Quantitative In-Depth Analysis of the Mouse Mast Cell Transcriptome Reveals Organ-Specific Mast Cell Heterogeneity


Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Generation of Bone Marrow-Derived MCs (BMMCs) and the effect of LPS Stimulation
2.3. Peritoneal Cell Extraction and Sorting of Peritoneal Mast Cells
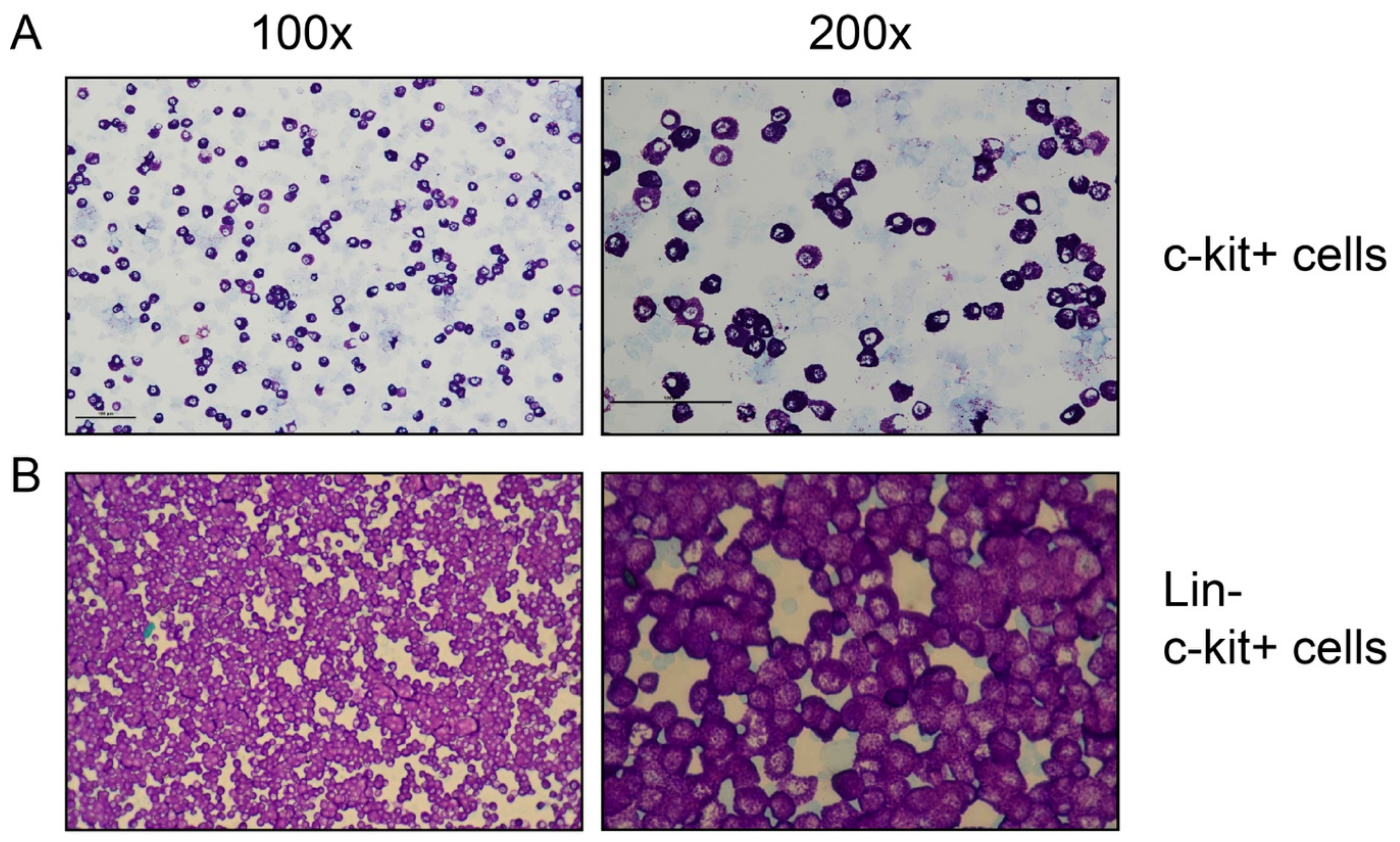
2.4. Image Analysis
2.5. RNA Isolation
2.6. Analysis of the Transcriptome by RNA-Seq and by the Thermo Fisher Ampliseq Chip and PCR-Based Method
3. Results
3.1. Preparation of RNA from Tissues and Purified Peritoneal Cell Fractions
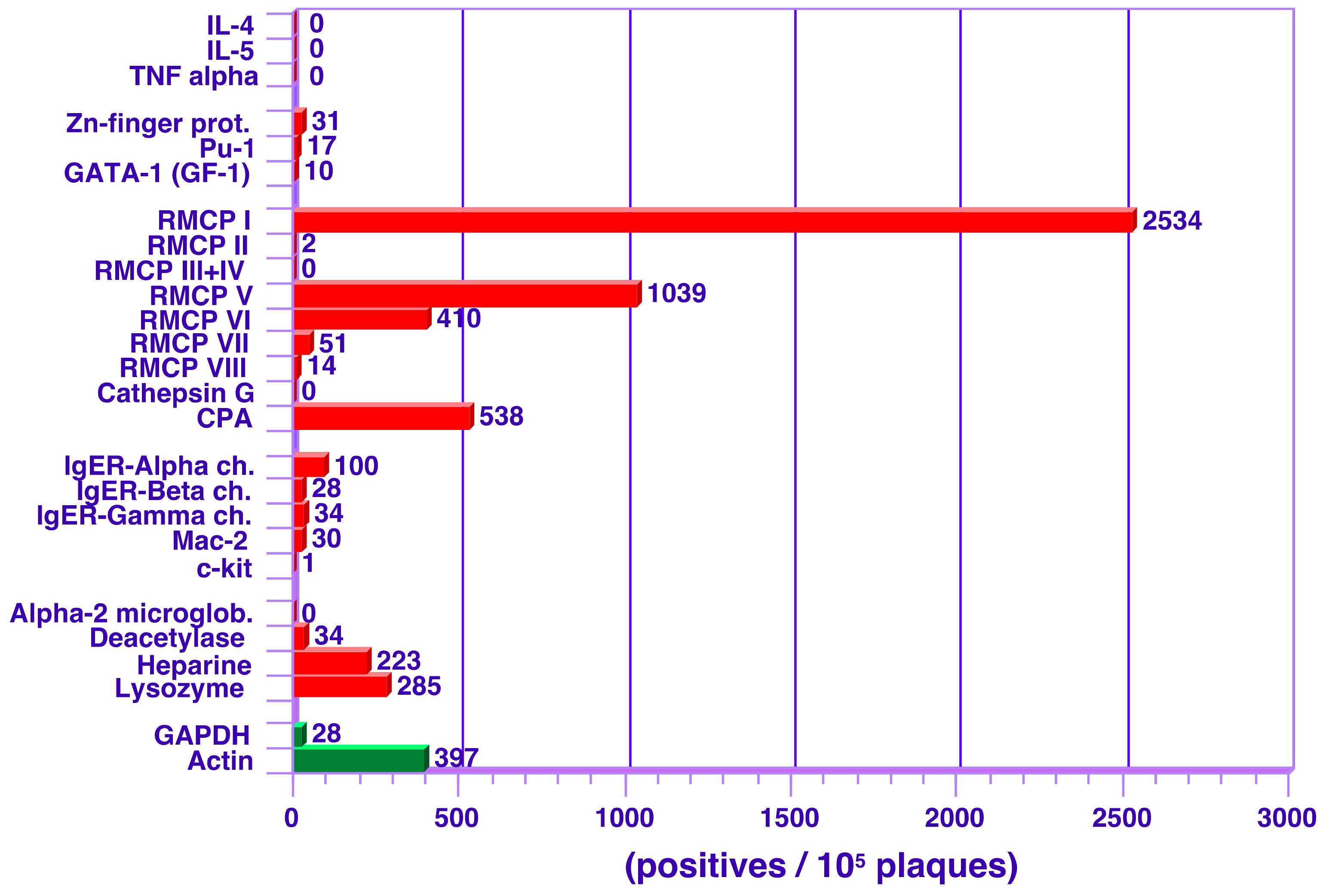
3.2. Analysis of Transcript Levels in Peritoneal MCs
3.3. Analysis of In Vitro Differentiated BMMCs
3.4. Analysis of the Transcript Levels in Mouse Ear with a Focus on MC Transcripts
3.5. Analysis of the Transcript Levels in Mouse Lungs with a Focus on MC Transcripts
3.6. Analysis of Transcript Levels in Peritoneal MCs of a Higher Purity to Obtain a More Complete Picture of the MC Transcriptome
3.7. Analysis of MC-Related Transcript Levels in Mouse Brain, Liver, Kidneys, Tongue, Heart, Pancreas, Duodenum, Proximal Part of the Colon, Total Spleen, and the Uterus.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Galli, S.J.; Starkl, P.; Marichal, T.; Tsai, M. Mast cells and IgE in defense against venoms: Possible “good side” of allergy? Allergol. Int. 2016, 65, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Hellman, L.T.; Akula, S.; Thorpe, M.; Fu, Z. Tracing the Origins of IgE, Mast Cells, and Allergies by Studies of Wild Animals. Front. Immunol. 2017, 8, 1749. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, M.C.; De Andrade, L.R.; Santos-Pinto, C.D.B.; Straus, A.H.; Takahashi, H.K.; Allodi, S.; Pavão, M.S. Colocalization of heparin and histamine in the intracellular granules of test cells from the invertebrate Styela plicata (Chordata-Tunicata). J. Struct. Boil. 2002, 137, 313–321. [Google Scholar] [CrossRef]
- Abe, T.; Swieter, M.; Imai, T.; Hollander, N.D.; Befus, A.D. Mast cell heterogeneity: Two-dimensional gel electrophoretic analyses of rat peritoneal and intestinal mucosal mast cell. Eur. J. Immunol. 1990, 20, 1941–1947. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.S.; Stevens, R.L.; Lane, W.S.; Carr, M.H.; Austen, K.F.; Serafin, W.E. Different mouse mast cell populations express various combinations of at least six distinct mast cell serine proteases. Proc. Natl. Acad. Sci. USA 1990, 87, 3230–3234. [Google Scholar] [CrossRef]
- Huang, R.; Blom, T.; Hellman, L. Cloning and structural analysis of MMCP-1, MMCP-4 and MMCP-5, three mouse mast cell-specific serine proteases. Eur. J. Immunol. 1991, 21, 1611–1621. [Google Scholar] [CrossRef]
