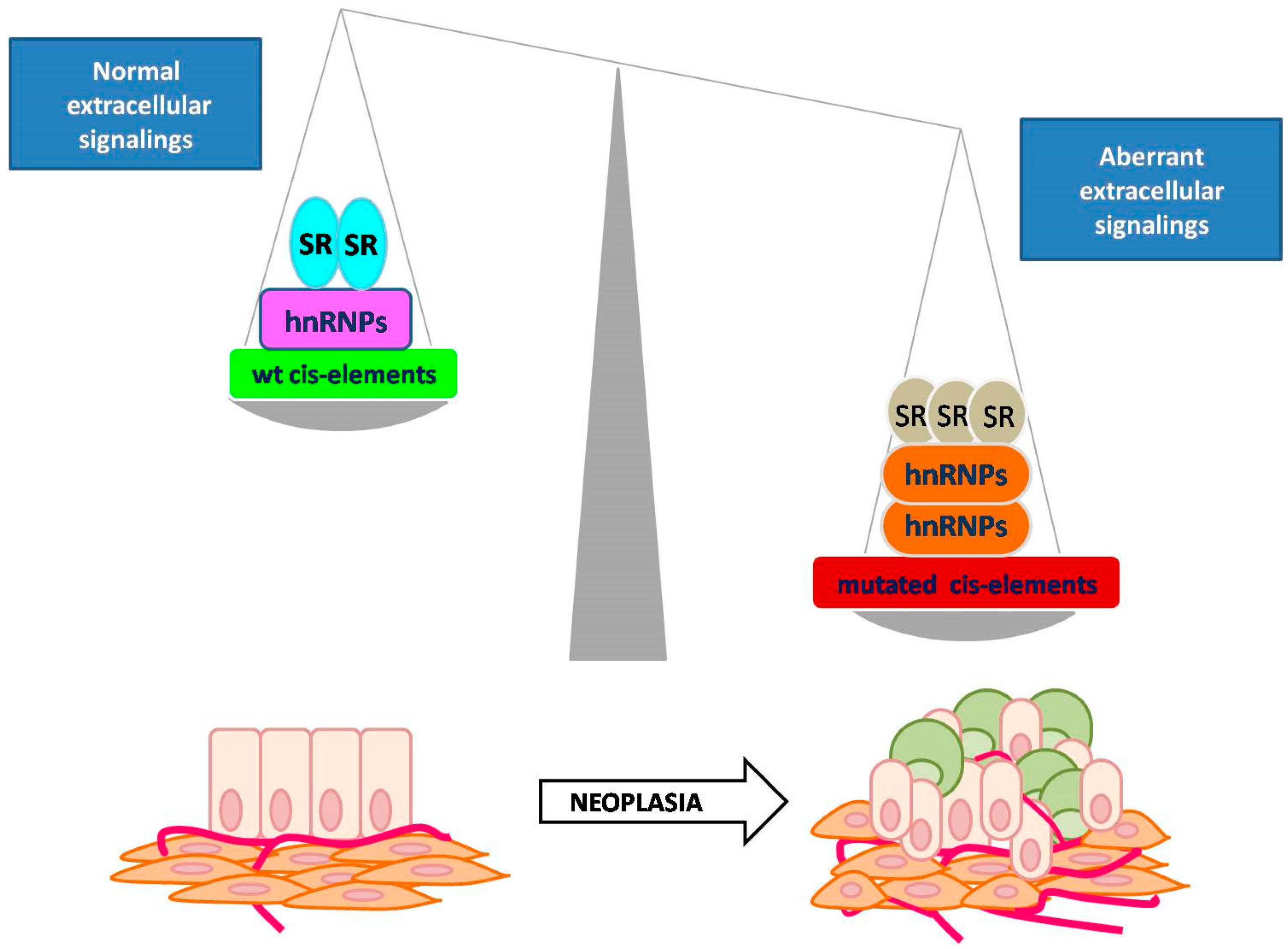
4. Contribution of AS to Tumorigenesis
4.1. Defective AS in Human Cancers
4.2. AS-Mediated Phenotypic Switch in Epithelial–Mesenchymal Transition
4.3. AS: An Important Player during Metabolic Stress and Neo-Angiogenesis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamdje, A.H.; Kamga, P.T.; Simo, R.T.; Vecchio, L.; Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Goel, R.K.; Amvene, J.M.; et al. Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol. Med. 2017, 14, 109–120. [Google Scholar]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehn, M.; Cho, R.W.; Clarke, M.F. Therapeutic implications of the cancer stem cell hypothesis. Semin. Radiat. Oncol. 2009, 19, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Invest. 2010, 120, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Becker, M.W.; Wicha, M.; Weissman, I.; Clarke, M.F. Therapeutic implications of cancer stem cells. Curr. Opin. Genet. Dev. 2004, 14, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anczukow, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Wahl, M.C.; Will, C.L.; Luhrmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef]
- Han, H.; Braunschweig, U.; Gonatopoulos-Pournatzis, T.; Weatheritt, R.J.; Hirsch, C.L.; Ha, K.C.H.; Radovani, E.; Nabeel-Shah, S.; Sterne-Weiler, T.; Wang, J.; et al. Multilayered Control of Alternative Splicing Regulatory Networks by Transcription Factors. Mol. Cell 2017, 65, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The alternative splicing side of cancer. Semin. Cell Dev. Biol. 2014, 32, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; Cartegni, L.; Jordan, P.; Paronetto, M.P. Posttranscriptional Regulation and RNA Binding Proteins in Cancer Biology. Biomed. Res. Int. 2015, 2015, 897821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, Z.; Castle, J.; Sun, S.; Johnson, J.; Krainer, A.R.; Zhang, M.Q. Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev. 2008, 22, 2550–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampietro, C.; Deflorian, G.; Gallo, S.; Matteo, D.A.; Pradella, D.; Bonomi, S.; Belloni, E.; Nyqvist, D.; Quaranta, V.; Confalonieri, S.; et al. The alternative splicing factor Nova2 regulates vascular development and lumen formation. Nat. Commun. 2015, 6, 8479. [Google Scholar] [CrossRef] [Green Version]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [Green Version]
- Lopez, A.J. Alternative splicing of pre-mRNA: Developmental consequences and mechanisms of regulation. Annu. Rev. Genet. 1998, 32, 279–305. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef]
- Naftelberg, S.; Schor, I.E.; Ast, G.; Kornblihtt, A.R. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem. 2015, 84, 165–198. [Google Scholar] [CrossRef]
- Herzel, L.; Ottoz, D.S.M.; Alpert, T.; Neugebauer, K.M. Splicing and transcription touch base: Co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 2017, 18, 637–650. [Google Scholar] [CrossRef]
- Lev, M.G.; Yearim, A.; Ast, G. The alternative role of DNA methylation in splicing regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar]
