Peroxiredoxin 5 Silencing Sensitizes Dopaminergic Neuronal Cells to Rotenone via DNA Damage-Triggered ATM/p53/PUMA Signaling-Mediated Apoptosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Animals and Treatment
2.3. Lentiviral Transduction
2.4. Sirna and Prx5-Expressing Plasmid Transfection
2.5. Immunocytochemistry
2.6. Immunohistochemistry
2.7. Cell Viability Assay
2.8. Apoptosis Assay
2.9. Flow Cytometry
2.10. Real-Time RT-PCR Analysis
2.11. Preparation of Whole-Cell Extracts and Subcellular Fractionations
2.12. Western Blotting
2.13. Caspase Activity Assay
2.14. Statistical Analysis
3. Results
3.1. Decrease of Prx5 Expression in DA Neurons in Rotenone-Induced Cellular and Rat Models of PD
3.2. Prx5 Depletion Sensitizes DA Neuronal Cells to Rotenone-Induced Apoptosis
3.3. Knockdown of Prx5 Exacerbates Mitochondria-Driven Apoptotic Pathway by Rotenone
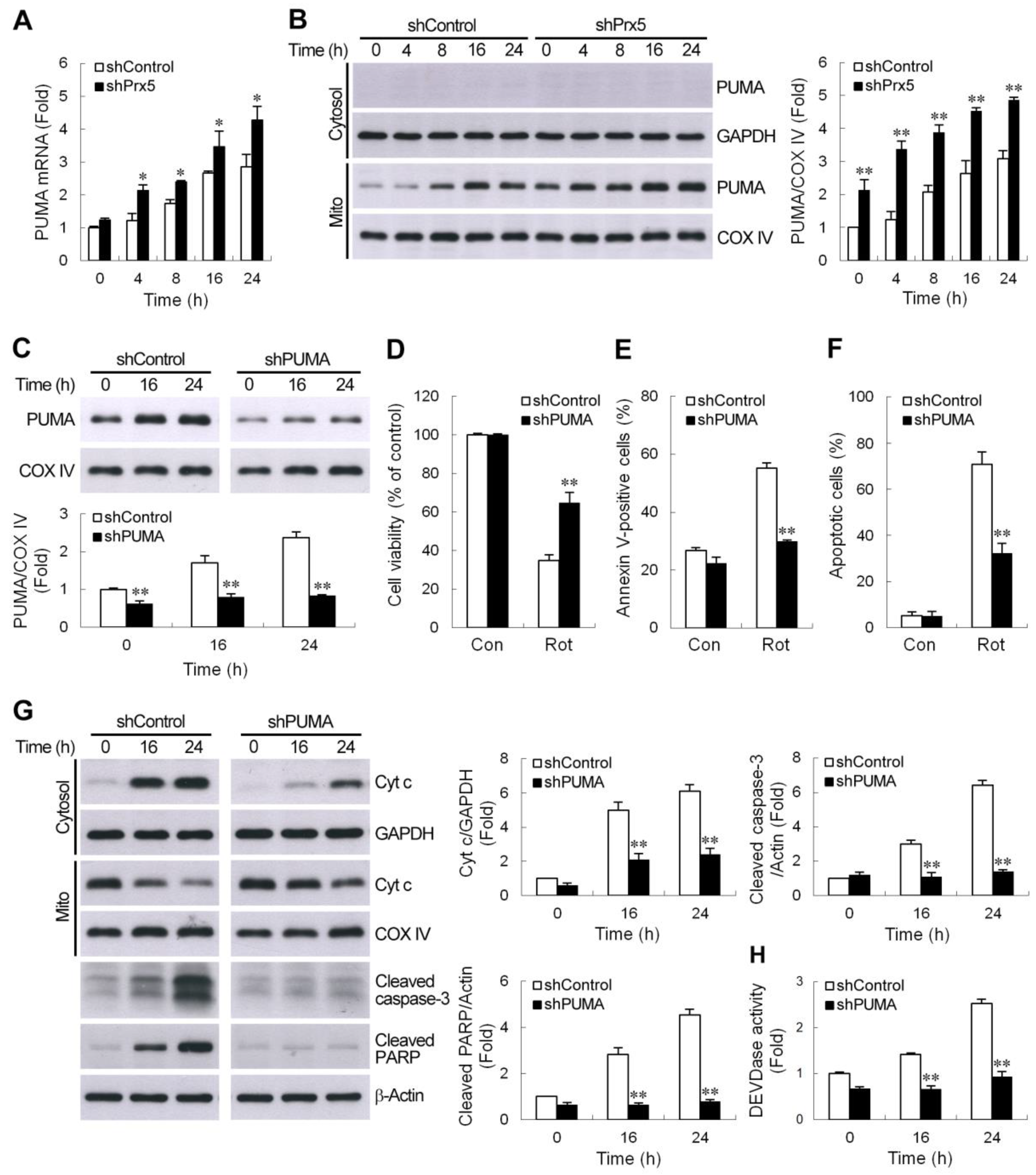
3.4. Silencing of Prx5 Enhances the Activation of PUMA-Mediated Mitochondrial Apoptotic Pathway
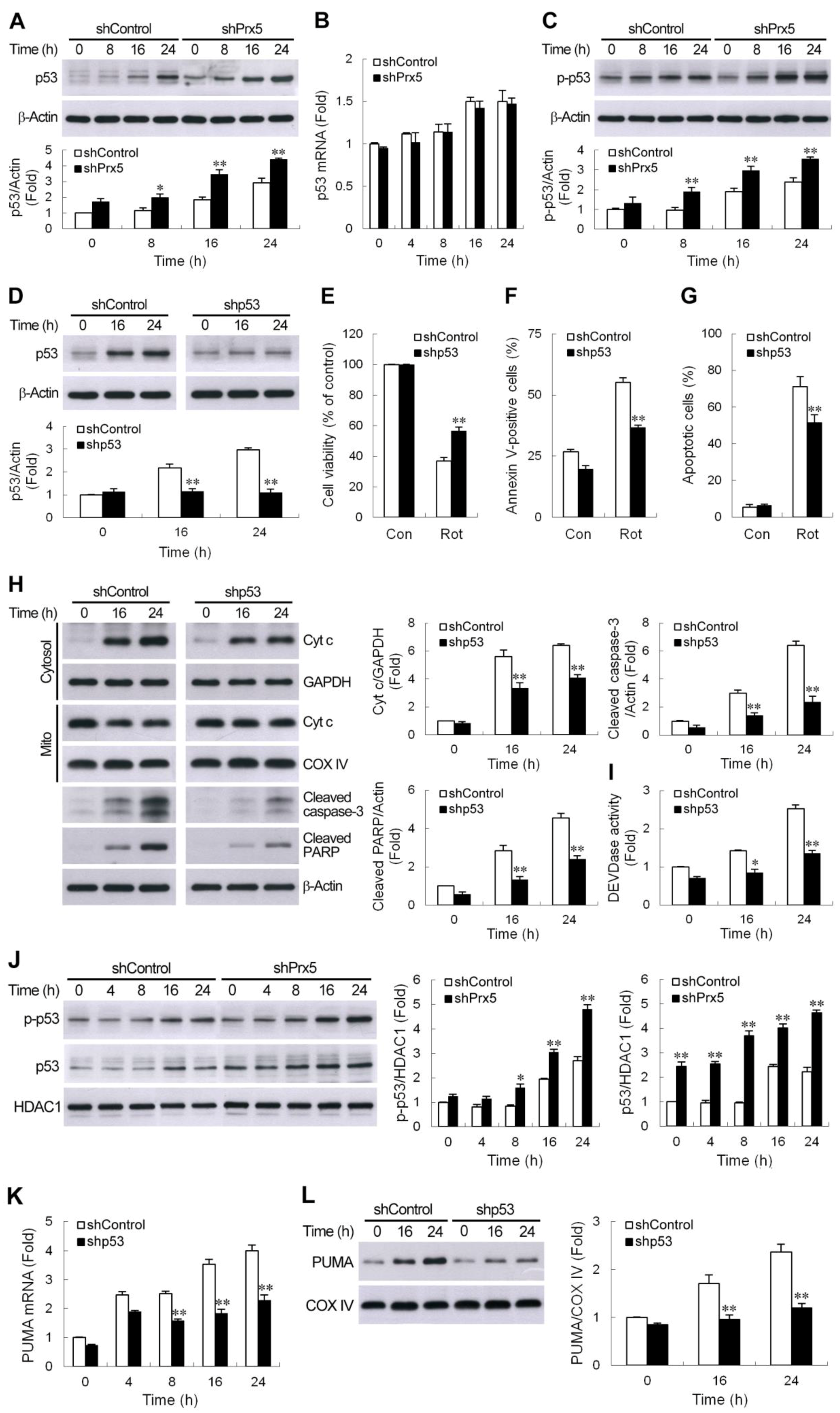
3.5. Prx5 Downregulation Strengthens the p53-PUMA-Mediated Apoptotic Pathway
3.6. Prx5 Loss Augments DNA Damage-Triggered ATM/p53 Signaling
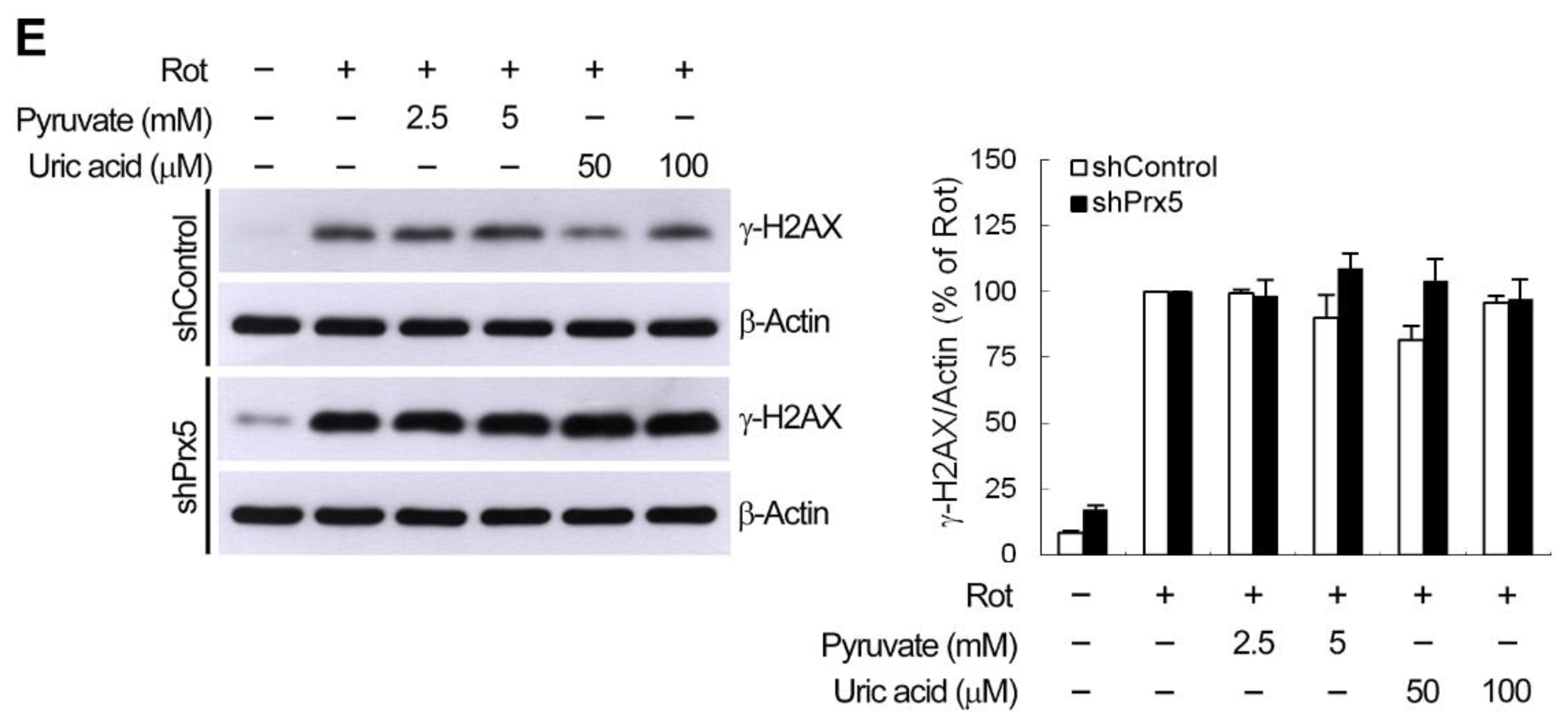
3.7. ROS Does Not Contribute to Prx5 Depletion-Increased γ-H2AX Induction by Rotenone
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and molecular basis of neurodegeneration in Parkinson disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Perry, G.; Smith, M.A.; Robertson, D.; Olson, S.J.; Graham, D.G.; Montine, T.J. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am. J. Pathol. 1999, 154, 1423–1429. [Google Scholar] [CrossRef]
- Hegde, M.L.; Gupta, V.B.; Anitha, M.; Harikrishna, T.; Shankar, S.K.; Muthane, U.; Subba Rao, K.; Jagannatha Rao, K.S. Studies on genomic DNA topology and stability in brain regions of Parkinson’s disease. Arch. Biochem. Biophys. 2006, 449, 143–156. [Google Scholar] [CrossRef]