- Lützelschwab, C.; Aveskogh, M.; Pejler, G.; Hellman, L. Secretory Granule Proteases in Rat Mast Cells. Cloning of 10 Different Serine Proteases and a Carboxypeptidase A from Various Rat Mast Cell Populations. J. Exp. Med. 1997, 185, 13–30. [Google Scholar] [CrossRef]
- Lützelschwab, C.; Huang, M.R.; Kullberg, M.C.; Aveskogh, M.; Hellman, L. Characterization of mouse mast cell protease-8, the first member of a novel subfamily of mouse mast cell serine proteases, distinct from both the classical chymases and tryptases. Eur. J. Immunol. 1998, 28, 1022–1033. [Google Scholar] [CrossRef]
- Lützelschwab, C.; Lunderius, C.; Enerbäck, L.; Hellman, L. A kinetic analysis of the expression of mast cell protease mRNA in the intestines of Nippostrongylus brasiliensis-infected rats. Eur. J. Immunol. 1998, 28, 3730–3737. [Google Scholar] [CrossRef]
- Pejler, G.; Rönnberg, E.; Waern, I.; Wernersson, S. Mast cell proteases: Multifaceted regulators of inflammatory disease. Blood 2010, 115, 4981–4990. [Google Scholar] [CrossRef]
- Caughey, G.H. Mast cell proteases as protective and inflammatory mediators. Single Mol. Single Cell Seq. 2011, 716, 212–234. [Google Scholar]
- Hellman, L.; Thorpe, M. Granule proteases of hematopoietic cells, a family of versatile inflammatory mediators – an update on their cleavage specificity, in vivo substrates, and evolution. Boil. Chem. 2014, 395, 15–49. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.; Thorpe, M.; Boinapally, V.; Hellman, L. Granule Associated Serine Proteases of Hematopoietic Cells—An Analysis of Their Appearance and Diversification during Vertebrate Evolution. PLoS ONE 2015, 10, e0143091. [Google Scholar]
- Schwartz, L.B.; Irani, A.M.; Roller, K.; Castells, M.C.; Schechter, N.M. Quantitation of histamine, tryptase, and chymase in dispersed human T and TC mast cells. J. Immunol. 1987, 138, 2611–2615. [Google Scholar]
- Chandrasekharan, U.M.; Sanker, S.; Glynias, M.J.; Karnik, S.S.; Husain, A. Angiotensin II-Forming Activity in a Reconstructed Ancestral Chymase. Science 1996, 271, 502–505. [Google Scholar] [CrossRef]
- Metz, M.; Piliponsky, A.M.; Chen, C.-C.; Lammel, V.; Abrink, M.; Pejler, G.; Tsai, M.; Galli, S.J. Mast Cells Can Enhance Resistance to Snake and Honeybee Venoms. Science 2006, 313, 526–530. [Google Scholar] [CrossRef]
- Akahoshi, M.; Song, C.H.; Piliponsky, A.M.; Metz, M.; Guzzetta, A.; Abrink, M.; Schlenner, S.M.; Feyerabend, T.B.; Rodewald, H.-R.; Pejler, G.; et al. Mast cell chymase reduces the toxicity of Gila monster venom, scorpion venom, and vasoactive intestinal polypeptide in mice. J. Clin. Investig. 2011, 121, 4180–4191. [Google Scholar] [CrossRef]