- Matlin, A.J.; Clark, F.; Smith, C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, W.; Olson, S.D.; Prabhakara, K.S.; Zhou, X. Alternative splicing links histone modifications to stem cell fate decision. Genome Biol. 2018, 19, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomonis, N.; Schlieve, C.R.; Pereira, L.; Wahlquist, C.; Colas, A.; Zambon, A.C.; Vranizan, K.; Spindler, M.J.; Pico, A.R.; Cline, M.S.; et al. Alternative splicing regulates mouse embryonic stem cell pluripotency and differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 10514–10519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunarso, G.; Wong, K.Y.; Stanton, L.W.; Lipovich, L. Detailed characterization of the mouse embryonic stem cell transcriptome reveals novel genes and intergenic splicing associated with pluripotency. BMC Genom. 2008, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, G.W.; Xu, X.; Liang, T.Y.; Muotri, A.R.; Carson, C.T.; Coufal, N.G.; Gage, F.H. Alternative splicing events identified in human embryonic stem cells and neural progenitors. PLoS Comput. Biol. 2007, 3, 1951–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Corbi, N.; Basilico, C.; Dailey, L. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 1995, 9, 2635–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosetti, D.C.; Basilico, C.; Dailey, L. Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol. Cell Biol. 1997, 17, 6321–6329. [Google Scholar] [CrossRef] [Green Version]
- Mayshar, Y.; Rom, E.; Chumakov, I.; Kronman, A.; Yayon, A.; Benvenisty, N. Fibroblast growth factor 4 and its novel splice isoform have opposing effects on the maintenance of human embryonic stem cell self-renewal. Stem Cells 2008, 26, 767–774. [Google Scholar] [CrossRef]
- Rao, S.; Zhen, S.; Roumiantsev, S.; McDonald, L.T.; Yuan, G.C.; Orkin, S.H. Differential roles of Sall4 isoforms in embryonic stem cell pluripotency. Mol. Cell Biol. 2010, 30, 5364–5380. [Google Scholar] [CrossRef] [Green Version]
- Atlasi, Y.; Mowla, S.J.; Ziaee, S.A.; Gokhale, P.J.; Andrews, P.W. OCT4 spliced variants are differentially expressed in human pluripotent and nonpluripotent cells. Stem Cells 2008, 26, 3068–3074. [Google Scholar] [CrossRef]
- Gabut, M.; Samavarchi-Tehrani, P.; Wang, X.; Slobodeniuc, V.; O’Hanlon, D.; Sung, H.K.; Alvarez, M.; Talukder, S.; Pan, Q.; Mazzoni, E.O.; et al. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell 2011, 147, 132–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Liu, L.; Lazarev, D.; Al-Zain, A.; Fomin, V.; Yeung, P.L.; Chambers, S.M.; Lu, C.W.; Studer, L.; Manley, J.L. TCF3 alternative splicing controlled by hnRNP H/F regulates E-cadherin expression and hESC pluripotency. Genes Dev. 2018, 32, 1161–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou, F.C.; Gazzeri, S.; Eymin, B. A VEGF-A/SOX2/SRSF2 network controls VEGFR1 pre-mRNA alternative splicing in lung carcinoma cells. Sci. Rep. 2019, 9, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tung, C.L.; Hou, P.H.; Kao, Y.L.; Huang, Y.W.; Shen, C.C.; Cheng, Y.H.; Wu, S.F.; Lee, M.S.; Li, C. SOX2 modulates alternative splicing in transitional cell carcinoma. Biochem. Biophys. Res. Commun. 2010, 393, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Vuong, C.K.; Black, D.L.; Zheng, S. The neurogenetics of alternative splicing. Nat. Rev. Neurosci. 2016, 17, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Forster, E.; Bock, H.H.; Herz, J.; Chai, X.; Frotscher, M.; Zhao, S. Emerging topics in Reelin function. Eur. J. Neurosci. 2010, 31, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Traunmuller, L.; Gomez, A.M.; Nguyen, T.; Scheiffele, P. Control of neuronal synapse specification by a highly dedicated alternative splicing program. Science 2016, 352, 982–986. [Google Scholar] [CrossRef]
- Traunmuller, L.; Bornmann, C.; Scheiffele, P. Alternative splicing coupled nonsense-mediated decay generates neuronal cell type-specific expression of SLM proteins. J. Neurosci. 2014, 34, 16755–16761. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [Green Version]
