Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
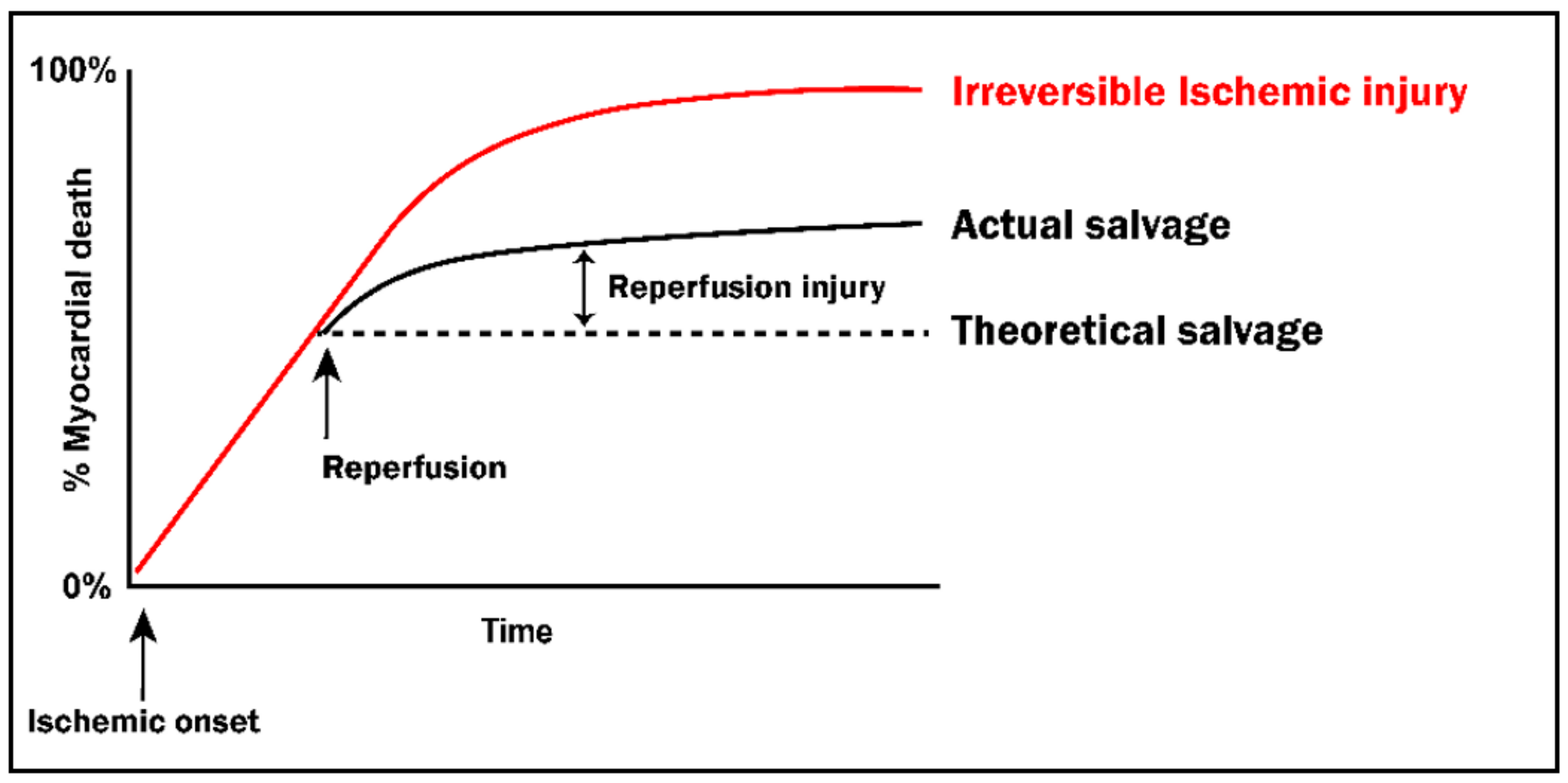
2. Lethal Ischemia-Reperfusion Injury
2.1. The Trigger: Myocardial Ischemia
2.2. Reintroduction of Oxygen: A ‘Double-Edged Sword’
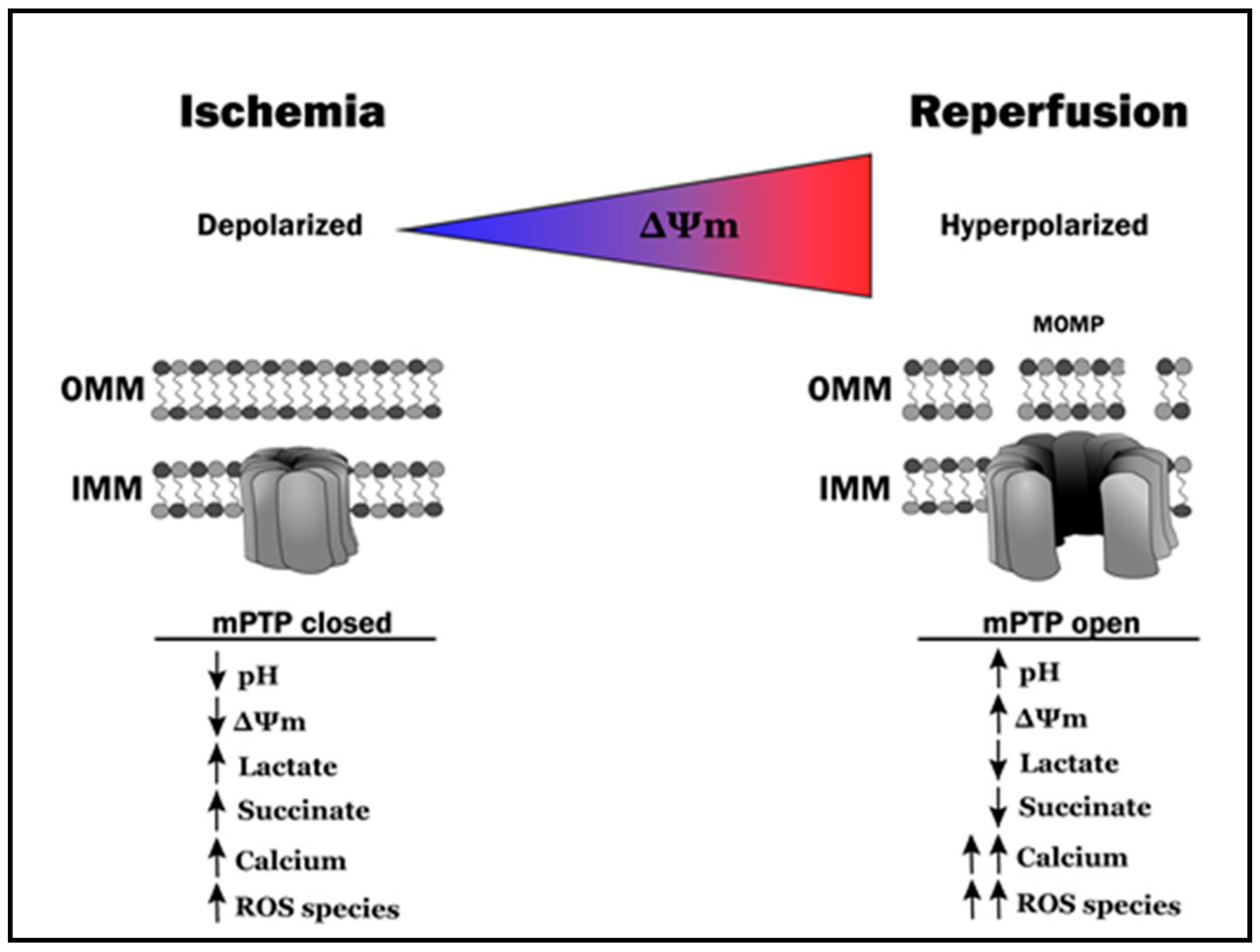
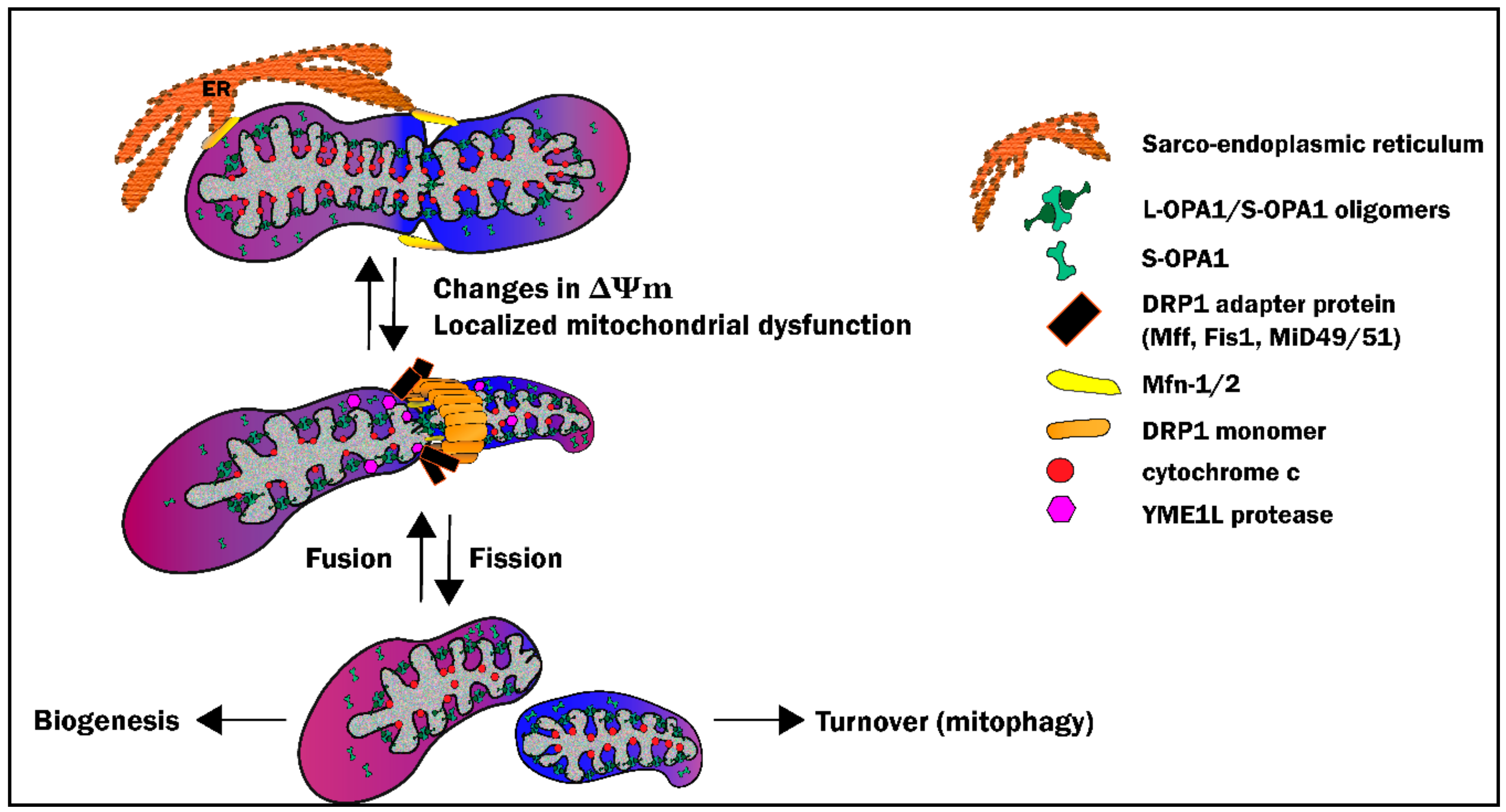
3. Mitochondrial Morphosis
3.1. Definitions and Key Players
3.2. Mitochondrial Fission: Sacrificing One for the Team
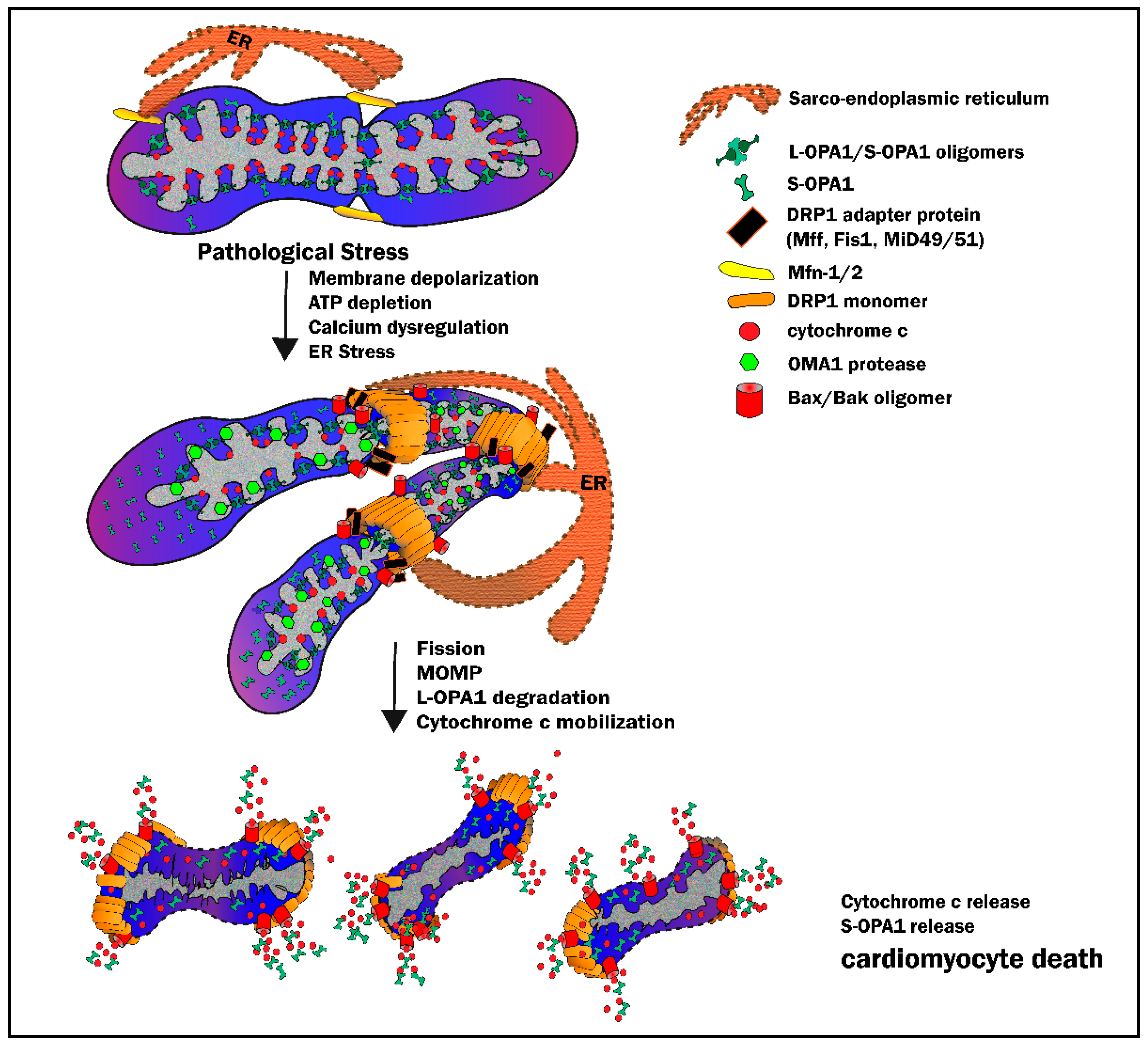
3.3. Mitochondrial Outer Membrane Permeabilization: When Fission Leads to Death
3.4. Mitochondrial Fusion: Safety in Numbers
3.5. Disruption of Cristae Architecture: Opening the Cytochrome C Flood Gates
3.6. A Complex Web: Ischemia-Reperfusion, Metabolic Dysfunction and Mitochondrial Morphosis
4. Mitochondrial Dynamics and Cardiomyocyte Fate
4.1. IR Injury and the Outer Mitochondrial Membrane—DRP1-Mediated Fission
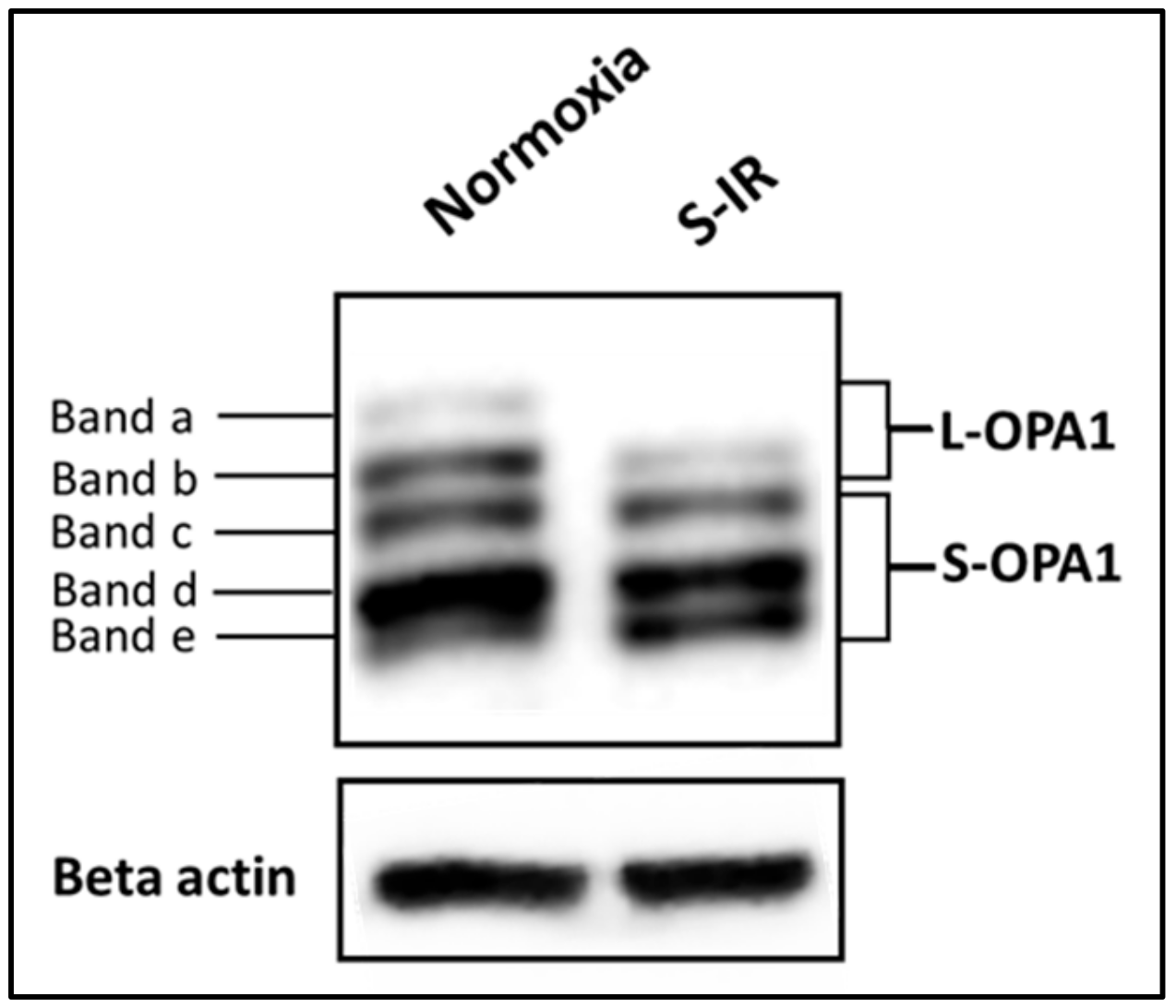
4.2. IR Injury and the Inner Mitochondrial Membrane—OPA1 and Cristae Integrity
4.3. Importance of Inner Versus Outer Mitochondrial Membrane Integrity in Lethal Ir Injury?
5. Pharmacologic Targeting of Mitochondrial Morphosis to Attenuate Lethal IR Injury
6. Mitophagy
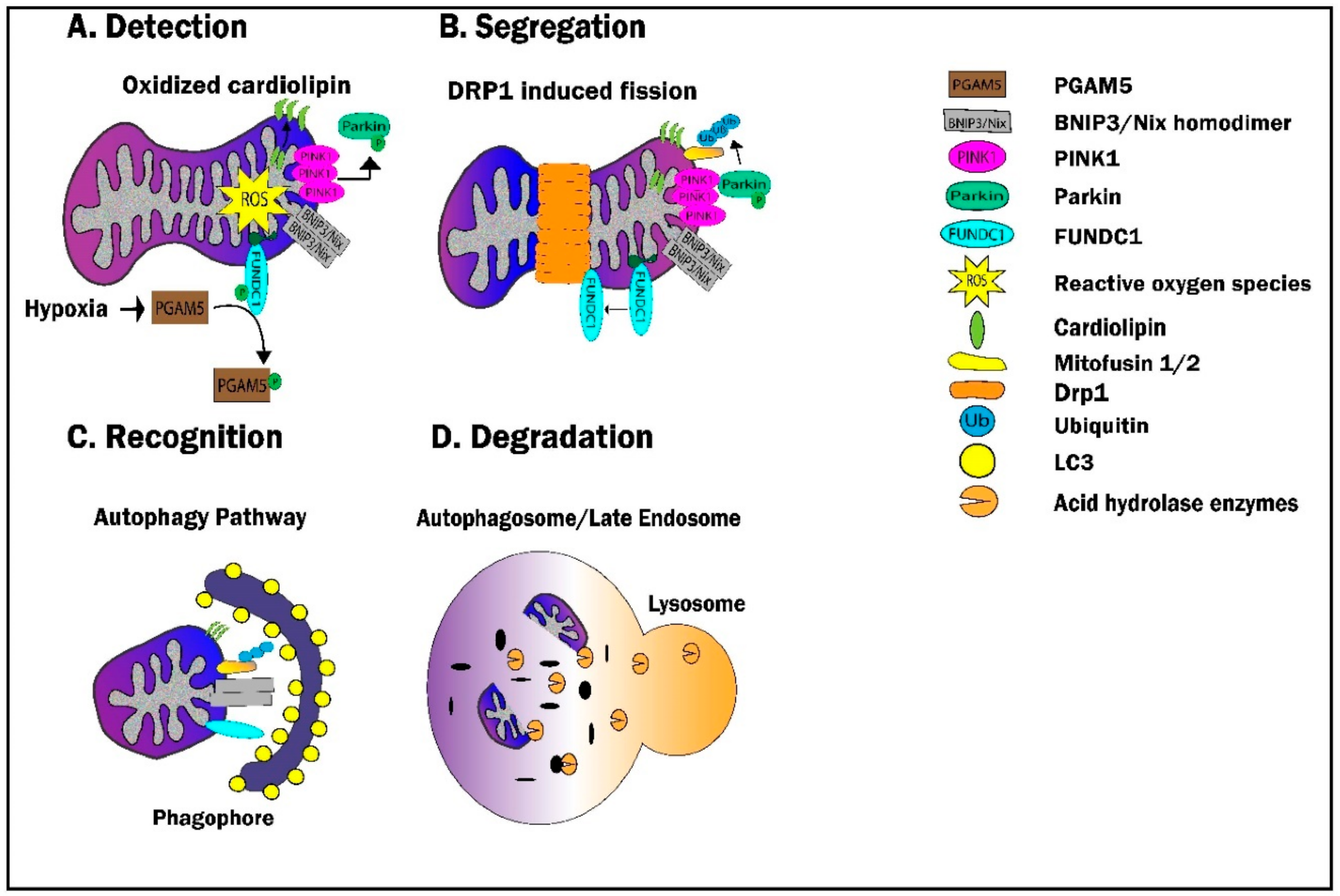
6.1. Definitions and Key Players
6.1.1. Detection
6.1.2. Segregation—The Link with Fission
6.1.3. Recognition
6.1.4. Degradation
6.2. PINK1/Parkin and the Ubiquitin-Proteasome System
7. Mitophagy and Cardiomyocyte Fate
7.1. Ischemia
7.2. Ischemia-Reperfusion: ‘Good Versus Evil’
8. Pharmacologic Targeting of Mitochondrial Morphosis to Attenuate Lethal IR Injury
9. Broad Relevance of the Paradigm: Mitochondrial Quality Control and IR Injury in Brain
10. Future Directions
11. Conclusions
Funding
Conflicts of Interest
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L. Timely PCI for STEMI—Still the treatment of choice. N. Engl. J. Med. 2013, 368, 14460–14477. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.R.; Bhatt, D.L. The state of periprocedural antiplatelet therapy after recent trials. JACC Cardiovasc. Interv. 2010, 3, 571–583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gersh, B.J.; Stone, G.W.; White, H.D.; Holmes, D.R.J. Pharmacological facilitation of primary percutaneous coronary intervention for acute myocardial infarction: Is the slope of the curve the shape of the future? JAMA 2005, 293, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Goodman, S.; Chang, W.C.; Van De Werf, F.; Granger, C.B.; Armstrong, P.W. Time to treatment influences the impact of ST-segment resolution on one-year prognosis: Insights from the assessment of the safety and efficacy of a new thrombolytic (ASSENT-2) trial. Circulation 2001, 104, 2653–2659. [Google Scholar] [CrossRef][Green Version]
- Ibanez, B.; Heusch, G.; Ovize, M.; Van de Werf, F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef]
- Eapen, Z.J.; Tang, W.H.; Felker, G.M.; Hernandez, A.F.; Mahaffey, K.W.; Lincoff, A.M.; Roe, M.T. Defining heart failure end points in ST-segment elevation myocardial infarction trials: Integrating past experiences to chart a path forward. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 594–600. [Google Scholar] [CrossRef]
- Jennings, R.B.; Reimer, K.A. The cell biology of acute myocardial ischemia. Annu. Rev. Med. 1991, 42, 225–246. [Google Scholar] [CrossRef]
- Jennings, R.B. Historical Perspective on the Pathology of Myocardial Ischemia/Reperfusion Injury. Circ. Res. 2013, 113, 428–438. [Google Scholar] [CrossRef]
- Bell, R.M.; Yellon, D.M. There is More to Life than Revascularization: Therapeutic Targeting of Myocardial Ischemia/Reperfusion Injury. Cardiovasc. Ther. 2011, 29, E67–E79. [Google Scholar] [CrossRef] [PubMed]
- Tullio, F.; Angotti, C.; Perrelli, M.G.; Penna, C.; Pagliaro, P. Redox balance and cardioprotection. Basic Res. Cardiol. 2013, 108, 392. [Google Scholar] [CrossRef] [PubMed]
- Windecker, S.; Bax, J.J.; Myat, A.; Stone, G.W.; Marber, M.S. Future treatment strategies in ST-segment elevation myocardial infarction. Lancet 2013, 382, 644–657. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Piper, H.M. Postconditioning: Reperfusion of “reperfusion injury” after hibernation. Cardiovasc. Res. 2006, 69, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell. Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed]
- Boengler, K.; Lochnit, G.; Schulz, R. Mitochondria “THE” target of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1215–H1231. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell. 2004, 15, 5001–5011. [Google Scholar] [CrossRef]
- Bialik, S.; Cryns, V.L.; Drincic, A.; Miyata, S.; Wollowick, A.L.; Srinivasan, A.; Kitsis, R.N. The mitochondrial apoptotic pathway is activated by serum and glucose deprivation in cardiac myocytes. Circ. Res. 1999, 85, 403–414. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Ong, S.B.; Yellon, D.M. The mitochondrial permeability transition pore as a target for preconditioning and postconditioning. Basic Res. Cardiol. 2009, 104, 189–202. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Walters, J.W.; Amos, D.; Ray, K.; Santanam, N. Mitochondrial redox status as a target for cardiovascular disease. Curr. Opin. Pharmacol. 2016, 27, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Argaud, L.; Gateau-Roesch, O.; Muntean, D.; Chalabreysse, L.; Loufouat, J.; Robert, D.; Ovize, M. Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Maneechote, C.; Palee, S.; Chattipakorn, S.C.; Chattipakorn, N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J. Cell. Mol. Med. 2017, 21, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Disatnik, M.H.; Ferreira, J.C.; Campos, J.C.; Gomes, K.S.; Dourado, P.M.; Qi, X.; Mochly-Rosen, D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013, 2, e000461. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-B.; Kalkhoran, S.B.; Hernandez-Resendiz, S.; Samangouei, P.; Ong, S.-G.; Hausenloy, D.J. Mitochondrial-Shaping Proteins in Cardiac Health and Disease-the Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef]
- Ong, S.-B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef]
- Dong, Y.; Undyala, V.V.R.; Przyklenk, K. Inhibition of mitochondrial fission as a molecular target for cardioprotection: Critical importance of the timing of treatment. Basic Res. Cardiol. 2016, 111, 59. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Ganote, C.E. Contraction band necrosis and irreversible myocardial injury. J. Mol. Cell. Cardiol. 1983, 15, 67–73. [Google Scholar] [CrossRef]
- McCully, J.D.; Wakiyama, H.; Hsieh, Y.J.; Jones, M.; Levitsky, S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1923–H1935. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Reimer, K.A. Lethal myocardial ischemic injury. Am. J. Pathol. 1981, 102, 241–255. [Google Scholar] [PubMed]
- Harden, W.R.; Barlow, C.H.; Simson, M.B.; Harken, A.H. Temporal relation between onset of cell anoxia and ischemic contractile failure. Myocardial ischemia and left ventricular failure in the isolated, perfused rabbit heart. Am. J. Cardiol. 1979, 44, 741–746. [Google Scholar] [CrossRef]