- Piliponsky, A.M.; Chen, C.-C.; Rios, E.J.; Treuting, P.M.; Lahiri, A.; Abrink, M.; Pejler, G.; Tsai, M.; Galli, S.J. The chymase mouse mast cell protease 4 degrades TNF, limits inflammation, and promotes survival in a model of sepsis. Am. J. Pathol. 2012, 181, 875–886. [Google Scholar] [CrossRef]
- Fu, Z.; Thorpe, M.; Alemayehu, R.; Roy, A.; Kervinen, J.; De Garavilla, L.; Åbrink, M.; Hellman, L. Highly Selective Cleavage of Cytokines and Chemokines by the Human Mast Cell Chymase and Neutrophil Cathepsin G. J. Immunol. 2017, 198, 1474–1483. [Google Scholar] [CrossRef]
- Fu, Z.; Akula, S.; Thorpe, M.; Hellman, L. Highly Selective Cleavage of TH2-Promoting Cytokines by the Human and the Mouse Mast Cell Tryptases, Indicating a Potent Negative Feedback Loop on TH2 Immunity. Int. J. Mol. Sci. 2019, 20, 5147. [Google Scholar] [CrossRef]
- Tchougounova, E.; Forsberg, E.; Angelborg, G.; Kjellen, L.; Pejler, G. Altered processing of fibronectin in mice lacking heparin. a role for heparin-dependent mast cell chymase in fibronectin degradation. J. Biol. Chem. 2001, 276, 3772–3777. [Google Scholar] [CrossRef] [PubMed]
- Tchougounova, E. Regulation of extravascular coagulation and fibrinolysis by heparin-dependent mast cell chymase. FASEB J. 2001, 15, 2763–2765. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Watts, G.F.M.; Oettgen, H.C.; Friend, D.S.; Pemberton, A.D.; Gurish, M.F.; Lee, D.M. Mouse Mast Cell Tryptase mMCP-6 Is a Critical Link between Adaptive and Innate Immunity in the Chronic Phase of Trichinella spiralis Infection1. J. Immunol. 2008, 180, 4885–4891. [Google Scholar] [CrossRef] [PubMed]
- Kjellén, L.; Pettersson, I.; Lillhager, P.; Steen, M.L.; Pettersson, U.; Lehtonen, P.; Karlsson, T.; Ruoslahti, E.; Hellman, L. Primary structure of a mouse mastocytoma proteoglycan core protein. Biochem. J. 1989, 263, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Angerth, T.; Huang, R.; Aveskogh, M.; Pettersson, I.; Kjellén, L.; Hellman, L. Cloning and structural analysis of a gene encoding a mouse mastocytoma proteoglycan core protein; analysis of its evolutionary relation to three cross hybridizing regions in the mouse genome. Gene 1990, 93, 235–240. [Google Scholar] [CrossRef]
- Rönnberg, E.; Melo, F.R.; Pejler, G. Mast Cell Proteoglycans. J. Histochem. Cytochem. 2012, 60, 950–962. [Google Scholar] [CrossRef]
- Ohtsu, H.; Tanaka, S.; Terui, T.; Hori, Y.; Makabe-Kobayashi, Y.; Pejler, G.; Tchougounova, E.; Hellman, L.; Gertsenstein, M.; Hirasawa, N.; et al. Mice lacking histidine decarboxylase exhibit abnormal mast cells. FEBS Lett. 2001, 502, 53–56. [Google Scholar] [CrossRef]
- Akula, S.; Mohammadamin, S.; Hellman, L. Fc Receptors for Immunoglobulins and Their Appearance during Vertebrate Evolution. PLoS ONE 2014, 9, e96903. [Google Scholar] [CrossRef]