- Kalsotra, A.; Cooper, T.A. Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 2011, 12, 715–729. [Google Scholar] [CrossRef]
- Manning, K.S.; Cooper, T.A. The roles of RNA processing in translating genotype to phenotype. Nat. Rev. Mol. Cell Biol. 2017, 18, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El, M.E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar]
- Taylor, M.D.; Gokgoz, N.; Andrulis, I.L.; Mainprize, T.G.; Drake, J.M.; Rutka, J.T. Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am. J. Hum. Genet. 2000, 66, 1403–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurahashi, H.; Takami, K.; Oue, T.; Kusafuka, T.; Okada, A.; Tawa, A.; Okada, S.; Nishisho, I. Biallelic inactivation of the APC gene in hepatoblastoma. Cancer Res. 1995, 55, 5007–5011. [Google Scholar]
- Tanko, Q.; Franklin, B.; Lynch, H.; Knezetic, J. A hMLH1 genomic mutation and associated novel mRNA defects in a hereditary non-polyposis colorectal cancer family. Mutat. Res. 2002, 503, 37–42. [Google Scholar] [CrossRef]
- Adler, A.S.; McCleland, M.L.; Yee, S.; Yaylaoglu, M.; Hussain, S.; Cosino, E.; Quinones, G.; Modrusan, Z.; Seshagiri, S.; Torres, E.; et al. An integrative analysis of colon cancer identifies an essential function for PRPF6 in tumor growth. Genes Dev. 2014, 28, 1068–1084. [Google Scholar] [CrossRef] [Green Version]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Daffada, A.A.; Chan, C.M.; Dowsett, M. Identification of an exon 3 deletion splice variant androgen receptor mRNA in human breast cancer. Int. J. Cancer 1997, 72, 574–580. [Google Scholar] [CrossRef]
- Misao, R.; Nakanishi, Y.; Fujimoto, J.; Tamaya, T. Expression of sex hormone-binding globulin exon VII splicing variant messenger RNA in human uterine endometrial cancers. Cancer Res. 1997, 57, 5579–5583. [Google Scholar] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Ratsch, G. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Munoz-Chapuli, R. Epithelial-mesenchymal transitions: A mesodermal cell strategy for evolutive innovation in Metazoans. Anat. Rec. 2002, 268, 343–351. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Invest 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, A.; Bruser, K.; Elfert, S.; Wallmen, B.; Wittel, Y.; Wohrle, S.; Hecht, A. Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 2010, 38, 1964–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Sandiford, S.; Wu, C.; Li, S.S. Numb regulates cell-cell adhesion and polarity in response to tyrosine kinase signalling. EMBO J. 2009, 28, 2360–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, D.; Zhang, Y.; Berry, D.M.; McGlade, C.J. Regulation of Numb isoform expression by activated ERK signaling. Oncogene 2016, 35, 5202–5213. [Google Scholar] [CrossRef] [PubMed]
- Warzecha, C.C.; Jiang, P.; Amirikian, K.; Dittmar, K.A.; Lu, H.; Shen, S.; Guo, W.; Xing, Y.; Carstens, R.P. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J. 2010, 29, 3286–3300. [Google Scholar] [CrossRef]