- Chen, C.M.; Liu, J.L.; Wu, Y.R.; Chen, Y.C.; Cheng, H.S.; Cheng, M.L.; Chiu, D.T. Increased oxidative damage in peripheral blood correlates with severity of Parkinson’s disease. Neurobiol. Dis. 2009, 33, 429–435. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kajitani, K.; Dan, Y.; Furuichi, M.; Ohno, M.; Sakumi, K.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine. Cell Death Differ. 2006, 13, 551–563. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Rhee, S.G.; Yang, K.S.; Kang, S.W.; Woo, H.A.; Chang, T.S. Controlled elimination of intracellular H2O2: Regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid. Redox Signal. 2005, 7, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Fujii, J.; Ikeda, Y. Advances in our understanding of peroxiredoxin, a multifunctional, mammalian redox protein. Redox Rep. 2002, 7, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Knoops, B.; Goemaere, J.; Van der Eecken, V.; Declercq, J.P. Peroxiredoxin 5: Structure, mechanism, and function of the mammalian atypical 2-Cys peroxiredoxin. Antioxid. Redox Signal. 2011, 15, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Kropotov, A.; Sedova, V.; Ivanov, V.; Sazeeva, N.; Tomilin, A.; Krutilina, R.; Oei, S.L.; Griesenbeck, J.; Buchlow, G.; Tomilin, N. A novel human DNA-binding protein with sequence similarity to a subfamily of redox proteins which is able to repress RNA-polymerase-III-driven transcription of the Alu-family retroposons in vitro. Eur. J. Biochem. 1999, 260, 336–346. [Google Scholar] [CrossRef]
- Kropotov, A.V.; Grudinkin, P.S.; Pleskach, N.M.; Gavrilov, B.A.; Tomilin, N.V.; Zhivotovsky, B. Downregulation of peroxiredoxin V stimulates formation of etoposide-induced double-strand DNA breaks. FEBS Lett. 2004, 572, 75–79. [Google Scholar] [CrossRef]
- De Simoni, S.; Linard, D.; Hermans, E.; Knoops, B.; Goemaere, J. Mitochondrial peroxiredoxin-5 as potential modulator of mitochondria-ER crosstalk in MPP+-induced cell death. J. Neurochem. 2013, 125, 473–485. [Google Scholar] [CrossRef]
- Kim, B.; Park, J.; Chang, K.T.; Lee, D.S. Peroxiredoxin 5 prevents amyloid-beta oligomer-induced neuronal cell death by inhibiting ERK-Drp1-mediated mitochondrial fragmentation. Free Radic. Biol. Med. 2016, 90, 184–194. [Google Scholar] [CrossRef]
- Park, J.; Kim, B.; Chae, U.; Lee, D.G.; Kam, M.K.; Lee, S.R.; Lee, S.; Lee, H.S.; Park, J.W.; Lee, D.S. Peroxiredoxin 5 decreases beta-amyloid-mediated cyclin-dependent kinase 5 activation through regulation of Ca2+-mediated calpain activation. Antioxid. Redox Signal. 2017, 27, 715–726. [Google Scholar] [CrossRef]