- Galli, S.J.; Zsebo, K.M.; Geissler, E.N. The kit ligand, stem cell factor. Adv. Immunol. 1994, 55, 1–96. [Google Scholar]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F. Immunological Genome Project C. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef]
- Tchougounova, E.; Pejler, G.; Åbrink, M. The Chymase, Mouse Mast Cell Protease 4, Constitutes the Major Chymotrypsin-like Activity in Peritoneum and Ear Tissue. A Role for Mouse Mast Cell Protease 4 in Thrombin Regulation and Fibronectin Turnover. J. Exp. Med. 2003, 198, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Karlson, U.; Pejler, G.; Tomasini-Johansson, B.; Hellman, L. Extended substrate specificity of rat mast cell protease 5, a rodent alpha-chymase with elastase-like primary specificity. J. Biol. Chem. 2003, 278, 39625–39631. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, D.S.; Gurley, D.S.; Austen, K.F.; Serafin, W.E. Cloning of the cDNA and gene of mouse mast cell protease-6. Transcription by progenitor mast cells and mast cells of the connective tissue subclass. J. Boil. Chem. 1991, 266, 3847–3853. [Google Scholar]
- Miller, H.R.P.; Pemberton, A.D. Tissue-specific expression of mast cell granule serine proteinases and their role in inflammation in the lung and gut. Immunology 2002, 105, 375–390. [Google Scholar] [CrossRef]
- Xing, W.; Austen, K.F.; Gurish, M.F.; Jones, T.G. Protease phenotype of constitutive connective tissue and of induced mucosal mast cells in mice is regulated by the tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 14210–14215. [Google Scholar] [CrossRef]
- Poorafshar, M.; Helmby, H.; Troye-Blomberg, M.; Hellman, L. MMCP-8, the first lineage-specific differentiation marker for mouse basophils. Elevated numbers of potent IL-4-producing and MMCP-8-positive cells in spleens of malaria-infected mice. Eur. J. Immunol. 2000, 30, 2660–2668. [Google Scholar] [CrossRef]
- Hitomi, K.; Tahara-Hanaoka, S.; Someya, S.; Fujiki, A.; Tada, H.; Sugiyama, T.; Shibayama, S.; Shibuya, K.; Shibuya, A. An immunoglobulin-like receptor, Allergin-1, inhibits immunoglobulin E–mediated immediate hypersensitivity reactions. Nat. Immunol. 2010, 11, 601–607. [Google Scholar] [CrossRef]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef]
- Porebski, G.; Kwiecien, K.; Pawica, M.; Kwitniewski, M. Mas-Related G Protein-Coupled Receptor-X2 (MRGPRX2) in Drug Hypersensitivity Reactions. Front. Immunol. 2018, 9, 3027. [Google Scholar] [CrossRef]
- Meixiong, J.; Anderson, M.; Limjunyawong, N.; Sabbagh, M.F.; Hu, E.; Mack, M.R.; Oetjen, L.K.; Wang, F.; Kim, B.S.; Dong, X. Activation of Mast-Cell-Expressed Mas-Related G-Protein-Coupled Receptors Drives Non-histaminergic Itch. Immunity 2019, 50, 1163–1171. [Google Scholar] [CrossRef]