- Brown, R.L.; Reinke, L.M.; Damerow, M.S.; Perez, D.; Chodosh, L.A.; Yang, J.; Cheng, C. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J. Clin. Invest. 2011, 121, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Brown, R.L.; Wei, Y.; Zhao, P.; Liu, S.; Liu, X.; Deng, Y.; Hu, X.; Zhang, J.; Gao, X.D.; et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019, 33, 166–179. [Google Scholar] [CrossRef]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakacs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valacca, C.; Bonomi, S.; Buratti, E.; Pedrotti, S.; Baralle, F.E.; Sette, C.; Ghigna, C.; Biamonti, G. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J. Cell Biol. 2010, 191, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Bielli, P.; Busa, R.; Paronetto, M.P.; Sette, C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr. Relat. Cancer 2011, 18, R91–R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisone, P.; Pradella, D.; Matteo, D.A.; Belloni, E.; Ghigna, C.; Paronetto, M.P. SAM68: Signal Transduction and RNA Metabolism in Human Cancer. Biomed. Res. Int. 2015, 2015, 528954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arman, E.; Haffner-Krausz, R.; Gorivodsky, M.; Lonai, P. Fgfr2 is required for limb outgrowth and lung-branching morphogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 11895–11899. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Chimenti, I.; Rosa, P.; Angelini, F.; Pagano, F.; Calogero, A.; Giacomello, A.; Messina, E. EMT/MET at the Crossroad of Stemness, Regeneration and Oncogenesis: The Ying-Yang Equilibrium Recapitulated in Cell Spheroids. Cancers 2017, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Scott, M.S.; Ono, M.; Yamada, K.; Endo, A.; Barton, G.J.; Lamond, A.I. Human box C/D snoRNA processing conservation across multiple cell types. Nucleic Acids Res. 2012, 40, 3676–3688. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Bernard, D.; Prasanth, K.V.; Tripathi, V.; Colasse, S.; Nakamura, T.; Xuan, Z.; Zhang, M.Q.; Sedel, F.; Jourdren, L.; Coulpier, F.; et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010, 29, 3082–3093. [Google Scholar] [CrossRef] [Green Version]
- Gordon, M.A.; Babbs, B.; Cochrane, D.R.; Bitler, B.G.; Richer, J.K. The long non-coding RNA MALAT1 promotes ovarian cancer progression by regulating RBFOX2-mediated alternative splicing. Mol. Carcinog. 2019, 58, 196–205. [Google Scholar] [CrossRef]
- Dang, C.V.; Semenza, G.L. Oncogenic alterations of metabolism. Trends Biochem. Sci. 1999, 24, 68–72. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biamonti, G.; Maita, L.; Montecucco, A. The Krebs Cycle Connection: Reciprocal Influence Between Alternative Splicing Programs and Cell Metabolism. Front. Oncol. 2018, 8, 408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paliwal, S.; Kovi, R.C.; Nath, B.; Chen, Y.W.; Lewis, B.C.; Grossman, S.R. The alternative reading frame tumor suppressor antagonizes hypoxia-induced cancer cell migration via interaction with the COOH-terminal binding protein corepressor. Cancer Res. 2007, 67, 9322–9329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013, 3, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Jurica, M.S.; Mesecar, A.; Heath, P.J.; Shi, W.; Nowak, T.; Stoddard, B.L. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 1998, 6, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Takenaka, M.; Noguchi, T.; Sadahiro, S.; Hirai, H.; Yamada, K.; Matsuda, T.; Imai, E.; Tanaka, T. Isolation and characterization of the human pyruvate kinase M gene. Eur. J. Biochem. 1991, 198, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zheng, Y.; Xia, Y.; Ji, H.; Chen, X.; Guo, F.; Lyssiotis, C.A.; Aldape, K.; Cantley, L.C.; Lu, Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012, 14, 1295–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlovski, I.; Siegfried, Z.; Amar-Schwartz, A.; Karni, R. The role of RNA alternative splicing in regulating cancer metabolism. Hum. Genet. 2017, 136, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Li, J.; Ho, J.C.; Chia, G.S.; Kato, H.; Jha, S.; Yang, H.; Poellinger, L.; Lee, K.L. Hypoxia is a Key Driver of Alternative Splicing in Human Breast Cancer Cells. Sci. Rep. 2017, 7, 4108. [Google Scholar] [CrossRef]