- Fuller, W.; Parmar, V.; Eaton, P.; Bell, J.R.; Shattock, M.J. Cardiac ischemia causes inhibition of the Na/K ATPase by a labile cytosolic compound whose production is linked to oxidant stress. Cardiovasc. Res. 2003, 57, 1044–1051. [Google Scholar] [CrossRef]
- Garcia-Dorado, D.; Ruiz-Meana, M.; Inserte, J.; Rodriguez-Sinovas, A.; Piper, H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012, 94, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Coccaro, E.; Fliegel, L. Sustained intracellular acidosis activates the myocardial Na(+)/H(+) exchanger independent of amino acid Ser(703) and p90(rsk). Biochim. Biophys. Acta 2010, 1798, 1565–1576. [Google Scholar] [CrossRef]
- Acosta, D.; Li, C.P. Actions of extracellular acidosis on primary cultures of rat myocardial cells deprived of oxygen and glucose. J. Mol. Cell. Cardiol. 1980, 12, 1459–1463. [Google Scholar] [CrossRef]
- Gambassi, G.; Capogrossi, M.C. Acidosis is associated with an intracellular accumulation of Ca2+. Its role in the modulation of myocardial contractility. Cardiologia 1992, 37, 587–589. [Google Scholar]
- Castaldo, P.; Macri, M.L.; Lariccia, V.; Matteucci, A.; Maiolino, M.; Gratteri, S.; Amoroso, S.; Magi, S. Na(+)/Ca(2+) exchanger 1 inhibition abolishes ischemic tolerance induced by ischemic preconditioning in different cardiac models. Eur. J. Pharmacol. 2017, 794, 246–256. [Google Scholar] [CrossRef]
- Reimer, K.A.; Vander Heide, R.S.; Richard, V.J. Reperfusion in acute myocardial infarction: Effect of timing and modulating factors in experimental models. Am. J. Cardiol. 1993, 72, 13G–21G. [Google Scholar] [CrossRef]
- Braunwald, E.; Kloner, R.A. Myocardial reperfusion: A double-edged sword? J. Clin. Investig. 1985, 76, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Buja, L.M.; Vander Heide, R.S. Pathobiology of Ischemic Heart Disease: Past, Present and Future. Cardiovasc. Pathol. 2016, 25, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Javadov, S.; Margreiter, R.; Grimm, M.; Hagenbuchner, J.; Ausserlechner, M.J. The Role of Mitochondria in the Mechanisms of Cardiac Ischemia-Reperfusion Injury. Antioxidants 2019, 8, 454. [Google Scholar] [CrossRef] [PubMed]
- Caccioppo, A.; Franchin, L.; Grosso, A.; Angelini, F.; D’Ascenzo, F.; Brizzi, M.F. Ischemia Reperfusion Injury: Mechanisms of Damage/Protection and Novel Strategies for Cardiac Recovery/Regeneration. Int. J. Mol. Sci. 2019, 20, 5024. [Google Scholar] [CrossRef] [PubMed]
- Escobales, N.; Nunez, R.E.; Jang, S.; Parodi-Rullan, R.; Ayala-Pena, S.; Sacher, J.R.; Skoda, E.M.; Wipf, P.; Frontera, W.; Javadov, S. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell. Cardiol. 2014, 77, 136–146. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Kloner, R.A.; Przyklenk, K.; Whittaker, P. Deleterious effects of oxygen radicals in ischemia/reperfusion. Resolved and unresolved issues. Circulation 1989, 80, 1115–1127. [Google Scholar] [CrossRef]
- Borutaite, V.; Jekabsone, A.; Morkuniene, R.; Brown, G.C. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J. Mol. Cell. Cardiol. 2003, 35, 357–366. [Google Scholar] [CrossRef]
- Lefer, D.J.; Bolli, R. Development of an NIH consortium for preclinicAl AssESsment of CARdioprotective therapies (CAESAR): A paradigm shift in studies of infarct size limitation. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 332–339. [Google Scholar] [CrossRef]
- Flaherty, J.T.; Pitt, B.; Gruber, J.W.; Heuser, R.R.; Rothbaum, D.A.; Burwell, L.R.; George, B.S.; Kereiakes, D.J.; Deitchman, D.; Gustafson, N.; et al. Recombinant human superoxide dismutase (h-SOD) fails to improve recovery of ventricular function in patients undergoing coronary angioplasty for acute myocardial infarction. Circulation 1994, 89, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guerin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in Patients with Acute Myocardial Infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Ottani, F.; Latini, R.; Staszewsky, L.; La Vecchia, L.; Locuratolo, N.; Sicuro, M.; Masson, S.; Barlera, S.; Milani, V.; Lombardi, M.; et al. Cyclosporine A in Reperfused Myocardial Infarction: The Multicenter, Controlled, Open-Label CYCLE Trial. J. Am. Coll. Cardiol. 2016, 67, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Chen-Scarabelli, C.; Scarabelli, T.M. Cyclosporine A Prior to Primary PCI in STEMI Patients: The Coup de Grace to Post-Conditioning? J. Am. Coll. Cardiol. 2016, 67, 375–378. [Google Scholar] [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef]
- Ong, S.B.; Hausenloy, D.J. Mitochondrial morphology and cardiovascular disease. Cardiovasc. Res. 2010, 88, 16–29. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell. Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef]
- Hayashi, J.; Ohta, S.; Kikuchi, A.; Takemitsu, M.; Goto, Y.; Nonaka, I. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 1991, 88, 10614–10618. [Google Scholar] [CrossRef]
- Nakada, K.; Inoue, K.; Chen, C.S.; Nonaka, I.; Goto, Y.; Ogura, A.; Hayashi, J.I. Correlation of functional and ultrastructural abnormalities of mitochondria in mouse heart carrying a pathogenic mutant mtDNA with a 4696-bp deletion. Biochem. Biophys. Res. Commun. 2001, 288, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Hom, J.; Sheu, S.S. Morphological dynamics of mitochondria-a special emphasis on cardiac muscle cells. J. Mol. Cell. Cardiol. 2009, 46, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Calo, L.; Dong, Y.; Kumar, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics: An emerging paradigm in ischemia-reperfusion injury. Curr. Pharm. Des. 2013, 19, 6848–6857. [Google Scholar] [CrossRef] [PubMed]
- Egner, A.; Jakobs, S.; Hell, S.W. Fast 100-nm resolution three-dimensional microscope reveals structural plasticity of mitochondria in live yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 3370–3375. [Google Scholar] [CrossRef]
- Jakobs, S.; Martini, N.; Schauss, A.C.; Egner, A.; Westermann, B.; Hell, S.W. Spatial and temporal dynamics of budding yeast mitochondria lacking the division component Fis1p. J. Cell. Sci. 2003, 116, 2005–2014. [Google Scholar] [CrossRef]
- Germain, M.; Mathai, J.P.; McBride, H.M.; Shore, G.C. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005, 24, 1546–1556. [Google Scholar] [CrossRef]
- Roe, N.D.; Ren, J. Oxidative activation of Ca(2+)/calmodulin-activated kinase II mediates ER stress-induced cardiac dysfunction and apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H828–H839. [Google Scholar] [CrossRef]
- Szegezdi, E.; Duffy, A.; O’Mahoney, M.E.; Logue, S.E.; Mylotte, L.A.; O’Brien, T.; Samali, A. ER stress contributes to ischemia-induced cardiomyocyte apoptosis. Biochem. Biophys Res. Commun. 2006, 349, 1406–1411. [Google Scholar] [CrossRef]