- Yoshida, K.; Ito, M.; Matsuoka, I. Divergent regulatory roles of extracellular ATP in the degranulation response of mouse bone marrow-derived mast cells. Int. Immunopharmacol. 2017, 43, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Obata, K.; Mukai, K.; Sato, S.; Takai, T.; Minegishi, Y.; Karasuyama, H. Mast cells and basophils are selectively activated in vitro and in vivo through CD200R3 in an IgE-independent manner. J. Immunol. 2007, 179, 7093–7100. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J. NTAL/LAB and LAT: A balancing act in mast-cell activation and function. Trends Immunol. 2005, 26, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Kurotaki, D.; Tamura, T. Regulation of basophil and mast cell development by transcription factors. Allergol. Int. 2016, 65, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Ohneda, K.; Ohmori, S.; Yamamoto, M. Mouse Tryptase Gene Expression is Coordinately Regulated by GATA1 and GATA2 in Bone Marrow-Derived Mast Cells. Int. J. Mol. Sci. 2019, 20, 4603. [Google Scholar] [CrossRef]
- Siraganian, R.P.; De Castro, R.O.; Barbu, E.A.; Zhang, J. Mast cell signaling: The role of protein tyrosine kinase Syk, its activation and screening methods for new pathway participants. FEBS Lett. 2010, 584, 4933–4940. [Google Scholar] [CrossRef]
- Åbrink, M.; Larsson, E.; Hellman, L. Demethylation of ERV3, an endogenous retrovirus regulating the Krüppel-related zinc finger gene H-plk, in several human cell lines arrested during early monocyte development. DNA Cell Boil. 1998, 17, 27–37. [Google Scholar] [CrossRef]
- Erjefält, J.S. Mast cells in human airways: The culprit? Eur. Respir. Rev. 2014, 23, 299–307. [Google Scholar] [CrossRef]
- Bankova, L.G.; Dwyer, D.F.; Liu, A.Y.; Austen, K.F.; Gurish, M.F. Maturation of mast cell progenitors to mucosal mast cells during allergic pulmonary inflammation in mice. Mucosal Immunol. 2015, 8, 596–606. [Google Scholar] [CrossRef]
- Zarnegar, B.; Mendez-Enriquez, E.; Westin, A.; Söderberg, C.; Dahlin, J.S.; Grönvik, K.-O.; Hallgren, J. Influenza Infection in Mice Induces Accumulation of Lung Mast Cells through the Recruitment and Maturation of Mast Cell Progenitors. Front. Immunol. 2017, 8, 9. [Google Scholar] [CrossRef]
- Andersson, C.K.; Shikhagaie, M.; Mori, M.; Al-Garawi, A.; Reed, J.L.; Humbles, A.A.; Welliver, R.; Mauad, T.; Bjermer, L.; Jordana, M.; et al. Distal respiratory tract viral infections in young children trigger a marked increase in alveolar mast cells. ERJ Open Res. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.R. Mucosal mast cells and the allergic response against nematode parasites. Veter Immunol. Immunopathol. 1996, 54, 331–336. [Google Scholar] [CrossRef]
