Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells


 ,
,  and
and 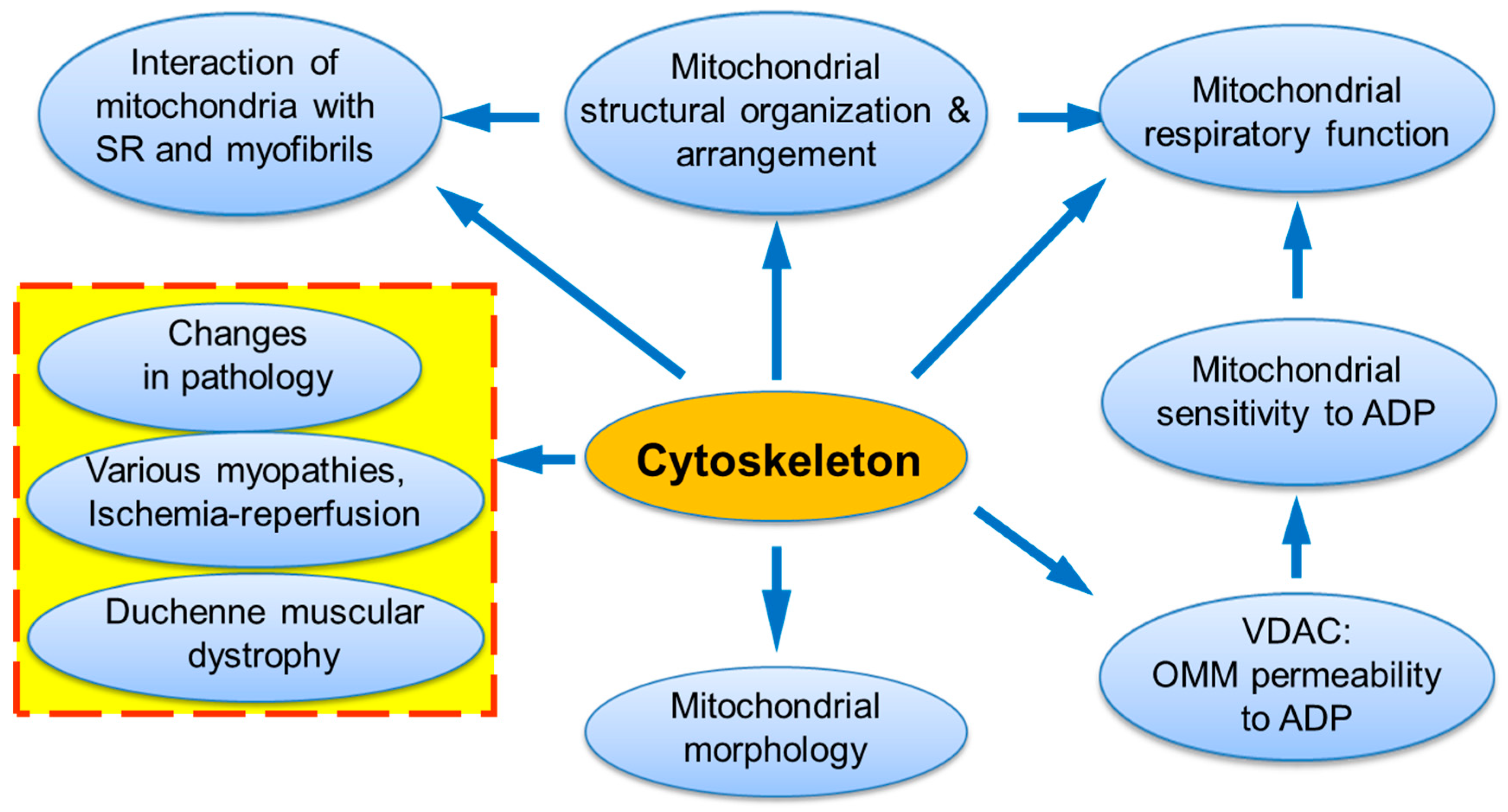{kind=link}
{kind=link}
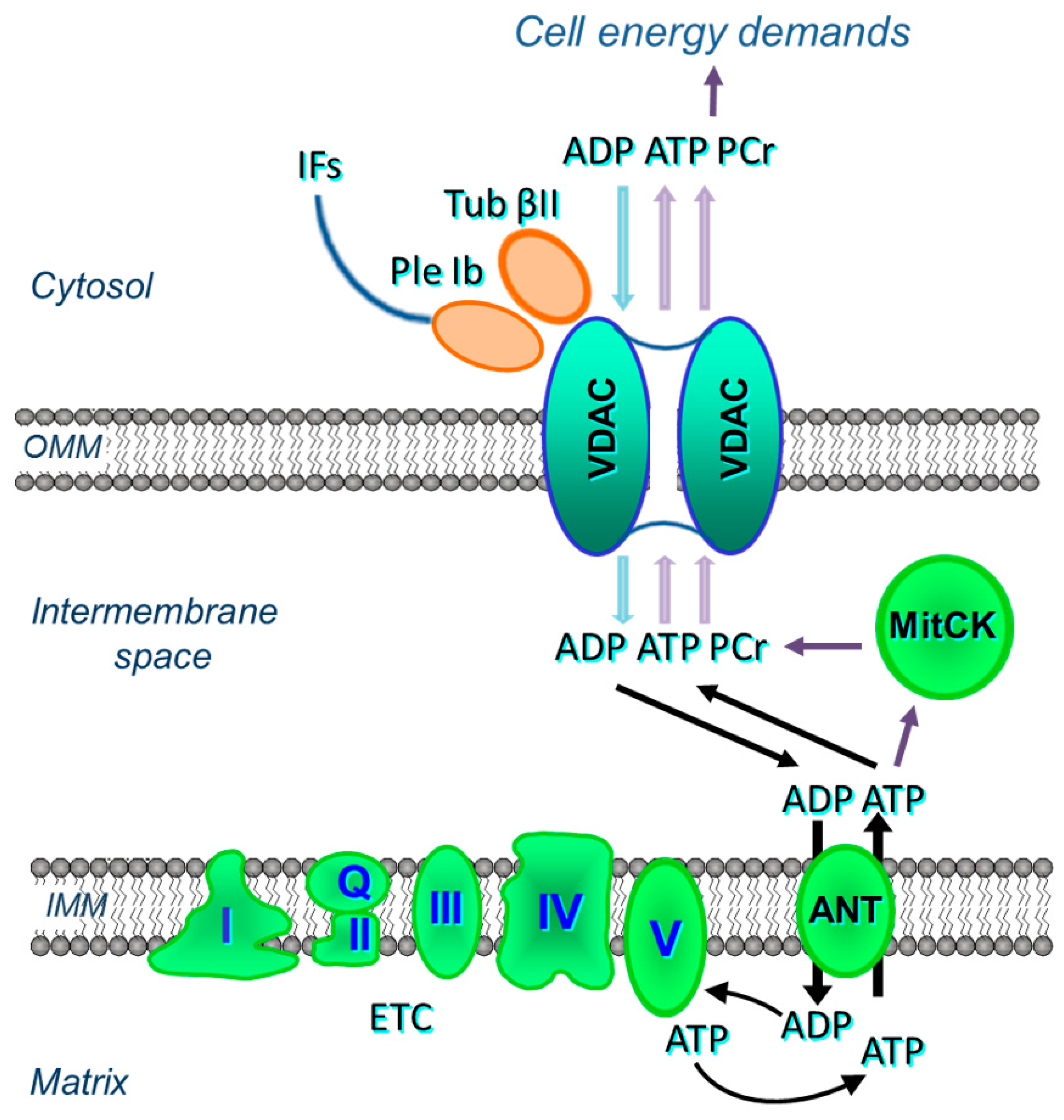{kind=link}
{kind=link}
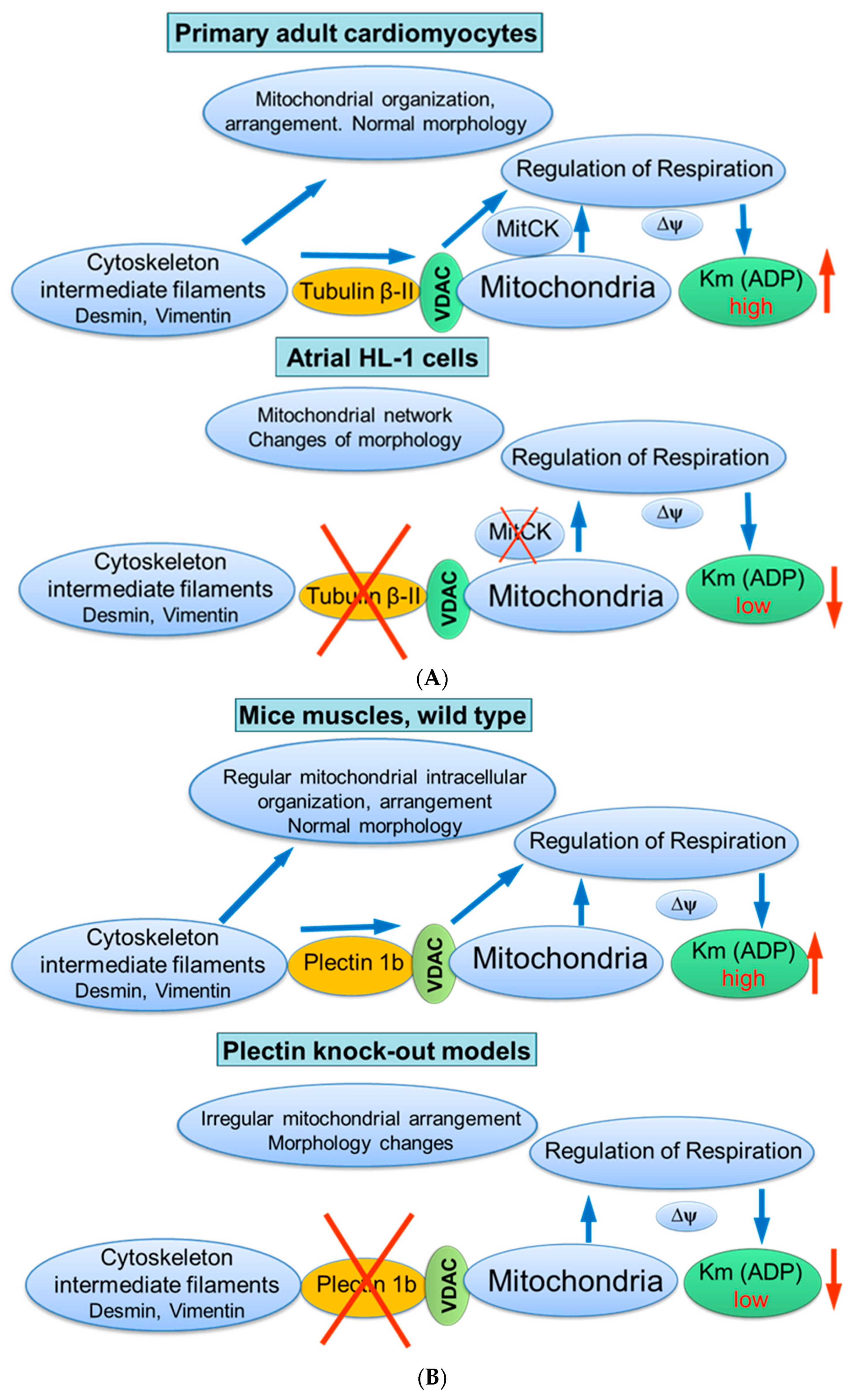{kind=link}
Abstract
1. Introduction
2. Historical Retrospective
3. The Role of Cytoskeleton in the Mitochondrial Intracellular Organization, Shape Morphology and Dynamics
4. Cytoskeleton and Mitochondria-SR Interactions
5. Possible Role of the Intermediate Filaments Proteins Desmin and Vimentin in the Regulation of Mitochondrial Bioenergetics
6. The Role of Tubulin in the Regulation of Mitochondrial Bioenergetics and Metabolism
7. Possible Role of Plectin in the Control of Mitochondrial Intracellular Organization and Function
8. Cytoskeletal-Mitochondria Interactions in Pathology
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Appaix, F.; Kuznetsov, A.V.; Usson, Y.; Kay, L.; Andrienko, T.; Olivares, J.; Kaambre, T.; Sikk, P.; Margreiter, R.; Saks, V. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Exp. Physiol. 2003, 88, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, L.; Oliviero, P.; Samuel, J.L. Cytoskeleton and mitochondrial morphology and function. Mol. Cell. Biochem. 1998, 184, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Valm, A.M.; Lippincott-Schwartz, J. Interacting organelles. Curr. Opin. Cell Biol. 2018, 53, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Andrienko, T.; Kuznetsov, A.V.; Kaambre, T.; Usson, Y.; Orosco, A.; Appaix, F.; Tiivel, T.; Sikk, P.; Vendelin, M.; Margreiter, R.; et al. Metabolic consequences of functional complexes of mitochondria; myofibrils and sarcoplasmic reticulum in muscle cells. J. Exp. Biol. 2003, 206, 2059–2072. [Google Scholar] [CrossRef]
- Kaasik, A.; Veksler, V.; Boehm, E.; Novotova, M.; Minajeva, A.; Ventura-Clapier, R. Energetic crosstalk between organelles: Architectural integration of energy production and utilization. Circ. Res. 2001, 89, 153–159. [Google Scholar] [CrossRef]
- Anesti, V.; Scorrano, L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta 2006, 1757, 692–699. [Google Scholar] [CrossRef]
- Kay, L.; Li, Z.; Mericskay, M.; Olivares, J.; Tranqui, L.; Fontaine, E.; Tiivel, T.; Sikk, P.; Kaambre, T.; Samuel, J.L.; et al. Study of regulation of mitochondrial respiration in vivo. An analysis of influence of ADP diffusion and possible role of cytoskeleton. Biochim. Biophys. Acta 1997, 1322, 41–59. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Guzun, R.; Grimm, M.; Saks, V.A. Cytoskeleton and regulation of mitochondrial function: The role of beta-tubulin II. Front. Physiol. 2013, 4, 82. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Csordás, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef]
- Lawrie, A.M.; Rizzuto, R.; Pozzan, T.; Simpson, A.W. A role for calcium influx in the regulation of mitochondrial calcium in endothelial cells. J. Biol. Chem. 1996, 271, 10753–10759. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Thomas, A.P.; Hajnoczky, G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999, 18, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.J.; Dalby, B.; Stewart, R.J.; Doxsey, S.J.; Goldstein, L.S. Mitochondrial association of a plus end-directed microtubule motor expressed during mitosis in Drosophila. J. Cell Biol. 1997, 136, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.P. Dynamic mitochondria. Nat. Cell Biol. 1999, 1, e149–e150. [Google Scholar] [CrossRef]
- Knowles, M.K.; Guenza, M.G.; Capaldi, R.A.; Marcus, A.H. Cytoskeletal-assisted dynamics of the mitochondrial reticulum in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14772–14777. [Google Scholar] [CrossRef]
- Thomson, M. The regulation of mitochondrial physiology by organelle-associated GTP-binding proteins. Cell Biochem. Funct. 2002, 20, 273–278. [Google Scholar] [CrossRef]
- Noskov, S.Y.; Rostovtseva, T.K.; Bezrukov, S.M. ATP transport through VDAC and the VDAC-tubulin complex probed by equilibrium and nonequilibrium MD simulations. Biochemistry 2013, 52, 9246–9256. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Chen, M.-Y.; Bezrukov, S.M. Membrane lipid composition regulates tubulin interaction with mitochondrial voltage-dependent anion channel. J. Biol. Chem. 2012, 287, 29589–29598. [Google Scholar] [CrossRef]
- Puurand, M.; Tepp, K.; Timohhina, N.; Aid, J.; Shevchuk, I.; Chekulayev, V.; Kaambre, T. Tubulin βII and βIII Isoforms as the Regulators of VDAC Channel Permeability in Health and Disease. Cells 2019, 8, 239. [Google Scholar] [CrossRef]