- Plaisant, F.; Clippe, A.; Vander Stricht, D.; Knoops, B.; Gressens, P. Recombinant peroxiredoxin 5 protects against excitotoxic brain lesions in newborn mice. Free Radic. Biol. Med. 2003, 34, 862–872. [Google Scholar] [CrossRef]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.; Lubec, G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J. Neural. Transm. Suppl. 2001, 61, 223–235. [Google Scholar]
- Basso, M.; Giraudo, S.; Corpillo, D.; Bergamasco, B.; Lopiano, L.; Fasano, M. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics 2004, 4, 3943–3952. [Google Scholar] [CrossRef] [PubMed]
- Qu, D.; Rashidian, J.; Mount, M.P.; Aleyasin, H.; Parsanejad, M.; Lira, A.; Haque, E.; Zhang, Y.; Callaghan, S.; Daigle, M.; et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 2007, 55, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Park, S.H.; Shin, D.I.; Hwang, J.Y.; Park, B.; Park, Y.J.; Lee, T.H.; Chae, H.Z.; Jin, B.K.; Oh, T.H.; et al. Oxidative modification of peroxiredoxin is associated with drug-induced apoptotic signaling in experimental models of Parkinson disease. J. Biol. Chem. 2008, 283, 9986–9998. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Lin, S.Z.; Kuo, J.S.; Chen, W.F.; Wang, M.J. G-CSF protects dopaminergic neurons from 6-OHDA-induced toxicity via the ERK pathway. Neurobiol. Aging 2007, 28, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hao, J.; Zheng, Y.; Su, R.; Liao, Y.; Gong, X.; Liu, L.; Wang, X. Fucoidan protects dopaminergic neurons by enhancing the mitochondrial function in a rotenone-induced rat model of Parkinson’s disease. Aging Dis. 2018, 9, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Chiu, T.L.; Chang, H.F.; Hsu, H.R.; Pang, C.Y.; Liew, H.K.; Wang, M.J. Epigenetic regulation contributes to urocortin-enhanced midbrain dopaminergic neuron differentiation. Stem Cells 2015, 33, 1601–1617. [Google Scholar] [CrossRef]
- Xiong, N.; Long, X.; Xiong, J.; Jia, M.; Chen, C.; Huang, J.; Ghoorah, D.; Kong, X.; Lin, Z.; Wang, T. Mitochondrial complex I inhibitor rotenone-induced toxicity and its potential mechanisms in Parkinson’s disease models. Crit. Rev. Toxicol. 2012, 42, 613–632. [Google Scholar] [CrossRef]