- Cildir, G.; Toubia, J.; Yip, K.H.; Zhou, M.; Pant, H.; Hissaria, P.; Zhang, J.; Hong, W.; Robinson, N.; Grimbaldeston, M.A.; et al. Genome-wide Analyses of Chromatin State in Human Mast Cells Reveal Molecular Drivers and Mediators of Allergic and Inflammatory Diseases. Immunity 2019, 51, 949–965. [Google Scholar] [CrossRef] [PubMed]
- Tchougounova, E.; Lundequist, A.; Fajardo, I.; Winberg, J.O.; Abrink, M.; Pejler, G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J. Biol. Chem. 2005, 280, 9291–9296. [Google Scholar] [CrossRef]
- Sundblad, V.; Morosi, L.G.; Geffner, J.R.; Rabinovich, G.A. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J. Immunol. 2017, 199, 3721–3730. [Google Scholar] [CrossRef]
- Michel, T.; Thérésine, M.; Poli, A.; Domingues, O.; Ammerlaan, W.; Brons, N.H.C.; Hentges, F.; Zimmer, J. Increased Th2 Cytokine Secretion, Eosinophilic Airway Inflammation, and Airway Hyperresponsiveness in Neurturin-Deficient Mice. J. Immunol. 2011, 186, 6497–6504. [Google Scholar] [CrossRef]
- Schneider, L.A.; Schlenner, S.M.; Feyerabend, T.B.; Wunderlin, M.; Rodewald, H.-R. Molecular mechanism of mast cell–mediated innate defense against endothelin and snake venom sarafotoxin. J. Exp. Med. 2007, 204, 2629–2639. [Google Scholar] [CrossRef]
- Shay, T.; Kang, J. Immunological Genome Project and systems immunology. Trends Immunol. 2013, 34, 602–609. [Google Scholar] [CrossRef]
- Motakis, E.; Guhl, S.; Ishizu, Y.; Itoh, M.; Kawaji, H.; De Hoon, M.; Lassmann, T.; Carninci, P.; Hayashizaki, Y.; Zuberbier, T.; et al. Redefinition of the human mast cell transcriptome by deep-CAGE sequencing. Blood 2014, 123, e58–e67. [Google Scholar] [CrossRef]
- Wong, G.W.; Zhuo, L.; Kimata, K.; Lam, B.K.; Satoh, N.; Stevens, R.L. Ancient origin of mast cells. Biochem. Biophys. Res. Commun. 2014, 451, 314–318. [Google Scholar] [CrossRef]
- Dobson, J.T.; Seibert, J.; Teh, E.M.; Da’As, S.; Fraser, R.B.; Paw, B.H.; Lin, T.-J.; Berman, J.N. Carboxypeptidase A5 identifies a novel mast cell lineage in the zebrafish providing new insight into mast cell fate determination. Blood 2008, 112, 2969–2972. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| GATC-RNA Seq | Ampliseq | ||
|---|---|---|---|
| Proteases | |||
| Cma1 (Mcpt5) | 17072 | 17116 | 35388 |
| Mcpt4 (mMCP-4) | 16668 | 16752 | 31292 |
| Tpsb2 (Mcpt6) | 14244 | 13548 | 39628 |
| Cpa3 (CPA-3) | 9564 | 9844 | 13892 |
| Tpsab1 (Mcpt7) | 352 | 348 (2%) | 96 |
| Mcpt9 (mMCP-9) | 12 | 12 | 0 |
| CtsG (CTS-G) | 216 | 204 (1%) | 512 |
| Mcpt8 (mMCP-8) | 8 | 12 | 12 |
| CtsC (DPP) | 148 | 152 (0.5%) | 876 |
| Gzm B | 68 | 72 (0.4%) | 236 |
| Gzm A | 8 | 12 | 12 |
| Gzm K | 0 | 0 | 2.4 |
| Gzm M | 2 | 4 | 1.6 |
| Gzm N | 0 | 0 | 1.2 |
| Gzm C | 0 | 0 | 0.8 |
| Gzm D | 0 | 0 | 0.4 |
| Gzm E, F, G | 0 | 0 | 0 |
| Receptors | |||
| FcεRI alpha | 480 | 500 (3%) | 252 |
| c-kit | 248 | 252 (1.5%) | 720 |
| IL-3R | 112 | 100 (0.5%) | 80 |