- Guzun, R.; Gonzalez-Granillo, M.; Karu-Varikmaa, M.; Grichine, A.; Usson, Y.; Kaambre, T.; Guerrero-Roesch, K.; Kuznetsov, A.; Schlattner, U.; Saks, V. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within Mitochondrial Interactosome. Biochim. Biophys. Acta 2012, 1818, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Bezrukov, S.M. VDAC inhibition by tubulin and its physiological implications. Biochim. Biophys. Acta 2012, 1818, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.R. Mitochondrial function in the heart. Annu. Rev. Physiol. 1979, 41, 485–506. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S. Regulation of oxidative phosphorylation in the mammalian cell. Am. J. Physiol. 1990, 258, C377–C389. [Google Scholar] [CrossRef] [PubMed]
- Saks, V.A.; Kupriyanov, V.V.; Kuznetsov, A.V.; Kapelko, V.I.; Sharov, V.G.; Veksler, V.I.; Javadov, S.A. Quantitative evaluation of relationship between cardiac energy metabolism and post-ischemic recovery of contractile function. J. Mol. Cell. Cardiol. 1989, 21, 67–78. [Google Scholar] [CrossRef]
- Saks, V.; Kuznetsov, A.; Andrienko, T.; Usson, Y.; Appaix, F.; Guerrero, K.; Kaambre, T.; Sikk, P.; Lemba, M.; Vendelin, M. Heterogeneity of ADP diffusion and regulation of respiration in cardiac cells. Biophys. J. 2003, 84, 3436–3456. [Google Scholar] [CrossRef]
- Saks, V.; Dzeja, P.; Schlattner, U.; Vendelin, M.; Terzic, A.; Wallimann, T. Cardiac system bioenergetics: Metabolic basis of the Frank-Starling law. J. Physiol. 2006, 571, 253–273. [Google Scholar] [CrossRef]
- Reipert, S.; Steinböck, F.; Fischer, I.; Bittner, R.E.; Zeöld, A.; Wiche, G. Association of mitochondria with plectin and desmin intermediate filaments in striated muscle. Exp. Cell Res. 1999, 252, 479–491. [Google Scholar] [CrossRef]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef]
- Timohhina, N.; Guzun, R.; Tepp, K.; Monge, C.; Varikmaa, M.; Vija, H.; Sikk, P.; Kaambre, T.; Sackett, D.; Saks, V. Direct measurement of energy fluxes from mitochondria into cytoplasm in permeabilized cardiac cells in situ: Some evidence for Mitochondrial Interactosome. J. Bioenerg. Biomembr. 2009, 41, 259–275. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Granillo, M.; Grichine, A.; Guzun, R.; Usson, Y.; Tepp, K.; Chekulayev, V.; Shevchuk, I.; Karu-Varikmaa, M.; Kuznetsov, A.V.; Grimm, M.; et al. Studies of the role of tubulin beta II isotype in regulation of mitochondrial respiration in intracellular energetic units in cardiac cells. J. Mol. Cell. Cardiol. 2012, 52, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Guzun, R.; Kaambre, T.; Bagur, R.; Grichine, A.; Usson, Y.; Varikmaa, M.; Anmann, T.; Tepp, K.; Timohhina, N.; Shevchuk, I.; et al. Modular organization of cardiac energy metabolism: Energy conversion; transfer and feedback regulation. Acta Physiol. 2015, 213, 84–106. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y. Desmin cytoskeleton: A potential regulator of muscle mitochondrial behavior and function. Trends Cardiovasc. Med. 2002, 12, 339–348. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The role of Ca2+ in the regulation of intramitochondrial energy production in heart. Biomed. Biochim. Acta 1987, 46, S487–S492. [Google Scholar]
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta 2009, 1787, 1324–1333. [Google Scholar] [CrossRef]
- Clark, J.F.; Kuznetsov, A.V.; Radda, G.K. ADP-regenerating enzyme systems in mitochondria of guinea pig myometrium and heart. Am. J. Physiol. 1997, 272 Pt 1, C399–C404. [Google Scholar] [CrossRef]
- Brdiczka, D. Function of the outer mitochondrial compartment in regulation of energy metabolism. Biochim. Biophys. Acta 1994, 1187, 264–269. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers; tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in human cells. Free Radic. Biol. Med. 2000, 29, 202–210. [Google Scholar] [CrossRef]
- Kummel, L. Ca, Mg-ATPase activity of permeabilised rat heart cells and its functional coupling to oxidative phosphorylation of the cells. Cardiovasc. Res. 1988, 22, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Veksler, V.I.; Kuznetsov, A.V.; Sharov, V.G.; Kapelko, V.I.; Saks, V.A. Mitochondrial respiratory parameters in cardiac tissue: A novel method of assessment by using saponin-skinned fibers. Biochim. Biophys. Acta 1987, 892, 191–196. [Google Scholar] [CrossRef]
- Saks, V.A.; Veksler, V.I.; Kuznetsov, A.V.; Kay, L.; Sikk, P.; Tiivel, T.; Tranqui, L.; Olivares, J.; Winkler, K.; Wiedemann, F.; et al. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol. Cell. Biochem. 1998, 184, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Saks, V.A.; Belikova, Y.O.; Kuznetsov, A.V. In vivo regulation of mitochondrial respiration in cardiomyocytes: Specific restrictions for intracellular diffusion of ADP. Biochim. Biophys. Acta 1991, 1074, 302–311. [Google Scholar] [CrossRef]
- Saks, V.A.; Vasil’eva, E.; Belikova, Y.O.; Kuznetsov, A.V.; Lyapina, S.; Petrova, L.; Perov, N.A. Retarded diffusion of ADP in cardiomyocytes: Possible role of mitochondrial outer membrane and creatine kinase in cellular regulation of oxidative phosphorylation. Biochim. Biophys. Acta 1993, 1144, 134–148. [Google Scholar] [CrossRef]