- Kubli, D.A.; Quinsay, M.N.; Huang, C.; Lee, Y.; Gustafsson, A.B. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2025–H2031. [Google Scholar] [CrossRef]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef]
- Chen, L.; Liu, T.; Tran, A.; Lu, X.; Tomilov, A.A.; Davies, V.; Cortopassi, G.; Chiamvimonvat, N.; Bers, D.M.; Votruba, M.; et al. OPA1 mutation and late-onset cardiomyopathy: Mitochondrial dysfunction and mtDNA instability. J. Am. Heart Assoc. 2012, 1, e003012. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Trincado, C.; Garcia-Carvajal, I.; Pennanen, C.; Parra, V.; Hill, J.A.; Rothermel, B.A.; Lavandero, S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016, 594, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.A.; Fangman, W.L. Mitochondrial DNA maintenance in yeast requires a protein containing a region related to the GTP-binding domain of dynamin. Genes. Dev. 1992, 6, 380–389. [Google Scholar] [CrossRef]
- Yoon, Y.; Krueger, E.W.; Oswald, B.J.; McNiven, M.A. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 2003, 23, 5409–5420. [Google Scholar] [CrossRef]
- Hermann, G.J.; Shaw, J.M. Mitochondrial dynamics in yeast. Annu. Rev. Cell. Dev. Biol. 1998, 14, 265–303. [Google Scholar] [CrossRef]
- Lasserre, J.P.; Dautant, A.; Aiyar, R.S.; Kucharczyk, R.; Glatigny, A.; Tribouillard-Tanvier, D.; Rytka, J.; Blondel, M.; Skoczen, N.; Reynier, P.; et al. Yeast as a system for modeling mitochondrial disease mechanisms and discovering therapies. Dis. Models Mech. 2015, 8, 509–526. [Google Scholar] [CrossRef]
- Guan, K.; Farh, L.; Marshall, T.K.; Deschenes, R.J. Normal mitochondrial structure and genome maintenance in yeast requires the dynamin-like product of the MGM1 gene. Curr. Genet. 1993, 24, 141–148. [Google Scholar] [CrossRef]
- Bleazard, W.; McCaffery, J.M.; King, E.J.; Bale, S.; Mozdy, A.; Tieu, Q.; Nunnari, J.; Shaw, J.M. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell. Biol. 1999, 1, 298–304. [Google Scholar] [CrossRef]
- Beraud, N.; Pelloux, S.; Usson, Y.; Kuznetsov, A.V.; Ronot, X.; Tourneur, Y.; Saks, V. Mitochondrial dynamics in heart cells: Very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J. Bioenerg. Biomembr. 2009, 41, 195–214. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. The regulation of mitochondrial morphology: Intricate mechanisms and dynamic machinery. Cell Signal. 2011, 23, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.C.; McBride, H.M.; Slack, R.S. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis 2007, 12, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell. Biol. 2006, 18, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Neuronal hypoxia disrupts mitochondrial fusion. Neuroscience 2015, 301, 71–78. [Google Scholar] [CrossRef]
- Owens, K.; Park, J.H.; Gourley, S.; Jones, H.; Kristian, T. Mitochondrial dynamics: Cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015, 47, 13–31. [Google Scholar] [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef]
- Chang, D.T.; Reynolds, I.J. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog. Neurobiol. 2006, 80, 241–268. [Google Scholar] [CrossRef]
- Otsuga, D.; Keegan, B.R.; Brisch, E.; Thatcher, J.W.; Hermann, G.J.; Bleazard, W.; Shaw, J.M. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J. Cell Biol. 1998, 143, 333–349. [Google Scholar] [CrossRef]
- Andres, A.M.; Stotland, A.; Queliconi, B.B.; Gottlieb, R.A. A time to reap, a time to sow: Mitophagy and biogenesis in cardiac pathophysiology. J. Mol. Cell. Cardiol. 2015, 78, 62–72. [Google Scholar] [CrossRef]
- Przyklenk, K.; Dong, Y.; Undyala, V.V.; Whittaker, P. Autophagy as a therapeutic target for ischaemia/reperfusion injury? Concepts, controversies, and challenges. Cardiovasc. Res. 2012, 94, 197–205. [Google Scholar] [CrossRef]
- Dong, Y.; Undyala, V.V.; Gottlieb, R.A.; Mentzer, R.M.J.; Przyklenk, K. Autophagy: Definition, molecular machinery, and potential role in myocardial ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Varadi, A.; Johnson-Cadwell, L.I.; Cirulli, V.; Yoon, Y.; Allan, V.J.; Rutter, G.A. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J. Cell. Sci. 2004, 117, 4389–4400. [Google Scholar] [CrossRef] [PubMed]
- Anesti, V.; Scorrano, L. The relationship between mitochondrial shape and function and the cytoskeleton. BBA Bioenerg. 2006, 1757, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Shurland, D.L.; Ryazantsev, S.N.; van der Bliek, A.M. A human dynamin-related protein controls the distribution of mitochondria. J. Cell Biol. 1998, 143, 351–358. [Google Scholar] [CrossRef]
- Shirihai, O.S.; Song, M.; Dorn, G.W. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015, 116, 1835–1849. [Google Scholar] [CrossRef]
- Dorn, G.W.; Kitsis, R.N. The mitochondrial dynamism-mitophagy-cell death interactome: Multiple roles performed by members of a mitochondrial molecular ensemble. Circ. Res. 2015, 116, 167–182. [Google Scholar] [CrossRef]
- Yoon, Y.; Pitts, K.R.; Dahan, S.; McNiven, M.A. A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J. Cell Biol. 1998, 140, 779–793. [Google Scholar] [CrossRef]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 2001, 12, 2245–2256. [Google Scholar] [CrossRef]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef]
- Zaja, I.; Bai, X.; Liu, Y.; Kikuchi, C.; Dosenovic, S.; Yan, Y.; Canfield, S.G.; Bosnjak, Z.J. Cdk1, PKCdelta and calcineurin-mediated Drp1 pathway contributes to mitochondrial fission-induced cardiomyocyte death. Biochem. Biophys Res. Commun. 2014, 453, 710–721. [Google Scholar] [CrossRef]
- Wang, J.X.; Jiao, J.Q.; Li, Q.; Long, B.; Wang, K.; Liu, J.P.; Li, Y.R.; Li, P.F. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat. Med. 2011, 17, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Van der Bliek, A.M. Functional diversity in the dynamin family. Trends Cell. Biol. 1999, 9, 96–102. [Google Scholar] [CrossRef]
- Kalia, R.; Wang, R.Y.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Lackner, L.; Nunnari, J. The machines that divide and fuse mitochondria. Annu. Rev. Biochem. 2007, 76, 751–780. [Google Scholar] [CrossRef]
- Pitts, K.R.; Yoon, Y.; Krueger, E.W.; McNiven, M.A. The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell. 1999, 10, 4403–4417. [Google Scholar] [CrossRef]
- Gandre-Babbe, S.; van der Bliek, A.M. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol. Biol. Cell. 2008, 19, 2402–2412. [Google Scholar] [CrossRef]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell. 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Palmer, C.S.; Elgass, K.D.; Parton, R.G.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J. Biol. Chem. 2013, 288, 27584–27593. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nister, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Otera, H.; Miyata, N.; Kuge, O.; Mihara, K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol. 2016, 212, 531–544. [Google Scholar] [CrossRef]