| Heparin and Histamine synthesis | |||
| Srgn (Serglycin) | (920) | (900)(22%) | (2800) |
| Ndst2 | 464 | 564 (2.7%) | 1688 |
| Ndst1 | 68 | 64 | 264 |
| Hdc (Histidine decarb.) | 300 | 292 (1.5%) | 600 |
| Cytokines & Chemokines | |||
| IL-4 | 12 | 12 | 9.2 |
| IL-5 | 0 | 0 | 0 |
| IL-15 | 12 | 8 | 48 |
| IL-18 | 52 | 52 | 0 |
| IL-6 | 8 | 8 | 8 |
| GATC-RNA Seq | Ampliseq | |||
|---|---|---|---|---|
| Normal | +4 h LPS | Normal | +4 h LPS | |
| Proteases | ||||
| Cma1 (Mcpt5) | 7102 | 7618 | 15683 | 17413 |
| Mcpt4 (mMCP-4) | 5 | 22 | 7 | 34 |
| Tpsb2 (mMCP-6) | 746 | 896 | 3119 | 3345 |
| Cpa3 (CPA-3) | 13430 | 11990 | 22478 | 17717 |
| Tpsab1 (Mcpt7) | 72 | 241 | 54 | 190 |
| Mcpt9 (mMCP-9) | 0.9 | 2.4 | 0 | 0 |
| CtsG (CTS-G) | 7 | 9 | 18 | 30 |
| Mcpt8 (mMCP-8) | 24 | 25 | 46 | 36 |
| Mcpt1 (mMCP-1) | 15 | 64 | 15 | 56 |
| Mcpt2 (mMCP-2) | 1 | 10 | 1.2 | 15 |
| CtsC (DPP) | 51 | 44 | 288 | 243 |
| Gzm B | 161 | 700 | 386 | 1900 (4-5x) |
| Gzm A | 0 | 0 | 0 | 0 |
| Gzm K | 0 | 0 | 0.3 | 0.9 |
| Gzm M | 0.2 | 0.2 | 0.3 | 0.6 |
| Gzm N | 0 | 0 | 0.1 | 0.1 |
| Gzm C | 0.1 | 106 | 0.1 | 110 (1000x) |
| Gzm D | 0 | 0 | 0 | 0.8 |
| Gzm E | 0 | 0 | 0.1 | 0.1 |
| Receptors | ||||
| FcεRIα | 4918 | 3280 | 1631 | 1342 (10–70%) |
| c-kit | 318 | 307 | 833 | 931 |
| IL-3R | 30 | 22 | 25 | 22 |
| IL1RL1 (ST2) | 1039 | 1310 | 4582 | 5859 |
| CsfRI2B (coβIL-3 GM-CSF) | 1851 | 1855 | 3485 | 3499 |
| FcεRIγ | 468 | 432 | 1815 | 1545 |
| CD23 | 0.3 | 0.5 | 0 | 0 |
| FcγRIII | 132 | 135 | 538 | 433 |
| FcγRIIb | 33 | 32 | 40 | 39 |
| FcγRI | 1 | 1 | 5 | 1 |
| CD63 | 1894 | 2153 | 2699 | 3624 |
| Integrin alpha2b | 495 | 474 | 2472 | 2003 |
| Heparin and Histamine synthesis + Transcr. Factors | ||||
| Srgn (Serglycin) | 2637 | 3139 | 8107 | 9990 |
| Ndst2 | 188 | 210 | 820 | 874 |
| Ndst1 | 13 | 7 | 60 | 30 |
| 5-Lipoxygenase (Alox5) | 1029 | 767 | 2757 | 2016 |
| Histidine decarb. (Hdc) | 19 | 99 | 48 | 279 |
| Tryp. Hydroxylase (Tph1) | 711 | 1368 | 2370 | 4148 |
| Cyp11a1 | 1840 | 2041 | 6422 | 6252 |
| Slc18a2 (Monoamine transp.) | 2218 | 2348 | ||
| GATA1 | 95 | 81 | 296 | 267 |
| GATA2 | 594 | 900 | 5205 | 7289 |
| GATA3 | 4 | 3 | 14 | 12 |
| GATC-RNA Seq | Ampliseq | ||
|---|---|---|---|
| Major skin transcripts | |||
| Keratin 10 | 9490 | 13724 | 14386 |
| Keratin 2 | 6121 | 15412 | 18444 |
| Keratin 14 | 3938 | 7855 | 7670 |
| Keratin 5 | 2726 | 5304 | 5637 |
| Keratin 15 | 1277 | 2897 | 2733 |
| Keratin 1 | 1093 | 2373 | 2673 |
| Keratin 77 | 617 | 2199 | 2430 |
| Keratin 79 | 525 | 2321 | 2362 |
| Keratin 17 | 492 | 878 | 780 |
| Keratin 80 | 171 | 1453 | 1574 |
| Loricrin (Lor) | 4029 | 14026 | 9840 |
| Calmodulin 4 | 3133 | 2895 | 3821 |
| Mast cell transcripts | |||
| Mcpt4 (mMCP-4) | 110 | 148 | 206 |
| Cma1 (Mcpt5) | 52 | 75 | 79 |