- Varikmaa, M.; Bagur, R.; Kaambre, T.; Grichine, A.; Timohhina, N.; Tepp, K.; Shevchuk, I.; Chekulayev, V.; Metsis, M.; Boucher, F.; et al. Role of mitochondria-cytoskeleton interactions in respiration regulation and mitochondrial organization in striated muscles. Biochim. Biophys. Acta 2014, 1837, 232–245. [Google Scholar] [CrossRef]
- Guzun, R.; Karu-Varikmaa, M.; Gonzalez-Granillo, M.; Kuznetsov, A.V.; Michel, L.; Cottet-Rousselle, C.; Saaremäe, M.; Kaambre, T.; Metsis, M.; Grimm, M.; et al. Mitochondria-cytoskeleton interaction: Distribution of β-tubulins in cardiomyocytes and HL-1 cells. Biochim. Biophys. Acta 2011, 1807, 458–469. [Google Scholar] [CrossRef]
- Winter, L.; Kuznetsov, A.V.; Grimm, M.; Zeöld, A.; Fischer, I.; Wiche, G. Plectin isoform P1b and P1d deficiencies differentially affect mitochondrial morphology and function in skeletal muscle. Hum. Mol. Genet. 2015, 24, 4530–4544. [Google Scholar] [CrossRef]
- Ball, E.H.; Singer, S.J. Mitochondria are associated with microtubules and not with intermediate filaments in cultured fibroblasts. Proc. Natl. Acad. Sci. USA 1982, 79, 123–126. [Google Scholar] [CrossRef]
- Mose-Larsen, P.; Bravo, R.; Fey, S.J.; Small, J.V.; Celis, J.E. Putative association of mitochondria with a subpopulation of intermediate-sized filaments in cultured human skin fibroblasts. Cell 1982, 31, 681–692. [Google Scholar] [CrossRef]
- Hirokawa, N. Cross-linker system between neurofilaments, microtubules and membranous organelles in frog axons revealed by quick-freeze, deep etching method. J. Cell Biol. 1982, 94, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Heggeness, M.H.; Simon, M.; Singer, S.J. Association of mitochondria with microtubules in cultured cells. Proc. Natl. Acad. Sci. USA 1978, 75, 3863–3866. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.G.; Simon, V.R.; O’Sullivan, H.; Pon, L.A. Organelle-cytoskeletal interactions: Actin mutations inhibit meiosis-dependent mitochondrial rearrangement in the budding yeast Saccharomyces Cerevisiae. Mol. Biol. Cell. 1995, 6, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.R.; Pon, L.A. Interactions of mitochondria with the actin cytoskeleton. Biochim. Biophys. Acta 2006, 1763, 450–462. [Google Scholar] [CrossRef]
- Morris, R.L.; Hollenbeck, P.J. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J. Cell. Biol. 1995, 131, 1315–1326. [Google Scholar] [CrossRef]
- Boldogh, I.R.; Yang, H.C.; Nowakowski, W.D.; Karmon, S.L.; Hays, L.G.; Yates, J.R., 3rd; Pon, L.A. Arp2/3 complex and actin dynamics are required for actin-based mitochondrial motility in yeast. Proc. Natl. Acad. Sci. USA 2001, 98, 3162–3167. [Google Scholar]
- Jensen, R.E. Control of mitochondrial shape. Curr. Opin. Cell Biol. 2005, 17, 384–388. [Google Scholar] [CrossRef]
- Burgess, S.M.; Delannoy, M.; Jensen, R.E. MMM1 encodes a mitochondrial outer membrane protein essential for establishing and maintaining the structure of yeast mitochondria. J. Cell Biol. 1994, 126, 1375–1391. [Google Scholar] [CrossRef]
- Berger, K.H.; Sogo, L.F.; Yaffe, M.P. Mdm12p, a component required for mitochondrial inheritance that is conserved between budding and fission yeast. J. Cell Biol. 1997, 136, 545–553. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Jakobs, S.; Vogel, F.; Altmann, K.; Westermann, B. Mdm31 and Mdm32 are inner membrane proteins required for maintenance of mitochondrial shape and stability of mitochondrial DNA nucleoids in yeast. J. Cell Biol. 2005, 168, 103–115. [Google Scholar] [CrossRef]
- Sogo, L.F.; Yaffe, M.P. Regulation of mitochondrial morphology and inheritance by Mdm10p, a protein of the mitochondrial outer membrane. J. Cell Biol. 1994, 126, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Youngman, M.J.; Hobbs, A.E.; Burgess, S.M.; Srinivasan, M.; Jensen, R.E. Mmm2p, a mitochondrial outer membrane protein required for yeast mitochondrial shape and maintenance of mtDNA nucleoids. J. Cell Biol. 2004, 164, 677–688. [Google Scholar] [CrossRef] [PubMed]
- John, G.B.; Shang, Y.; Li, L.; Renken, C.; Mannella, C.A.; Selker, J.M.; Rangell, L.; Bennett, M.J.; Zha, J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell 2005, 16, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Frederick, R.L.; McCaffery, J.M.; Cunningham, K.W.; Okamoto, K.; Shaw, J.M. Yeast Miro GTPase, Gem1p, regulates mitochondrial morphology via a novel pathway. J. Cell Biol. 2004, 167, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brethes, D.; Di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Sesaki, H.; Jensen, R.E. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J. Cell Biol. 1999, 147, 699–706. [Google Scholar] [CrossRef]