- Chen, K.H.; Dasgupta, A.; Lin, J.; Potus, F.; Bonnet, S.; Iremonger, J.; Fu, J.; Mewburn, J.; Wu, D.; Dunham-Snary, K.; et al. Epigenetic Dysregulation of the Dynamin-Related Protein 1 Binding Partners MiD49 and MiD51 Increases Mitotic Mitochondrial Fission and Promotes Pulmonary Arterial Hypertension: Mechanistic and Therapeutic Implications. Circulation 2018, 138, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Qvit, N.; Haileselassie, B.; Shamloo, M.; Bernardi, P.; Mochly-Rosen, D. Interaction of mitochondrial fission factor with dynamin related protein 1 governs physiological mitochondrial function in vivo. Sci. Rep. 2018, 8, 14034. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell. Sci. 2016, 129, 2170–2181. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 2001, 1, 515–525. [Google Scholar] [CrossRef]
- Grosse, L.; Wurm, C.A.; Bruser, C.; Neumann, D.; Jans, D.C.; Jakobs, S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016, 35, 402–413. [Google Scholar] [CrossRef]
- Sheridan, C.; Delivani, P.; Cullen, S.P.; Martin, S.J. Bax- or Bak-induced mitochondrial fission can be uncoupled from cytochrome C release. Mol. Cell. 2008, 31, 570–585. [Google Scholar] [CrossRef]
- Landes, T.; Martinou, J.C. Mitochondrial outer membrane permeabilization during apoptosis: The role of mitochondrial fission. Biochim. Biophys Acta 2011, 1813, 540–545. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell. Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Er, E.; Oliver, L.; Cartron, P.F.; Juin, P.; Manon, S.; Vallette, F.M. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim. Biophys Acta 2006, 1757, 1301–1311. [Google Scholar] [CrossRef]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005, 15, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basanez, G.; Meda, P.; et al. Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 2010, 142, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Zepeda, R.; Kuzmicic, J.; Parra, V.; Troncoso, R.; Pennanen, C.; Riquelme, J.A.; Pedrozo, Z.; Chiong, M.; Sanchez, G.; Lavandero, S. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 2014, 63, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, H.; Zheng, H.; Zhai, M.; Lu, F.; Dong, S.; Fang, T.; Zhang, W. CaSR activates PKCdelta to induce cardiomyocyte apoptosis via ER stressassociated apoptotic pathways during ischemia/reperfusion. Int. J. Mol. Med. 2019, 44, 1117–1126. [Google Scholar] [PubMed]
- Deniaud, A.; Sharaf el dein, O.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to Metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef]
- Bohovych, I.; Fernandez, M.R.; Rahn, J.J.; Stackley, K.D.; Bestman, J.E.; Anandhan, A.; Franco, R.; Claypool, S.M.; Lewis, R.E.; Chan, S.S.; et al. Metalloprotease OMA1 Fine-tunes Mitochondrial Bioenergetic Function and Respiratory Supercomplex Stability. Sci. Rep. 2015, 5, 13989. [Google Scholar] [CrossRef] [PubMed]
- Ngoh, G.A.; Papanicolaou, K.N.; Walsh, K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. J. Biol. Chem. 2012, 287, 20321–20332. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Zheng, M.; Cao, C.; Chen, C.; Tang, J.; Zhang, W.; Cheng, H.; Chen, K.H.; Xiao, R.P. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 23354–23361. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell. 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Ngoh, G.A.; Dabkowski, E.R.; O’Connell, K.A.; Ribeiro, R.F., Jr.; Stanley, W.C.; Walsh, K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H167–H179. [Google Scholar] [CrossRef]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Kalkhoran, S.B.; Dyson, A.; Vicencio, J.M.; Dorn, G.W.; Yellon, D.M.; Hausenloy, D.J. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016, 7, e2238. [Google Scholar] [CrossRef]
- Lebiedzinska, M.; Szabadkai, G.; Jones, A.W.; Duszynski, J.; Wieckowski, M.R. Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles. Int. J. Biochem. Cell. Biol. 2009, 41, 1805–1816. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Papanicolaou, K.N.; Phillippo, M.M.; Walsh, K. Mitofusins and the mitochondrial permeability transition: The potential downside of mitochondrial fusion. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H243–H255. [Google Scholar] [CrossRef] [PubMed]
- DeVay, R.M.; Dominguez-Ramirez, L.; Lackner, L.L.; Hoppins, S.; Stahlberg, H.; Nunnari, J. Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion. J. Cell Biol. 2009, 186, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, S.; DeVay, R.; Block, J.; Cassidy-Stone, A.; Wayson, S.; McCaffery, J.M.; Nunnari, J. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 2006, 127, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- MacVicar, T.; Langer, T. OPA1 processing in cell death and disease-the long and short of it. J. Cell. Sci. 2016, 129, 2297–2306. [Google Scholar] [CrossRef]
- Griparic, L.; Kanazawa, T.; van der Bliek, A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007, 178, 757–764. [Google Scholar] [CrossRef]
- Baricault, L.; Segui, B.; Guegand, L.; Olichon, A.; Valette, A.; Larminat, F.; Lenaers, G. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp. Cell Res. 2007, 313, 3800–3808. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Lechuga-Vieco, A.V.; Del Mar Munoz, M.; Nieto-Arellano, R.; Torroja, C.; Sanchez-Cabo, F.; Jimenez, C.; Gonzalez-Guerra, A.; Carrascoso, I.; Beninca, C.; et al. Ablation of the stress protease OMA1 protects against heart failure in mice. Sci. Transl. Med. 2018, 10, eaan4935. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gong, Q.; Stice, J.P.; Knowlton, A.A. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc. Res. 2009, 84, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Lebeau, J.; Puchades, C.; Wiseman, R.L. Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep. 2016, 14, 2041–2049. [Google Scholar] [CrossRef] [PubMed]
- Kaser, M.; Kambacheld, M.; Kisters-Woike, B.; Langer, T. Oma1, a novel membrane-bound metallopeptidase in mitochondria with activities overlapping with the m-AAA protease. J. Biol. Chem. 2003, 278, 46414–46423. [Google Scholar] [CrossRef]
- Ruan, Y.; Li, H.; Zhang, K.; Jian, F.; Tang, J.; Song, Z. Loss of Yme1L perturbates mitochondrial dynamics. Cell Death Dis. 2013, 4, e896. [Google Scholar] [CrossRef]
- Mopert, K.; Hajek, P.; Frank, S.; Chen, C.; Kaufmann, J.; Santel, A. Loss of Drp1 function alters OPA1 processing and changes mitochondrial membrane organization. Exp. Cell Res. 2009, 315, 2165–2180. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; van der Bliek, A.M. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef]
- Jiang, X.; Jiang, H.; Shen, Z.; Wang, X. Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc. Natl. Acad. Sci. USA 2014, 111, 14782–14787. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Lartigue, L.; Perkins, G.; Scott, R.T.; Dixit, A.; Kushnareva, Y.; Kuwana, T.; Ellisman, M.H.; Newmeyer, D.D. Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol. Cell 2008, 31, 557–569. [Google Scholar] [CrossRef]