- Luo, D.; Wang, Z.; Wu, J.; Jiang, C.; Wu, J. The role of hypoxia inducible factor-1 in hepatocellular carcinoma. Biomed. Res. Int. 2014, 2014, 409272. [Google Scholar] [CrossRef]
- Dales, J.P.; Garcia, S.; Meunier-Carpentier, S.; Andrac-Meyer, L.; Haddad, O.; Lavaut, M.N.; Allasia, C.; Bonnier, P.; Charpin, C. Overexpression of hypoxia-inducible factor HIF-1alpha predicts early relapse in breast cancer: Retrospective study in a series of 745 patients. Int. J. Cancer 2005, 116, 734–739. [Google Scholar] [CrossRef]
- Chia, S.K.; Wykoff, C.C.; Watson, P.H.; Han, C.; Leek, R.D.; Pastorek, J.; Gatter, K.C.; Ratcliffe, P.; Harris, A.L. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J. Clin. Oncol. 2001, 19, 3660–3668. [Google Scholar] [CrossRef]
- Watnick, R.S. The role of the tumor microenvironment in regulating angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006676. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2019. [CrossRef] [PubMed] [Green Version]
- Ergorul, C.; Ray, A.; Huang, W.; Darland, D.; Luo, Z.K.; Grosskreutz, C.L. Levels of vascular endothelial growth factor-A165b (VEGF-A165b) are elevated in experimental glaucoma. Mol. Vis. 2008, 14, 1517–1524. [Google Scholar] [PubMed]
- Schumacher, V.A.; Jeruschke, S.; Eitner, F.; Becker, J.U.; Pitschke, G.; Ince, Y.; Miner, J.H.; Leuschner, I.; Engers, R.; Everding, A.S.; et al. Impaired glomerular maturation and lack of VEGF165b in Denys-Drash syndrome. J. Am. Soc. Nephrol. 2007, 18, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merdzhanova, G.; Gout, S.; Keramidas, M.; Edmond, V.; Coll, J.L.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene 2010, 29, 5392–5403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.O.; Cui, T.G.; Doughty, J.M.; Winkler, M.; Sugiono, M.; Shields, J.D.; Peat, D.; Gillatt, D.; Harper, S.J. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002, 62, 4123–4131. [Google Scholar] [PubMed]
- Rennel, E.; Waine, E.; Guan, H.; Schuler, Y.; Leenders, W.; Woolard, J.; Sugiono, M.; Gillatt, D.; Kleinerman, E.; Bates, D.; et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br. J. Cancer 2008, 98, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Pritchard-Jones, R.O.; Dunn, D.B.; Qiu, Y.; Varey, A.H.; Orlando, A.; Rigby, H.; Harper, S.J.; Bates, D.O. Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br. J. Cancer 2007, 97, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biamonti, G.; Infantino, L.; Gaglio, D.; Amato, A. An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer. Cells 2020, 9, 34. https://doi.org/10.3390/cells9010034
Biamonti G, Infantino L, Gaglio D, Amato A. An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer. Cells. 2020; 9(1):34. https://doi.org/10.3390/cells9010034
Chicago/Turabian StyleBiamonti, Giuseppe, Lucia Infantino, Daniela Gaglio, and Angela Amato. 2020. "An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer" Cells 9, no. 1: 34. https://doi.org/10.3390/cells9010034
APA StyleBiamonti, G., Infantino, L., Gaglio, D., & Amato, A. (2020). An Intricate Connection between Alternative Splicing and Phenotypic Plasticity in Development and Cancer. Cells, 9(1), 34. https://doi.org/10.3390/cells9010034