- Griparic, L.; Van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; Van der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet. 2005, 14, R283–R289. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; Van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Bleazard, W.; McCaffery, J.M.; King, E.J.; Bale, S.; Mozdy, A.; Tieu, Q.; Nunnari, J.; Shaw, J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999, 1, 298. [Google Scholar] [CrossRef]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.C. hFis1, a novel component of the mammalian mitochondrial fission machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.J.; Thatcher, J.W.; Mills, J.P.; Hales, K.G.; Fuller, M.T.; Nunnari, J.; Shaw, J.M. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 1998, 143, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Scorrano, L. The many shapes of mitochondrial death. Oncogene 2006, 25, 4717–4724. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Rajapurohitam, V.; Kilić, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: The effect of NHE-1 inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Adaniya, S.M.; O-Uchi, J.; Cypress, M.W.; Kusakari, Y.; Jhun, B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. Cell Physiol. 2019, 316, C583–C604. [Google Scholar] [CrossRef]
- Olichon, A.; Guillou, E.; Delettre, C.; Landes, T.; Arnaune-Pelloquin, L.; Emorine, L.J.; Mils, V.; Daloyau, M.; Hamel, C.; Amati-Bonneau, P.; et al. Mitochondrial dynamics and disease. Biochim. Biophys. Acta 2006, 1763, 500–509. [Google Scholar] [CrossRef]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef]
- Karbowski, M.; Youle, R.J. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003, 10, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.L.; Roumier, T.; Kroemer, G. Mitochondrial fusion and fission in the control of apoptosis. Trends Cell Biol. 2005, 15, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Vale, R.D.; Funatsu, T.; Pierce, D.W.; Romberg, L.; Harada, Y.; Yanagida, T. Direct observation of single kinesin molecules moving along microtubules. Nature 1996, 380, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Vale, R.D. The molecular motor toolbox for intracellular transport. Cell 2003, 112, 467–480. [Google Scholar] [CrossRef]
- Hollenbeck, P.J.; Saxton, W.M. The axonal transport of mitochondria. J. Cell Sci. 2005, 118, 5411–5419. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Zhai, X.Y.; Ohsato, K.; Futamata, H.; Shimada, O.; Atsumi, S. Mitochondrial accumulation in the distal part of the initial segment of chicken spinal motoneurons. Brain Res. 2004, 1026, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Chada, S.R.; Hollenbeck, P.J. Mitochondrial movement and positioning in axons: The role of growth factor signaling. J. Exp. Biol. 2003, 206, 1985–1992. [Google Scholar] [CrossRef]
- Boldogh, I.R.; Pon, L.A. Mitochondria on the move. Trends Cell Biol. 2007, 17, 502–510. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Hermann, M.; Saks, V.; Hengster, P.; Margreiter, R. The cell type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 1928–1939. [Google Scholar] [CrossRef]
- Hudder, A.; Nathanson, L.; Deutscher, M.P. Organization of mammalian cytoplasm. Mol. Cell Biol. 2003, 23, 9318–9326. [Google Scholar] [CrossRef]
- Toivola, D.M.; Tao, G.Z.; Habtezion, A.; Liao, J.; Omary, M.B. Cellular integrity plus: Organelle-related and protein-targeting functions of intermediate filaments. Trends Cell Biol. 2005, 15, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Mado, K.; Chekulayev, V.; Shevchuk, I.; Puurand, M.; Tepp, K.; Kaambre, T. On the role of tubulin; plectin; desmin; and vimentin in the regulation of mitochondrial energy fluxes in muscle cells. Am. J. Physiol. Cell Physiol. 2019, 316, C657–C667. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Martone, M.E.; Yu, Z.; Thor, A.; Doi, M.; Holst, M.J.; Ellisman, M.H.; Hoshijima, M. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J. Cell Sci. 2009, 122, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Lebiedzinska, M.; Szabadkai, G.; Jones, A.W.; Duszynski, J.; Wieckowski, M.R. Interactions between the endoplasmic reticulum; mitochondria; plasma membrane and other subcellular organelles. Int. J. Biochem. Cell Biol. 2009, 41, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Hool, L.C. How does calcium regulate mitochondrial energetics in the heart?—New insights. Heart Lung Circ. 2014, 23, 602–609. [Google Scholar] [CrossRef]
- Van Vliet, A.R.; Agostinis, P. Mitochondria-Associated Membranes and ER Stress. Curr. Top. Microbiol. Immunol. 2018, 414, 73–102. [Google Scholar]
- Zhu, L.; Ling, S.; Yu, X.-D.; Venkatesh, L.K.; Subramanian, T.; Chinnadurai, G.; Kuo, T.H. Modulation of Mitochondrial Ca2+ Homeostasis by Bcl-2. J. Biol. Chem. 1999, 274, 33267–33273. [Google Scholar] [CrossRef]