- Arnoult, D.; Grodet, A.; Lee, Y.J.; Estaquier, J.; Blackstone, C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J. Biol. Chem. 2005, 280, 35742–35750. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Raghunayakula, S.; Kumar, R. Release of mitochondrial Opa1 following oxidative stress in HT22 cells. Mol. Cell. Neurosci. 2015, 64, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Ryu, S.W.; Choi, K.; Yoon, J.; Kim, S.; Choi, C. Endoplasmic reticulum-specific BH3-only protein BNIP1 induces mitochondrial fragmentation in a Bcl-2- and Drp1-dependent manner. J. Cell. Physiol. 2012, 227, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Shintani-Ishida, K.; Yoshida, K. Ischemia induces phospholamban dephosphorylation via activation of calcineurin, PKC-alpha, and protein phosphatase 1, thereby inducing calcium overload in reperfusion. Biochim. Biophys. Acta 2011, 1812, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Huttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Huttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The role of intramitochondrial Ca2+ in the regulation of oxidative phosphorylation in mammalian tissues. Biochem. Soc. Trans. 1993, 21, 793–799. [Google Scholar] [CrossRef]
- Valls-Lacalle, L.; Barba, I.; Miro-Casas, E.; Alburquerque-Bejar, J.J.; Ruiz-Meana, M.; Fuertes-Agudo, M.; Rodriguez-Sinovas, A.; Garcia-Dorado, D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc. Res. 2016, 109, 374–384. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, H.; Song, Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014, 15, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Alavian, K.N.; Beutner, G.; Lazrove, E.; Sacchetti, S.; Park, H.A.; Licznerski, P.; Li, H.; Nabili, P.; Hockensmith, K.; Graham, M.; et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. USA 2014, 111, 10580–10585. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Andrienko, T.N.; Pasdois, P.; Pereira, G.C.; Ovens, M.J.; Halestrap, A.P. The role of succinate and ROS in reperfusion injury—A critical appraisal. J. Mol. Cell. Cardiol. 2017, 110, 1–14. [Google Scholar] [CrossRef]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B.; et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef]
- Jourdain, A.; Martinou, J.C. Mitochondrial outer-membrane permeabilization and remodelling in apoptosis. Int. J. Biochem. Cell Biol. 2009, 41, 1884–1889. [Google Scholar] [CrossRef]
- Braschi, E.; Zunino, R.; McBride, H.M. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009, 10, 748–754. [Google Scholar] [CrossRef]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef]
- Wasiak, S.; Zunino, R.; McBride, H.M. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 2007, 177, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Logue, S.E.; Sayen, M.R.; Jinno, M.; Kirshenbaum, L.A.; Gottlieb, R.A.; Gustafsson, A.B. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007, 14, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Scimia, M.C.; Wilkinson, D.; Trelles, R.D.; Wood, M.R.; Bowtell, D.; Dillin, A.; Mercola, M.; Ronai, Z.A. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 2011, 44, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef]
- Tian, L.; Neuber-Hess, M.; Mewburn, J.; Dasgupta, A.; Dunham-Snary, K.; Wu, D.; Chen, K.H.; Hong, Z.; Sharp, W.W.; Kutty, S.; et al. Ischemia-induced Drp1 and Fis1-mediated mitochondrial fission and right ventricular dysfunction in pulmonary hypertension. J. Mol. Med. 2017, 95, 381–393. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell. 2017, 40, 583–594. [Google Scholar] [CrossRef]
- Ong, S.B.; Kwek, X.Y.; Katwadi, K.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Ismail, N.I.; Lin, Y.H.; Yap, E.P.; Lim, S.Y.; Ja, K.; et al. Targeting Mitochondrial Fission Using Mdivi-1 in A Clinically Relevant Large Animal Model of Acute Myocardial Infarction: A Pilot Study. Int. J. Mol. Sci. 2019, 20, 3972. [Google Scholar] [CrossRef]
- Nan, J.; Nan, C.; Ye, J.; Qian, L.; Geng, Y.; Xing, D.; Rahman, M.S.U.; Huang, M. EGCG protects cardiomyocytes against hypoxia-reperfusion injury through inhibition of OMA1 activation. J. Cell Sci. 2019, 132, jcs220871. [Google Scholar] [CrossRef]
- Le Page, S.; Niro, M.; Fauconnier, J.; Cellier, L.; Tamareille, S.; Gharib, A.; Chevrollier, A.; Loufrani, L.; Grenier, C.; Kamel, R.; et al. Increase in Cardiac Ischemia-Reperfusion Injuries in Opa1+/- Mouse Model. PLoS ONE 2016, 11, e0164066. [Google Scholar] [CrossRef]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef]
- Karbowski, M.; Lee, Y.J.; Gaume, B.; Jeong, S.Y.; Frank, S.; Nechushtan, A.; Santel, A.; Fuller, M.; Smith, C.L.; Youle, R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 2002, 159, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Anzell, A.R.; Maizy, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial Quality Control and Disease: Insights into Ischemia-Reperfusion Injury. Mol. Neurobiol. 2018, 55, 2547–2564. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [PubMed]
- Sciarretta, S.; Maejima, Y.; Zablocki, D.; Sadoshima, J. The Role of Autophagy in the Heart. Annu. Rev. Physiol. 2018, 80, 1–26. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Pearson, T.; Lightfoot, A.P.; Nye, G.A.; Wells, N.; Giakoumaki, I.I.; Vasilaki, A.; Griffiths, R.D.; Jackson, M.J.; McArdle, A. Mitochondrial ROS regulate oxidative damage and mitophagy but not age-related muscle fiber atrophy. Sci. Rep. 2016, 6, 33944. [Google Scholar] [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476. [Google Scholar] [CrossRef]
- Yuan, Y.; Zheng, Y.; Zhang, X.; Chen, Y.; Wu, X.; Wu, J.; Shen, Z.; Jiang, L.; Wang, L.; Yang, W.; et al. BNIP3L/NIX-mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 2017, 13, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.T.; Szweda, L.I. Cardiac reperfusion injury: Aging, lipid peroxidation, and mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 1998, 95, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef]
- Kazlauskaite, A.; Muqit, M.M. PINK1 and Parkin-mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J. 2015, 282, 215–223. [Google Scholar] [CrossRef]
- Okatsu, K.; Koyano, F.; Kimura, M.; Kosako, H.; Saeki, Y.; Tanaka, K.; Matsuda, N. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 2015, 209, 111–128. [Google Scholar] [CrossRef]
- Okatsu, K.; Kimura, M.; Oka, T.; Tanaka, K.; Matsuda, N. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 2015, 128, 964–978. [Google Scholar] [CrossRef] [PubMed]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the heart of mitochondrial metabolism. Cell. Mol. Life Sci. 2008, 65, 2493–2506. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef]
- Kagan, V.E.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Desbourdes, C.; Cottet-Rousselle, C.; Dar, H.H.; Verma, M.; Tyurin, V.A.; Kapralov, A.A.; et al. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 2016, 23, 1140–1151. [Google Scholar] [CrossRef]