- Hu, X.; Weng, Z.; Chu, C.T.; Zhang, L.; Cao, G.; Gao, Y.; Signore, A.; Zhu, J.; Hastings, T.; Greenamyre, J.T.; et al. Peroxiredoxin-2 protects against 6-hydroxydopamine- induced dopaminergic neurodegeneration via attenuation of the apoptosis signal- regulating kinase (ASK1) signaling cascade. J. Neurosci. 2011, 31, 247–261. [Google Scholar] [CrossRef]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P., Jr. Chronic, low dose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21. [Google Scholar] [CrossRef]
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death Dis. 2018, 9, 818. [Google Scholar] [CrossRef]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Betarbet, R.; Sherer, T.B. The rotenone model of Parkinson’s disease: Genes, environment and mitochondria. Parkinsonism Relat. Disord. 2003, 9, S59–S64. [Google Scholar] [CrossRef]
- Sherer, T.B.; Richardson, J.R.; Testa, C.M.; Seo, B.B.; Panov, A.V.; Yagi, T.; Matsuno-Yagi, A.; Miller, G.W.; Greenamyre, J.T. Mechanism of toxicity of pesticides acting at complex I: Relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 2007, 100, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Bouillet, P.; Strasser, A. BH3-only proteins - evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 2002, 115, 1567–1574. [Google Scholar] [PubMed]
- Yu, J.; Zhang, L. PUMA, a potent killer with or without p53. Oncogene 2008, 27, S71–S83. [Google Scholar] [CrossRef] [PubMed]
- Cregan, S.P.; Arbour, N.A.; Maclaurin, J.G.; Callaghan, S.M.; Fortin, A.; Cheung, E.C.; Guberman, D.S.; Park, D.S.; Slack, R.S. P53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J. Neurosci. 2004, 24, 10003–10012. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Tolkovsky, A.M. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J. Neurochem. 2006, 96, 1213–1226. [Google Scholar] [CrossRef]
- Uo, T.; Kinoshita, Y.; Morrison, R.S. Apoptotic actions of p53 require transcriptional activation of PUMA and do not involve a direct mitochondrial/cytoplasmic site of action in postnatal cortical neurons. J. Neurosci. 2007, 27, 12198–12210. [Google Scholar] [CrossRef]
- Steckley, D.; Karajgikar, M.; Dale, L.B.; Fuerth, B.; Swan, P.; Drummond-Main, C.; Poulter, M.O.; Ferguson, S.S.; Strasser, A.; Cregan, S.P. Puma is a dominant regulator of oxidative stress induced Bax activation and neuronal apoptosis. J. Neurosci. 2007, 27, 12989–12999. [Google Scholar] [CrossRef]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.J.; Yang, L.X. Gamma-H2AX- a novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar] [PubMed]