- Pinton, P.; Ferrari, D.; Rapizzi, E.; Virgilio, F.D.; Pozzan, T.; Rizzuto, R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: Significance for the molecular mechanism of Bcl-2 action. EMBO J. 2001, 20, 2690–2701. [Google Scholar] [CrossRef]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis; Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- Simmen, T.; Tagaya, M. Organelle Communication at Membrane Contact Sites (MCS): From Curiosity to Center Stage in Cell Biology and Biomedical Research. Adv. Exp. Med. Biol. 2017, 997, 1–12. [Google Scholar]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the Muscle Cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar] [PubMed]
- Costa, M.L.; Escaleira, R.; Cataldo, A.; Oliveira, F.; Mermelstein, C.S. Desmin: Molecular interactions and putative functions of the muscle intermediate filament protein. Braz. J. Med. Biol. Res. 2004, 37, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Lung, H.L.; Wu, K.C.; Le, A.H.P.; Tang, H.M.; Fung, M.C. Vimentin Supports Mitochondrial Morphology and Organization. Biochem. J. 2008, 410, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Leube, R.E. Intermediate Filaments as Organizers of Cellular Space: How They Affect Mitochondrial Structure and Function. Cells 2016, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Thornell, L.-E.; Carlsson, L.; Li, Z.; Mericskay, M.; Paulin, D. Null Mutation in the Desmin Gene Gives Rise to a Cardiomyopathy. J. Mol. Cell. Cardiol. 1997, 29, 2107–2124. [Google Scholar] [CrossRef] [PubMed]
- Kay, L.; Nicolay, K.; Wieringa, B.; Saks, V.; Wallimann, T. Direct evidence for the control of mitochondrial respiration by mitochondrial creatine kinase in oxidative muscle cells in situ. J. Biol. Chem. 2000, 275, 6937–6944. [Google Scholar] [CrossRef]
- Dolder, M.; Wendt, S.; Wallimann, T. Mitochondrial creatine kinase in contact sites: Interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol. Signals Recept. 2001, 10, 93–111. [Google Scholar] [CrossRef]
- Dolder, M.; Walzel, B.; Speer, O.; Schlattner, U.; Wallimann, T. Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. J. Biol. Chem. 2003, 278, 17760–17766. [Google Scholar] [CrossRef]
- Matveeva, E.A.; Venkova, L.S.; Chernoivanenko, I.S.; Minin, A.A. Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 2015, 4, 1290–1297. [Google Scholar] [CrossRef]
- Chernoivanenko, I.S.; Matveeva, E.A.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB J. 2015, 29, 820–827. [Google Scholar] [CrossRef]
- Seppet, E.K.; Eimre, M.; Anmann, T.; Seppet, E.; Peet, N.; Käämbre, T.; Paju, K.; Piirsoo, A.; Kuznetsov, A.V.; Vendelin, M.; et al. Intracellular energetic units in healthy and diseased hearts. Exp. Clin. Cardiol. 2005, 10, 173–183. [Google Scholar] [PubMed]
- Saks, V.; Kuznetsov, A.V.; Gonzalez-Granillo, M.; Tepp, K.; Timohhina, N.; Karu-Varikmaa, M.; Kaambre, T.; Dos Santos, P.; Boucher, F.; Guzun, R. Intracellular Energetic Units regulate metabolism in cardiac cells. J. Mol. Cell. Cardiol. 2012, 52, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Summerhayes, I.C.; Wong, D.; Chen, L.B. Effect of microtubules and intermediate filaments on mitochondrial distribution. J. Cell Sci. 1983, 61, 87–105. [Google Scholar] [PubMed]
- Saetersdal, T.; Greve, G.; Dalen, H. Associations between beta-tubulin and mitochondria in adult isolated heart myocytes as shown by immunofluorescence and immunoelectron microscopy. Histochemistry 1990, 95, 1–10. [Google Scholar] [CrossRef]
- Winter, L.; Abrahamsberg, C.; Wiche, G. Plectin isoform 1b mediates mitochondrion-intermediate lament network linkage and controls organelle shape. J. Cell Biol. 2008, 181, 903–911. [Google Scholar] [CrossRef]
- Redeker, V. Mass spectrometry analysis of C-terminal posttranslational modifications of tubulins. Methods Cell Biol. 2010, 95, 77–103. [Google Scholar]
- Hein, S.; Kostin, S.; Heling, A.; Maeno, Y.; Schaper, J. The role of the cytoskeleton in heart failure. Cardiovasc. Res. 2000, 45, 273–278. [Google Scholar] [CrossRef]
- Kostin, S.; Hein, S.; Arnon, E.; Scholz, D.; Schaper, J. The cytoskeleton and related proteins in the human failing heart. Heart Fail. Rev. 2000, 5, 271–280. [Google Scholar] [CrossRef]
- Tagawa, H.; Koide, M.; Sato, H.; Zile, M.R.; Carabello, B.A.; Cooper, G. Cytoskeletal role in the transition from compensated to decompensated hypertrophy during adult canine left ventricular pressure overloading. Circ. Res 1998, 82, 751–761. [Google Scholar] [CrossRef]
- Guerrero, K.; Monge, C.; Bruckner, A.; Puurand, U.; Kadaja, L.; Kaambre, T.; Seppet, E.; Saks, V. Study of possible interactions of tubulin; microtubular network; and STOP protein with mitochondria in muscle cells. Mol. Cell. Biochem. 2010, 337, 239–249. [Google Scholar] [CrossRef]