- Bruick, R.K. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 9082–9087. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Henson, E.S.; Cizeau, J.; McMillan-Ward, E.; Israels, S.J.; Gibson, S.B. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008, 4, 195–204. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Scorrano, L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta 2008, 1777, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Sun, Y.; Guo, S.; Lu, B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 2011, 20, 3227–3240. [Google Scholar] [CrossRef] [PubMed]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef]
- Kubli, D.A.; Ycaza, J.E.; Gustafsson, A.B. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem. J. 2007, 405, 407–415. [Google Scholar] [CrossRef]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, A.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef]
- Prieto, J.; Leon, M.; Ponsoda, X.; Sendra, R.; Bort, R.; Ferrer-Lorente, R.; Raya, A.; Lopez-Garcia, C.; Torres, J. Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 2016, 7, 11124. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar]
- Moyzis, A.G.; Sadoshima, J.; Gustafsson, A.B. Mending a broken heart: The role of mitophagy in cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H183–H192. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef]
- Von Muhlinen, N.; Akutsu, M.; Ravenhill, B.J.; Foeglein, A.; Bloor, S.; Rutherford, T.J.; Freund, S.M.; Komander, D.; Randow, F. LC3C, bound selectively by a noncanonical LIR motif in NDP52, is required for antibacterial autophagy. Mol. Cell 2012, 48, 329–342. [Google Scholar] [CrossRef]
- Thurston, T.L.; Ryzhakov, G.; Bloor, S.; von Muhlinen, N.; Randow, F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009, 10, 1215–1221. [Google Scholar] [CrossRef]
- Yussman, M.G.; Toyokawa, T.; Odley, A.; Lynch, R.A.; Wu, G.; Colbert, M.C.; Aronow, B.J.; Lorenz, J.N.; Dorn, G.W. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat. Med. 2002, 8, 725–730. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Schulman, B.A. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat. Struct. Mol. Biol. 2014, 21, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Nair, U.; Jotwani, A.; Geng, J.; Gammoh, N.; Richerson, D.; Yen, W.L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T.; et al. SNARE proteins are required for macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef]
- Razi, M.; Chan, E.Y.; Tooze, S.A. Early endosomes and endosomal coatomer are required for autophagy. J. Cell Biol. 2009, 185, 305–321. [Google Scholar] [CrossRef]
- Rusten, T.E.; Vaccari, T.; Lindmo, K.; Rodahl, L.M.; Nezis, I.P.; Sem-Jacobsen, C.; Wendler, F.; Vincent, J.P.; Brech, A.; Bilder, D.; et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol. 2007, 17, 1817–1825. [Google Scholar] [CrossRef]
- Nickerson, D.P.; Brett, C.L.; Merz, A.J. Vps-C complexes: Gatekeepers of endolysosomal traffic. Curr. Opin Cell Biol. 2009, 21, 543–551. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; Von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef]
- Jager, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.C.; Chan, D.C. Parkin uses the UPS to ship off dysfunctional mitochondria. Autophagy 2011, 7, 771–772. [Google Scholar] [CrossRef] [PubMed]
- Rakovic, A.; Ziegler, J.; Martensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; Konig, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2019, 26, 1428–1441. [Google Scholar] [CrossRef] [PubMed]
- Ardley, H.C.; Robinson, P.A. E3 ubiquitin ligases. Essays Biochem. 2005, 41, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Robbins, J. Heart failure and protein quality control. Circ. Res. 2006, 99, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Robbins, J. Proteotoxicity: An underappreciated pathology in cardiac disease. J. Mol. Cell Cardiol. 2014, 71, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Robbins, J. Proteasomal and lysosomal protein degradation and heart disease. J. Mol. Cell. Cardiol. 2014, 71, 16–24. [Google Scholar] [CrossRef]
- Zheng, Q.; Su, H.; Tian, Z.; Wang, X. Proteasome malfunction activates macroautophagy in the heart. Am. J. Cardiovasc. Dis. 2011, 1, 214–226. [Google Scholar]
- Wang, Y.; Le, W.D. Autophagy and Ubiquitin-Proteasome System. Adv. Exp. Med. Biol. 2019, 1206, 527–550. [Google Scholar]
- Zheng, Q.; Su, H.; Ranek, M.J.; Wang, X. Autophagy and p62 in cardiac proteinopathy. Circ. Res. 2011, 109, 296–308. [Google Scholar] [CrossRef]
- Kyrychenko, V.O.; Nagibin, V.S.; Tumanovska, L.V.; Pashevin, D.O.; Gurianova, V.L.; Moibenko, A.A.; Dosenko, V.E.; Klionsky, D.J. Knockdown of PSMB7 induces autophagy in cardiomyocyte cultures: Possible role in endoplasmic reticulum stress. Pathobiology 2014, 81, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Jia, C.; Zhang, S.; Fan, G.; Li, Y.; Shan, P.; Sun, L.; Xiao, W.; Li, L.; Zheng, Y.; et al. The REGgamma proteasome regulates hepatic lipid metabolism through inhibition of autophagy. Cell Metab. 2013, 18, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Gorbea, C.; Rechsteiner, M.; Vallejo, J.G.; Bowles, N.E. Depletion of the 26S proteasome adaptor Ecm29 increases Toll-like receptor 3 signaling. Sci. Signal. 2013, 6, ra86. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Dunner, K.J.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xu, Q.; Yuan, Q.; Jia, M.; Niu, H.; Liu, X.; Zhang, J.; Young, C.Y.; Yuan, H. Proteasome inhibition boosts autophagic degradation of ubiquitinated-AGR2 and enhances the antitumor efficiency of bevacizumab. Oncogene 2019, 38, 3458–3474. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell. Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Liu, J.; Tang, M.; Mestril, R.; Wang, X. Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 40, 451–454. [Google Scholar] [CrossRef]
- Fielitz, J.; van Rooij, E.; Spencer, J.A.; Shelton, J.M.; Latif, S.; van der Nagel, R.; Bezprozvannaya, S.; de Windt, L.; Richardson, J.A.; Bassel-Duby, R.; et al. Loss of muscle-specific RING-finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc. Natl. Acad. Sci. USA 2007, 104, 4377–4382. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Z.; He, X.R.; Michael, L.H.; Patterson, C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2836–H2842. [Google Scholar] [CrossRef]
- Li, J.; Horak, K.M.; Su, H.; Sanbe, A.; Robbins, J.; Wang, X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J. Clin. Investig. 2011, 121, 3689–3700. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zheng, H.; Li, J.; Li, Y.; Su, H.; Wang, X. Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-reperfusion injury in mice. Circ. Res. 2012, 111, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Divald, A.; Kivity, S.; Wang, P.; Hochhauser, E.; Roberts, B.; Teichberg, S.; Gomes, A.V.; Powell, S.R. Myocardial ischemic preconditioning preserves postischemic function of the 26S proteasome through diminished oxidative damage to 19S regulatory particle subunits. Circ. Res. 2010, 106, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Tian, Y.; Xu, H.; Pan, B.; Terpstra, E.M.; Wu, P.; Wang, H.; Li, F.; Liu, J.; Wang, X. Inadequate ubiquitination-proteasome coupling contributes to myocardial ischemia-reperfusion injury. J. Clin. Investig. 2018, 128, 5294–5306. [Google Scholar] [CrossRef]