- Yuan, J.; Adamski, R.; Chen, J. Focus on histone variant H2AX: To be or not to be. FEBS Lett. 2010, 584, 3717–3724. [Google Scholar] [CrossRef] [PubMed]
- Katsube, T.; Mori, M.; Tsuji, H.; Shiomi, T.; Wang, B.; Liu, Q.; Nenoi, M.; Onoda, M. Most hydrogen peroxide-induced histone H2AX phosphorylation is mediated by ATR and is not dependent on DNA double-strand breaks. J. Biochem. 2014, 156, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Vafeiadou, K.; Rice-Evans, C.; Cadenas, E.; Spencer, J.P. Inhibition of cellular proliferation by the genistein metabolite 5,7,3′,4′-tetrahydroxyisoflavone is mediated by DNA damage and activation of the ATR signalling pathway. Arch. Biochem. Biophys. 2007, 468, 159–166. [Google Scholar] [CrossRef]
- Hattori, F.; Oikawa, S. Peroxiredoxins in the central nervous system. Subcell. Biochem. 2007, 44, 357–374. [Google Scholar]
- Goemaere, J.; Knoops, B. Peroxiredoxin distribution in the mouse brain with emphasis on neuronal populations affected in neurodegenerative disorders. J. Comp. Neurol. 2012, 520, 258–280. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef]
- Allen, S.; Heath, P.R.; Kirby, J.; Wharton, S.B.; Cookson, M.R.; Menzies, F.M.; Banks, R.E.; Shaw, P.J. Analysis of the cytosolic proteome in a cell culture model of familial amyotrophic lateral sclerosis reveals alterations to the proteasome, antioxidant defenses, and nitric oxide synthetic pathways. J. Biol. Chem. 2003, 278, 6371–6383. [Google Scholar] [CrossRef]
- Strey, C.W.; Spellman, D.; Stieber, A.; Gonatas, J.O.; Wang, X.; Lambris, J.D.; Gonatas, N.K. Dysregulation of stathmin, a microtubule-destabilizing protein, and up-regulation of Hsp25, Hsp27, and the antioxidant peroxiredoxin 6 in a mouse model of familial amyotrophic lateral sclerosis. Am. J. Pathol. 2004, 165, 1701–1718. [Google Scholar] [CrossRef]