| Tpsb2 (Mcpt6) | 38 | 92 | 93 |
| Cpa3 (CPA-3) | 34 | 48 | 42 |
| Mcpt7 (mMCP-7) | 8 | 1.4 | 1.5 |
| Mcpt2 (mMCP-2) | 0.2 | 0 | 0 |
| Mcpt1 (mMCP-1) | 0.1 | 0 | 0 |
| Mcpt8 (mMCP-8) | 0 | 0 | 0 |
| CtsG (CTS-G) | 2 | 3 | 6 |
| Srgn (Serglycin) | 18 | 38 | 48 |
| FcεRIα | 0.6 | 0.1 | 0.5 |
| FcεRIγ | 18 | 61 | 56 |
| Ndst2 | 7 | 41 | 45 |
| Ndst1 | 13 | 76 | 83 |
| Granzymes | |||
| Gzm C | 2 | 0.95 | 1.7 |
| Gzm M | 3 | 0.95 | 1.2 |
| Gzm A | 0.5 | 0.2 | 0.2 |
| Gzm B | 0.15 | 0.1 | 0.4 |
| Gzm D | 0 | 0 | 0 |
| GATC-RNA Seq | Ampliseq | ||
|---|---|---|---|
| Major Lung transcripts | |||
| Scgb1a1 (Uteroglobin) | 16 | 31234 | 35193 |
| Scgb3a2 | 1011 | 1263 | 1288 |
| Scgb3a1 | 260 | 522 | 522 |
| Scgb1c1 | 23 | 39 | 30 |
| Sftpc (Surfactant) | 21382 | 20059 | 23191 |
| Sftpa1 | 1073 | 9154 | 9384 |
| Sftpd | 434 | 73 | 67 |
| Tmsb4x (Thymosin beta 4) | 6430 | 5597 | 4519 |
| Lyz2 (P-Lysozyme) | 4843 | 10629 | 10721 |
| Lyz1 (M-Lysozyme) | 1131 | 990 | 962 |
| Cbr2 (Carbonyl reductase, NADPH2) | 2911 | 6181 | 4566 |
| Sparc (Osteonectin, BM40) | 652 | 4577 | 4276 |
| Inmt (Amine-N-methyltransferase) | 1947 | 4363 | 4730 |
| Sptbn1 (Spectrin beta chain) | 280 | 3742 | 3432 |
| Epas1 (Endothelial PAS domain cont. prot 1) | 642 | 3710 | 3439 |
| Cyp2f2 (Cytochrome P450 2F2) | 1378 | 3437 | 2651 |
| Sec14l3 (Sec 14 like lipid transport) | 431 | 1640 | 1583 |
| Mast cell transcripts | |||
| Cma1 (Mcpt5) | 3.7 | 8 | 8 |
| Cpa3 (CPA-3) | 3.6 | 1 | 1 |
| Mcpt4 (mMCP-4) | 1 | 2 | 3 |
| Tpsb2 (Mcpt6) | 0.8 | 2 | 2 |
| Mcpt7 (mMCP-7) | 0.16 | 0.1 | 0 |
| Mcpt8 (mMCP-8) | 0.7 | 3 | 3 |
| Mcpt1 (mMCP-1) | 0.15 | 0.3 | 0.4 |
| Mcpt2 (mMCP-2) | 0.15 | 0.4 | 0.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akula, S.; Paivandy, A.; Fu, Z.; Thorpe, M.; Pejler, G.; Hellman, L. Quantitative In-Depth Analysis of the Mouse Mast Cell Transcriptome Reveals Organ-Specific Mast Cell Heterogeneity. Cells 2020, 9, 211. https://doi.org/10.3390/cells9010211
Akula S, Paivandy A, Fu Z, Thorpe M, Pejler G, Hellman L. Quantitative In-Depth Analysis of the Mouse Mast Cell Transcriptome Reveals Organ-Specific Mast Cell Heterogeneity. Cells. 2020; 9(1):211. https://doi.org/10.3390/cells9010211
Chicago/Turabian StyleAkula, Srinivas, Aida Paivandy, Zhirong Fu, Michael Thorpe, Gunnar Pejler, and Lars Hellman. 2020. "Quantitative In-Depth Analysis of the Mouse Mast Cell Transcriptome Reveals Organ-Specific Mast Cell Heterogeneity" Cells 9, no. 1: 211. https://doi.org/10.3390/cells9010211
APA StyleAkula, S., Paivandy, A., Fu, Z., Thorpe, M., Pejler, G., & Hellman, L. (2020). Quantitative In-Depth Analysis of the Mouse Mast Cell Transcriptome Reveals Organ-Specific Mast Cell Heterogeneity. Cells, 9(1), 211. https://doi.org/10.3390/cells9010211