- Carré, M.; André, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Bezrukov, S.M. VDAC regulation: Role of cytosolic proteins and mitochondrial lipids. J. Bioenerg. Biomembr. 2008, 40, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Gurnev, P.A.; Queralt-Martin, M.; Aguilella, V.M.; Rostovtseva, T.K.; Bezrukov, S.M. Probing tubulin-blocked state of VDAC by varying membrane surface charge. Biophys. J. 2012, 102, 2070–2076. [Google Scholar] [CrossRef] [PubMed]
- Monge, C.; Beraud, N.; Kuznetsov, A.V.; Rostovtseva, T.; Sackett, D.; Schlattner, U.; Vendelin, M.; Saks, V.A. Regulation of respiration in brain mitochondria and synaptosomes: Restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol. Cell. Biochem. 2008, 318, 147–165. [Google Scholar] [CrossRef]
- Janke, C.; Kneussel, M. Tubulin post-translational modifications: Encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010, 33, 362–372. [Google Scholar] [CrossRef]
- Luduena, R.F. Multiple forms of tubulin: Different gene products and covalent modifications. Int. Rev. Cytol. 1998, 178, 207–275. [Google Scholar]
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Tiivel, T.; Sikk, P.; Kaambre, T.; Kay, L.; Daneshrad, Z.; Rossi, A.; Kadaja, L.; Peet, N.; Seppet, E.; et al. Striking difference between slow and fast twitch muscles in the kinetics of regulation of respiration by ADP in the cells in vivo. Eur. J. Biochem. 1996, 241, 909–915. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar]
- Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. The Warburg effect and mitochondrial stability in cancer cells. Mol. Asp. Med. 2010, 31, 60–74. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Mayboroda, O.; Kunz, D.; Winkler, K.; Schubert, W.; Kunz, W.S. Functional imaging of mitochondria in saponin-permeabilized mice muscle fibers. J. Cell Biol. 1998, 140, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Usson, Y.; Leverve, X.; Margreiter, R. Subcellular heterogeneity of mitochondrial function and dysfunction: Evidence obtained by confocal imaging. Mol. Cell. Biochem. 2004, 256, 359–365. [Google Scholar] [CrossRef]
- Romashko, D.N.; Marban, E.; O’Rourke, B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1618–1623. [Google Scholar] [CrossRef]
- Anmann, T.; Guzun, R.; Beraud, N.; Pelloux, S.; Kuznetsov, A.V.; Kogerman, L.; Kaambre, T.; Sikk, P.; Paju, K.; Peet, N.; et al. Different kinetics of the regulation of respiration in permeabilized cardiomyocytes and in HL-1 cardiac cells. Importance of cell structure/organization for respiration regulation. Biochim. Biophys. Acta 2006, 1757, 1597–1606. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta 2015, 1853, 276–284. [Google Scholar] [CrossRef]
- Konieczny, P.; Fuchs, P.; Reipert, S.; Kunz, W.S.; Zeöld, A.; Fischer, I.; Paulin, D.; Schröder, R.; Wiche, G. Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J. Cell Biol. 2008, 181, 667–681. [Google Scholar] [CrossRef]
- Fuchs, E.; Yang, Y. Crossroads on cytoskeletal highways. Cell 1999, 98, 547–550. [Google Scholar] [CrossRef]
- Rezniczek, G.A.; Abrahamsberg, C.; Fuchs, P.; Spazierer, D.; Wiche, G. Plectin 5’-transcript diversity: Short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum. Mol. Genet. 2003, 12, 3181–3194. [Google Scholar] [CrossRef]
- Rezniczek, G.A.; Konieczny, P.; Nikolic, B.; Reipert, S.; Schneller, D.; Abrahamsberg, C.; Davies, K.E.; Winder, S.J.; Wiche, G. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J. Cell Biol. 2007, 176, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Reimann, J.; Kunz, W.S.; Vielhaber, S.; Kappes-Horn, K.; Schröder, R. Mitochondrial dysfunction in myofibrillar myopathy. Neuropathol. Appl. Neurobiol. 2003, 29, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Schröder, R.; Kunz, W.S.; Rouan, F.; Pfendner, E.; Tolksdorf, K.; Kappes-Horn, K.; Altenschmidt-Mehring, M.; Knoblich, R.; Van der Ven, P.F.M.; Reimann, J.; et al. Disorganization of the desmin cytoskeleton and mitochondrial dysfunction in plectin-related epidermolysis bullosa simplex with muscular dystrophy. J. Neuropathol. Exp. Neurol. 2002, 61, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef]
- Miragoli, M.; Cabassi, A. Mitochondrial Mechanosensor Microdomains in Cardiovascular Disorders. Adv. Exp. Med. Biol. 2017, 982, 247–264. [Google Scholar]
- Iribe, G.; Kaihara, K.; Yamaguchi, Y.; Nakaya, M.; Inoue, R.; Naruse, K. Mechano-sensitivity of mitochondrial function in mouse cardiac myocytes. Prog. Biophys. Mol. Biol. 2017, 130 Pt B, 315–322. [Google Scholar] [CrossRef]
- Bartolák-Suki, E.; Imsirovic, J.; Nishibori, Y.; Krishnan, R.; Suki, B. Regulation of Mitochondrial Structure and Dynamics by the Cytoskeleton and Mechanical Factors. Int. J. Mol. Sci. 2017, 18, 1812. [Google Scholar] [CrossRef]
- Kaasik, A.; Kuum, M.; Joubert, F.; Wilding, J.; Ventura-Clapier, R.; Veksler, V. Mitochondria as a source of mechanical signals in cardiomyocytes. Cardiovasc. Res. 2010, 87, 83–91. [Google Scholar] [CrossRef]