- Zaha, V.G.; Young, L.H. AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 2012, 111, 800–814. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Kundu, M.; Lindsten, T.; Yang, C.Y.; Wu, J.; Zhao, F.; Zhang, J.; Selak, M.A.; Ney, P.A.; Thompson, C.B. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008, 112, 1493–1502. [Google Scholar] [CrossRef]
- Diwan, A.; Krenz, M.; Syed, F.M.; Wansapura, J.; Ren, X.; Koesters, A.G.; Li, H.; Kirshenbaum, L.A.; Hahn, H.S.; Robbins, J.; et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investig. 2007, 117, 2825–2833. [Google Scholar] [CrossRef]
- Sciarretta, S.; Hariharan, N.; Monden, Y.; Zablocki, D.; Sadoshima, J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr. Cardiol. 2011, 32, 275–281. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef]
- Hoshino, A.; Matoba, S.; Iwai-Kanai, E.; Nakamura, H.; Kimata, M.; Nakaoka, M.; Katamura, M.; Okawa, Y.; Ariyoshi, M.; Mita, Y.; et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J. Mol. Cell. Cardiol. 2012, 52, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Sun, J.; Yoon, J.S.; Zhang, Y.; Zheng, L.; Murphy, E.; Mattson, M.P.; Lenardo, M.J. Mitochondrial Protein PGAM5 Regulates Mitophagic Protection against Cell Necroptosis. PLoS ONE 2016, 11, e0147792. [Google Scholar] [CrossRef]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R.A. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef] [PubMed]
- Hamacher-Brady, A.; Brady, N.R.; Gottlieb, R.A. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J. Biol. Chem. 2006, 281, 29776–29787. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ye, M.; Liu, D.; Yang, J.; Ding, J.W.; Zhang, J.; Wang, X.A.; Dong, W.S.; Fan, Z.X.; Yang, J. UCP2 protect the heart from myocardial ischemia/reperfusion injury via induction of mitochondrial autophagy. J. Cell. Biochem. 2019, 120, 15455–15466. [Google Scholar] [CrossRef]
- Bi, W.; Jia, J.; Pang, R.; Nie, C.; Han, J.; Ding, Z.; Liu, B.; Sheng, R.; Xu, J.; Zhang, J. Thyroid hormone postconditioning protects hearts from ischemia/reperfusion through reinforcing mitophagy. Biomed. Pharmacother. 2019, 118, 109220. [Google Scholar] [CrossRef]
- Siddall, H.K.; Yellon, D.M.; Ong, S.B.; Mukherjee, U.A.; Burke, N.; Hall, A.R.; Angelova, P.R.; Ludtmann, M.H.; Deas, E.; Davidson, S.M.; et al. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS ONE 2013, 8, e62400. [Google Scholar] [CrossRef]
- Sun, T.; Ding, W.; Xu, T.; Ao, X.; Yu, T.; Li, M.; Liu, Y.; Zhang, X.; Hou, L.; Wang, J. Parkin Regulates Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury by Targeting Cyclophilin-D. Antioxid. Redox Signal. 2019, 31, 1177–1193. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Liu, H.; Foyil, S.R.; Godar, R.J.; Weinheimer, C.J.; Hill, J.A.; Diwan, A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 2012, 125, 3170–3181. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Undyala, V.V.; Wider, J.; Sala-Mercado, J.A.; Gottlieb, R.A.; Mentzer, R.M.J. Acute induction of autophagy as a novel strategy for cardioprotection: Getting to the heart of the matter. Autophagy 2011, 7, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wei, S.; Hao, P.; Xing, J.; Yuan, Q.; Wang, J.; Xu, F.; Chen, Y. Aldehyde Dehydrogenase 2 Has Cardioprotective Effects on Myocardial Ischaemia/Reperfusion Injury via Suppressing Mitophagy. Front. Pharmacol. 2016, 7, 101. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.N.; Kyoi, S.; Hariharan, N.; Takagi, H.; Sadoshima, J.; Gottlieb, R.A. Novel methods for measuring cardiac autophagy in vivo. Methods Enzymol. 2009, 453, 325–342. [Google Scholar] [PubMed]
- Hariharan, N.; Zhai, P.; Sadoshima, J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 2011, 14, 2179–2190. [Google Scholar] [CrossRef]
- Huang, C.; Liu, W.; Perry, C.N.; Yitzhaki, S.; Lee, Y.; Yuan, H.; Tsukada, Y.T.; Hamacher-Brady, A.; Mentzer, R.M.J.; Gottlieb, R.A. Autophagy and protein kinase C are required for cardioprotection by sulfaphenazole. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H570–H579. [Google Scholar] [CrossRef]
- Giricz, Z.; Varga, Z.V.; Koncsos, G.; Nagy, C.T.; Gorbe, A.; Mentzer, R.M.J.; Gottlieb, R.A.; Ferdinandy, P. Autophagosome formation is required for cardioprotection by chloramphenicol. Life Sci. 2017, 186, 11–16. [Google Scholar] [CrossRef]
- Filippone, S.M.; Samidurai, A.; Roh, S.K.; Cain, C.K.; He, J.; Salloum, F.N.; Kukreja, R.C.; Das, A. Reperfusion Therapy with Rapamycin Attenuates Myocardial Infarction through Activation of AKT and ERK. Oxid. Med. Cell. Longev. 2017, 2017, 4619720. [Google Scholar] [CrossRef]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Zhu, X.; Ladenheim, B.; Yu, Q.S.; Guo, Z.; Oyler, J.; Cutler, R.G.; Cadet, J.L.; Greig, N.H.; Mattson, M.P. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol. 2002, 52, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.N.; Baughman, J.M.; Phu, L.; Tea, J.S.; Yu, C.; Coons, M.; Kirkpatrick, D.S.; Bingol, B.; Corn, J.E. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat. Cell Biol. 2015, 17, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Hwang, E.S. Nicotinamide enhances mitochondria quality through autophagy activation in human cells. Aging Cell 2009, 8, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Sebori, R.; Kuno, A.; Hosoda, R.; Hayashi, T.; Horio, Y. Resveratrol Decreases Oxidative Stress by Restoring Mitophagy and Improves the Pathophysiology of Dystrophin-Deficient mdx Mice. Oxid. Med. Cell. Longev. 2018, 2018, 9179270. [Google Scholar] [CrossRef]
- Kuno, A.; Hosoda, R.; Sebori, R.; Hayashi, T.; Sakuragi, H.; Tanabe, M.; Horio, Y. Resveratrol Ameliorates Mitophagy Disturbance and Improves Cardiac Pathophysiology of Dystrophin-deficient mdx Mice. Sci. Rep. 2018, 8, 15555. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Zhu, G.; Zhang, Y.; He, M.; Zhang, J. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp. Cell Res. 2016, 341, 42–53. [Google Scholar] [CrossRef]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef]
- Jia, S.; Xu, X.; Zhou, S.; Chen, Y.; Ding, G.; Cao, L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis. 2019, 10, 142. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef] [PubMed]
- East, D.A.; Fagiani, F.; Crosby, J.; Georgakopoulos, N.D.; Bertrand, H.; Schaap, M.; Fowkes, A.; Wells, G.; Campanella, M. PMI: A DeltaPsim independent pharmacological regulator of mitophagy. Chem. Biol. 2014, 21, 1585–1596. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.R.; Wang, X.; Hu, W.W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef]