- Banmeyer, I.; Marchand, C.; Verhaeghe, C.; Vucic, B.; Rees, J.F.; Knoops, B. Overexpression of human peroxiredoxin 5 in subcellular compartments of Chinese hamster ovary cells: Effects on cytotoxicity and DNA damage caused by peroxides. Free Radic. Biol. Med. 2004, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Walbrecq, G.; Wang, B.; Becker, S.; Hannotiau, A.; Fransen, M.; Knoops, B. Antioxidant cytoprotection of peroxisomal peroxiredoxin 5. Free Radic. Biol. Med. 2015, 84, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Hattori, F.; Murayama, N.; Noshita, T.; Oikawa, S. Mitochondrial peroxiredoxin-3 protects hippocampal neurons from excitotoxic injury in vivo. J. Neurochem. 2003, 86, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Salzano, S.; Checconi, P.; Hanschmann, E.M.; Lillig, C.H.; Bowler, L.D.; Chan, P.; Vaudry, D.; Mengozzi, M.; Coppo, L.; Sacre, S.; et al. Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. USA 2014, 111, 12157–12162. [Google Scholar] [CrossRef] [PubMed]
- Mullen, L.; Hanschmann, E.M.; Lillig, C.H.; Herzenberg, L.A.; Ghezzi, P. Cysteine oxidation targets peroxiredoxins 1 and 2 for exosomal release through a novel mechanism of redox-dependent secretion. Mol. Med. 2015, 21, 98–108. [Google Scholar] [CrossRef]
- Tiedemann, K.; Sadvakassova, G.; Mikolajewicz, N.; Juhas, M.; Sabirova, Z.; Tabariès, S.; Gettemans, J.; Siegel, P.M.; Komarova, S.V. Exosomal release of L-plastin by breast cancer cells facilitates metastatic bone osteolysis. Transl. Oncol. 2019, 12, 462–474. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef]
- Perier, C.; Bove, J.; Vila, M. Mitochondria and programmed cell death in Parkinson’s disease: Apoptosis and beyond. Antioxid. Redox Signal. 2012, 16, 883–895. [Google Scholar] [CrossRef]
- Kountouras, J.; Zavos, C.; Polyzos, S.A.; Deretzi, G.; Vardaka, E.; Giartza-Taxidou, E.; Katsinelos, P.; Rapti, E.; Chatzopoulos, D.; Tzilves, D.; et al. Helicobacter pylori infection and Parkinson’s disease: Apoptosis as an underlying common contributor. Eur. J. Neurol. 2012, 19, e56. [Google Scholar] [CrossRef]
- Perier, C.; Bové, J.; Wu, D.C.; Dehay, B.; Choi, D.K.; Jackson-Lewis, V.; Rathke-Hartlieb, S.; Bouillet, P.; Strasser, A.; Schulz, J.B.; et al. Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 8161–8166. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.K.; Lee, K.J.; Rhee, S.; Seo, S.B.; Pak, J.H. Protective effects of peroxiredoxin 6 overexpression on amyloid β-induced apoptosis in PC12 cells. Free Radic. Res. 2013, 47, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle 2009, 8, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, A.I.; Garrison, S.P.; Zambetti, G.P.; O’Malley, K.L. 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol. Neurodegener. 2011, 6, 2. [Google Scholar] [CrossRef]