- Khurana, T.S.; Davies, K.E. Pharmacological strategies for muscular dystrophy. Nat. Rev. Drug Discov. 2003, 2, 379–390. [Google Scholar] [CrossRef]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.; Von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef]
- Randazzo, D.; Khalique, U.; Belanto, J.J.; Kenea, A.; Talsness, D.M.; Olthoff, J.T.; Tran, M.D.; Zaal, K.J.; Pak, K.; Pinal-Fernandez, I.; et al. Persistent upregulation of the β-tubulin tubb6, linked to muscle regeneration, is a source of microtubule disorganization in dystrophic muscle. Hum. Mol. Genet. 2019, 28, 1117–1135. [Google Scholar] [CrossRef] [PubMed]
- Ganote, C.; Armstrong, S. Ischaemia and the myocyte cytoskeleton: Review and speculation. Cardiovasc. Res. 1993, 27, 1387–1403. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Tanguy, S.; Foriel, S.; Grichine, A.; Sanchez, C.; Pernet-Gallay, K.; Kaambre, T.; Kuznetsov, A.V.; Usson, Y.; Boucher, F.; et al. The impact of cardiac ischemia/reperfusion on the mitochondria-cytoskeleton interactions. Biochim. Biophys. Acta 2016, 1862, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Perricone, A.J.; Vander Heide, R.S. Novel therapeutic strategies for ischemic heart disease. Pharm. Res. 2014, 89, 36–45. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef]
- Saks, V.A.; Belikova, Y.O.; Kuznetsov, A.V.; Khuchua, Z.A.; Branishte, T.; Semenovsky, M.L.; Naumov, V.G. Phosphocreatine pathway for intracellular energy transport: Facilitation of restricted diffusion of ADP in cardiomyocytes and alterations in cardiomyopathy. Am. J. Physiol. 1991, 261, 30–38. [Google Scholar]
- Khuchua, Z.A.; Kuznetsov, A.V.; Grishin, M.N.; Ventura-Clapier, R.; Saks, V.A. Alterations in the creatine kinase system in the cardiomyopathic hamsters. Biochem. Biophys. Res. Commun. 1989, 165, 748–757. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V.; Joubert, F. Bioenergetics of the failing heart. Biochim. Biophys. Acta 2011, 1813, 1360–1372. [Google Scholar] [CrossRef]
- Cao, F.; Zervou, S.; Lygate, C.A. The creatine kinase system as a therapeutic target for myocardial ischaemia-reperfusion injury. Biochem. Soc. Trans. 2018, 46, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Pucar, D.; Dzeja, P.P.; Bast, P.; Juranic, N.; Macura, S.; Terzic, A. Cellular energetics in the preconditioned state: Protective role for phosphotransfer reactions captured by 18O-assisted 31P NMR. J. Biol. Chem. 2001, 276, 44812–44819. [Google Scholar] [CrossRef] [PubMed]
- Devillard, L.; Vandroux, D.; Tissier, C.; Brochot, A.; Voisin, S.; Rochette, L.; Athias, P. Tubulin ligands suggest a microtubule-NADPH oxidase relationship in postischemic cardiomyocytes. Eur. J. Pharm. 2006, 548, 64–73. [Google Scholar] [CrossRef]
- Devillard, L.; Vandroux, D.; Tissier, C.; Dumont, L.; Borgeot, J.; Rochette, L.; Athias, P. Involvement of microtubules in the tolerance of cardiomyocytes to cold ischemia-reperfusion. Mol. Cell. Biochem. 2008, 307, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Decker, R.S.; Decker, M.L.; Nakamura, S.; Zhao, Y.-S.; Hedjbeli, S.; Harris, K.R.; Klocke, F.J. HSC73-tubulin complex formation during low-flow ischemia in the canine myocardium. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1322–H1333. [Google Scholar] [CrossRef]
- Kay, L.; Rossi, A.; Saks, V. Detection of early ischemic damage by analysis of mitochondrial function in skinned fibers. Mol. Cell. Biochem. 1997, 174, 79–85. [Google Scholar] [CrossRef]
- Boudina, S.; Laclau, M.N.; Tariosse, L.; Daret, D.; Gouverneur, G.; Bonoron-Adèle, S.; Saks, V.A.; Dos Santos, P. Alteration of mitochondrial function in a model of chronic ischemia in vivo in rat heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H821–H831. [Google Scholar] [CrossRef][Green Version]
- Ichas, F.; Mazat, J.P. From calcium signaling to cell death: Two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta 1998, 1366, 33–50. [Google Scholar] [CrossRef]
- Halestrap, A.P. Regulation of mitochondrial metabolism through changes in matrix volume. Biochem. Soc. Trans. 1994, 22, 522–529. [Google Scholar] [CrossRef]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M. Mitochondrial channels: Ion fluxes and more. Physiol. Rev. 2014, 94, 519–608. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Kerr, P.M.; Javadov, S.; Woodfield, K.Y. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim. Biophys. Acta 1998, 1366, 79–94. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharm. Exp. 2009, 330, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef]
- Javadov, S.; Jang, S.; Parodi-Rullán, R.; Khuchua, Z.; Kuznetsov, A.V. Mitochondrial permeability transition in cardiac ischemia-reperfusion: Whether cyclophilin D is a viable target for cardioprotection? Cell. Mol. Life Sci. 2017, 74, 2795–2813. [Google Scholar] [CrossRef]