- Qi, X.; Davis, B.; Chiang, Y.H.; Filichia, E.; Barnett, A.; Greig, N.H.; Hoffer, B.; Luo, Y. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson’s disease model. J. Neurochem. 2016, 138, 746–757. [Google Scholar] [CrossRef]
- Nair, V.D. Activation of p53 signaling initiates apoptotic death in a cellular model of Parkinson’s disease. Apoptosis 2006, 11, 955–966. [Google Scholar] [CrossRef]
- Bernstein, A.I.; O’Malley, K.L. MPP+-induces PUMA- and p53-dependent, but ATF3- independent cell death. Toxicol. Lett. 2013, 219, 93–98. [Google Scholar] [CrossRef]
- Wu, L.; Tian, Y.Y.; Shi, J.P.; Xie, W.; Shi, J.Q.; Lu, J.; Zhang, Y.D. Inhibition of endoplasmic reticulum stress is involved in the neuroprotective effects of candesartan cilexitil in the rotenone rat model of Parkinson’s disease. Neurosci. Lett. 2013, 548, 50–55. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Wu, F.; Wang, Z.; Gu, J.H.; Ge, J.B.; Liang, Z.Q.; Qin, Z.H. p38(MAPK)/p53-Mediated Bax induction contributes to neurons degeneration in rotenone-induced cellular and rat models of Parkinson’s disease. Neurochem. Int. 2013, 63, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liu, T.; Dong, S.Y.; Guo, Y.J.; Jankovic, J.; Xu, H.; Wu, Y.C. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J. Neurochem. 2015, 134, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.D.; McNaught, K.S.; González-Maeso, J.; Sealfon, S.C.; Olanow, C.W. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J. Biol. Chem. 2006, 281, 39550–39560. [Google Scholar] [CrossRef] [PubMed]
- Konopka, A.; Atkin, J.D. The emerging role of DNA damage in the pathogenesis of the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2018, 19, 3137. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.J.; Keramaris, E.; Rideout, H.J.; Slack, R.S.; Dyson, N.J.; Stefanis, L.; Park, D.S. Cyclin-dependent kinases and p53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J. Neurosci. 2001, 21, 5017–5026. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Liu, Z.; Pipino, J.; Chestnut, B.; Landek, M.A. Molecular regulation of DNA damage- induced apoptosis in neurons of cerebral cortex. Cereb. Cortex 2009, 19, 1273–1293. [Google Scholar] [CrossRef]
- Hong, Y.; Nie, H.; Wei, X.; Fu, S.; Ying, W. NAD+ treatment can prevent rotenone-induced increases in DNA damage, Bax levels and nuclear translocation of apoptosis-inducing factor in differentiated PC12 cells. Nuerochem. Res. 2015, 40, 837–842. [Google Scholar] [CrossRef]
- Xia, N.; Zhang, Q.; Wang, S.T.; Gu, L.; Yang, H.M.; Liu, L.; Bakshi, R.; Yang, H.; Zhang, H. Blockade of metabotropic glutamate receptor 5 protects against DNA damage in a rotenone-induced Parkinson’s disease model. Free Radic. Biol. Med. 2015, 89, 567–580. [Google Scholar] [CrossRef]
- Kropotov, A.; Serikov, V.; Suh, J.; Smirnova, A.; Bashkirov, V.; Zhivotovsky, B.; Tomilin, N. Constitutive expression of the human peroxiredoxin V gene contributes to protection of the genome from oxidative DNA lesions and to suppression of transcription of noncoding DNA. FEBS J. 2006, 273, 2607–2617. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.-J.; Huang, H.-Y.; Chiu, T.-L.; Chang, H.-F.; Wu, H.-R. Peroxiredoxin 5 Silencing Sensitizes Dopaminergic Neuronal Cells to Rotenone via DNA Damage-Triggered ATM/p53/PUMA Signaling-Mediated Apoptosis. Cells 2020, 9, 22. https://doi.org/10.3390/cells9010022
Wang M-J, Huang H-Y, Chiu T-L, Chang H-F, Wu H-R. Peroxiredoxin 5 Silencing Sensitizes Dopaminergic Neuronal Cells to Rotenone via DNA Damage-Triggered ATM/p53/PUMA Signaling-Mediated Apoptosis. Cells. 2020; 9(1):22. https://doi.org/10.3390/cells9010022
Chicago/Turabian StyleWang, Mei-Jen, Hsin-Yi Huang, Tsung-Lang Chiu, Hui-Fen Chang, and Hsin-Rong Wu. 2020. "Peroxiredoxin 5 Silencing Sensitizes Dopaminergic Neuronal Cells to Rotenone via DNA Damage-Triggered ATM/p53/PUMA Signaling-Mediated Apoptosis" Cells 9, no. 1: 22. https://doi.org/10.3390/cells9010022
APA StyleWang, M.-J., Huang, H.-Y., Chiu, T.-L., Chang, H.-F., & Wu, H.-R. (2020). Peroxiredoxin 5 Silencing Sensitizes Dopaminergic Neuronal Cells to Rotenone via DNA Damage-Triggered ATM/p53/PUMA Signaling-Mediated Apoptosis. Cells, 9(1), 22. https://doi.org/10.3390/cells9010022