- Ramos, S.V.; Hughes, M.C.; Perry, C.G.R. Altered skeletal muscle microtubule-mitochondrial VDAC2 binding is related to bioenergetic impairments after paclitaxel but not vinblastine chemotherapies. Am. J. Physiol. Cell Physiol. 2019, 316, C449–C455. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Shanmughapriya, S.; Ahooyi, T.M.; Knezevic, T.; Gupta, M.K.; Kontos, C.D.; McClung, J.M.; Madesh, M.; Gordon, J.; Feldman, A.M.; et al. Dysregulation of mitochondrial bioenergetics and quality control by HIV-1 Tat in cardiomyocytes. J. Cell. Physiol. 2018, 233, 748–758. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, J.P.; Zhao, X.Y.; Guo, W.S. A chemical kinetic model for Ca2+ induced spontaneous oscillatory contraction of myocardium. Biophys. Chem. 2019, 253, 106221. [Google Scholar]
- Yang, Y.; Tian, Y.; Hu, S.; Bi, S.; Li, S.; Hu, Y.; Kou, J.; Qi, J.; Yu, B. Extract of Sheng-Mai-San Ameliorates Myocardial Ischemia-Induced Heart Failure by Modulating Ca2+-Calcineurin-Mediated Drp1 Signaling Pathways. Int. J. Mol. Sci. 2017, 18, 1825. [Google Scholar] [CrossRef] [PubMed]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.-T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Woollard, J.R.; Jordan, K.L.; Lerman, A.; Lerman, L.O.; Eirin, A. Mitochondrial targeted peptides preserve mitochondrial organization and decrease reversible myocardial changes in early swine metabolic syndrome. Cardiovasc. Res. 2018, 114, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Trimarco, B.; Iaccarino, G.; Santulli, G. New Insights in Cardiac Calcium Handling and Excitation-Contraction Coupling. Adv. Exp. Med. Biol. 2018, 1067, 373–385. [Google Scholar]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Mark, W.; Steurer, W.; Saks, V.; Usson, Y.; Margreiter, R.; Gnaiger, E. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am. J. Physiol. 2004, 286, H1633–H1641. [Google Scholar] [CrossRef]
- Beraud, N.; Pelloux, S.; Usson, Y.; Kuznetsov, A.V.; Ronot, X.; Tourneur, Y.; Saks, V. Mitochondrial dynamics in heart cells: Very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non-beating Hl-1 cells. J. Bioenerg. Biomembr. 2009, 41, 195–214. [Google Scholar] [CrossRef]
- Yancey, D.M.; Guichard, J.L.; Ahmed, M.I.; Zhou, L.; Murphy, M.P.; Johnson, M.S.; Benavides, G.A.; Collawn, J.; Darley-Usmar, V.; Dell’Italia, L.J. Cardiomyocyte mitochondrial oxidative stress and cytoskeletal breakdown in the heart with a primary volume overload. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H651–H663. [Google Scholar] [CrossRef]
- Tsutsui, H.; Tagawa, H.; Kent, R.L.; McCollam, P.L.; Ishihara, K.; Nagatsu, M.; Cooper, G. Role of microtubules in contractile dysfunction of hypertrophied cardiocytes. Circulation 1994, 90, 533–555. [Google Scholar] [CrossRef]
- Koide, M.; Hamawaki, M.; Narishige, T.; Sato, H.; Nemoto, S.; DeFreyte, G.; Zile, M.R.; Cooper, G., IV; Carabello, B.A. Microtubule depolymerization normalizes in vivo myocardial contractile function in dogs with pressure-overload left ventricular hypertrophy. Circulation. 2000, 102, 1045–1052. [Google Scholar] [CrossRef]
- Nederlof, R.; Eerbeek, O.; Hollmann, M.W.; Southworth, R.; Zuurbier, C.J. Targeting hexokinase II to mitochondria to modulate energy metabolism and reduce ischaemia-reperfusion injury in heart. Br. J. Pharmacol. 2014, 171, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Belmadani, S.; Poüs, C.; Fischmeister, R.; Méry, P.F. Post-translational modifications of tubulin and microtubule stability in adult rat ventricular myocytes and immortalized HL-1 cardiomyocytes. Mol. Cell. Biochem. 2004, 258, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Schaper, J.; Kostin, S.; Hein, A.; Elsasser, E.; Arnon, D.; Zimmermann, R. Structural remodelling in heart failure. Exp. Clin. Cardiol. 2002, 7, 64–68. [Google Scholar] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsov, A.V.; Javadov, S.; Grimm, M.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells 2020, 9, 222. https://doi.org/10.3390/cells9010222
Kuznetsov AV, Javadov S, Grimm M, Margreiter R, Ausserlechner MJ, Hagenbuchner J. Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells. 2020; 9(1):222. https://doi.org/10.3390/cells9010222
Chicago/Turabian StyleKuznetsov, Andrey V., Sabzali Javadov, Michael Grimm, Raimund Margreiter, Michael J. Ausserlechner, and Judith Hagenbuchner. 2020. "Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells" Cells 9, no. 1: 222. https://doi.org/10.3390/cells9010222
APA StyleKuznetsov, A. V., Javadov, S., Grimm, M., Margreiter, R., Ausserlechner, M. J., & Hagenbuchner, J. (2020). Crosstalk between Mitochondria and Cytoskeleton in Cardiac Cells. Cells, 9(1), 222. https://doi.org/10.3390/cells9010222