- Yamamoto, A.; Tagawa, Y.; Yoshimori, T.; Moriyama, Y.; Masaki, R.; Tashiro, Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998, 23, 33–42. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Hou, H.; Zhang, Y.; Huang, Y.; Yi, Q.; Lv, L.; Zhang, T.; Chen, D.; Hao, Q.; Shi, Q. Inhibitors of phosphatidylinositol 3’-kinases promote mitotic cell death in HeLa cells. PLoS ONE 2012, 7, e35665. [Google Scholar] [CrossRef]
- Wang, R.; Dong, Y.; Lu, Y.; Zhang, W.; Brann, D.W.; Zhang, Q. Photobiomodulation for Global Cerebral Ischemia: Targeting Mitochondrial Dynamics and Functions. Mol. Neurobiol. 2019, 56, 1852–1869. [Google Scholar] [CrossRef]
- Kumari, S.; Anderson, L.; Farmer, S.; Mehta, S.L.; Li, P.A. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl. Stroke Res. 2012, 3, 296–304. [Google Scholar] [CrossRef]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Cui, M.; Chen, S.F.; Dong, Q.; Liu, X.Y. Amelioration of ischemic mitochondrial injury and Bax-dependent outer membrane permeabilization by Mdivi-1. CNS Neurosci. Ther. 2014, 20, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Baburamani, A.A.; Hurling, C.; Stolp, H.; Sobotka, K.; Gressens, P.; Hagberg, H.; Thornton, C. Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2015, 16, 22509–22526. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tian, F.; Kurata, T.; Morimoto, N.; Abe, K. Dynamic changes of mitochondrial fusion and fission proteins after transient cerebral ischemia in mice. J. Neurosci. Res. 2012, 90, 1183–1189. [Google Scholar] [CrossRef]
- Wang, H.; Zheng, S.; Liu, M.; Jia, C.; Wang, S.; Wang, X.; Xue, S.; Guo, Y. The Effect of Propofol on Mitochondrial Fission during Oxygen-Glucose Deprivation and Reperfusion Injury in Rat Hippocampal Neurons. PLoS ONE 2016, 11, e0165052. [Google Scholar] [CrossRef]
- Grohm, J.; Kim, S.W.; Mamrak, U.; Tobaben, S.; Cassidy-Stone, A.; Nunnari, J.; Plesnila, N.; Culmsee, C. Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 2012, 19, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, S.; Li, Y.; Che, L.; Zhao, Q. A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci. Lett. 2013, 535, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Rao, W.; Zhang, L.; Wang, K.; Hui, H.; Wang, L.; Su, N.; Luo, P.; Hao, Y.L.; Tu, Y.; et al. Mitofusin 2 ameliorates hypoxia-induced apoptosis via mitochondrial function and signaling pathways. Int. J. Biochem. Cell. Biol. 2015, 69, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Slupe, A.M.; Merrill, R.A.; Flippo, K.H.; Lobas, M.A.; Houtman, J.C.; Strack, S. A calcineurin docking motif (LXVP) in dynamin-related protein 1 contributes to mitochondrial fragmentation and ischemic neuronal injury. J. Biol. Chem. 2013, 288, 12353–12365. [Google Scholar] [CrossRef]
- Zuo, W.; Zhang, S.; Xia, C.Y.; Guo, X.F.; He, W.B.; Chen, N.H. Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: The role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology 2014, 86, 103–115. [Google Scholar] [CrossRef]
- Sheng, R.; Zhang, L.S.; Han, R.; Liu, X.Q.; Gao, B.; Qin, Z.H. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010, 6, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Ulamek-Koziol, M.; Kocki, J.; Bogucka-Kocka, A.; Januszewski, S.; Bogucki, J.; Czuczwar, S.J.; Pluta, R. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol. Rep. 2017, 69, 1289–1294. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, T.; Wang, J.; Zhang, Z.; Zhai, Y.; Yang, G.Y.; Sun, X. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem. Biophys Res. Commun. 2014, 444, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zheng, Y.; Wu, J.; Chen, Y.; Wu, X.; Zhou, Y.; Yuan, Y.; Lu, S.; Jiang, L.; Qin, Z.; et al. PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window. Autophagy 2017, 13, 473–485. [Google Scholar] [CrossRef]
- Di, Y.; He, Y.L.; Zhao, T.; Huang, X.; Wu, K.W.; Liu, S.H.; Zhao, Y.Q.; Fan, M.; Wu, L.Y.; Zhu, L.L. Methylene Blue Reduces Acute Cerebral Ischemic Injury via the Induction of Mitophagy. Mol. Med. 2015, 21, 420–429. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Y.; Jiang, L.; Zhang, J.; Gao, J.; Shen, Z.; Zheng, Y.; Deng, T.; Yan, H.; Li, W.; et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014, 10, 1801–1813. [Google Scholar] [CrossRef]
- Shi, R.Y.; Zhu, S.H.; Li, V.; Gibson, S.B.; Xu, X.S.; Kong, J.M. BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef]
- Ahsan, A.; Zheng, Y.R.; Wu, X.L.; Tang, W.D.; Liu, M.R.; Ma, S.J.; Jiang, L.; Hu, W.W.; Zhang, X.N.; Chen, Z. Urolithin A-activated autophagy but not mitophagy protects against ischemic neuronal injury by inhibiting ER stress in vitro and in vivo. CNS Neurosci. Ther. 2019, 25, 976–986. [Google Scholar] [CrossRef]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Przyklenk, K. Ischaemic conditioning: Pitfalls on the path to clinical translation. Br. J. Pharmacol. 2015, 172, 1961–1973. [Google Scholar] [CrossRef]
- Shirakabe, A.; Ikeda, Y.; Sciarretta, S.; Zablocki, D.K.; Sadoshima, J. Aging and Autophagy in the Heart. Circ. Res. 2016, 118, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Guberman, M.; Kirshenbaum, L.A. Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc. Med. 2018, 28, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Kobayashi, S. Mitochondrial quality control in the diabetic heart. J. Mol. Cell. Cardiol. 2016, 95, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Yan, W.Y.; Lei, Y.H.; Wan, Z.; Hou, Y.Y.; Sun, L.K.; Zhou, J.P. SIRT3 Regulation of Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Aging Neurosci. 2019, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F.; et al. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Samant, S.A.; Zhang, H.J.; Hong, Z.; Pillai, V.B.; Sundaresan, N.R.; Wolfgeher, D.; Archer, S.L.; Chan, D.C.; Gupta, M.P. SIRT3 deacetylates and activates OPA1 to regulate mitochondrial dynamics during stress. Mol. Cell. Biol. 2014, 34, 807–819. [Google Scholar] [CrossRef]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell. Longev. 2014, 2014, 345105. [Google Scholar] [CrossRef]
- Porter, G.A.; Urciuoli, W.R.; Brookes, P.S.; Nadtochiy, S.M. SIRT3 deficiency exacerbates ischemia-reperfusion injury: Implication for aged hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1602–H1609. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Y.; Song, L.; Yu, L.; Zhang, L.; Zhang, Y.; Xing, Y.; Yin, Y.; Ma, H. SIRT3 deficiency exacerbates p53/Parkinmediated mitophagy inhibition and promotes mitochondrial dysfunction: Implication for aged hearts. Int. J. Mol. Med. 2018, 41, 3517–3526. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulek, A.R.; Anzell, A.; Wider, J.M.; Sanderson, T.H.; Przyklenk, K. Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury. Cells 2020, 9, 214. https://doi.org/10.3390/cells9010214
Kulek AR, Anzell A, Wider JM, Sanderson TH, Przyklenk K. Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury. Cells. 2020; 9(1):214. https://doi.org/10.3390/cells9010214
Chicago/Turabian StyleKulek, Andrew R., Anthony Anzell, Joseph M. Wider, Thomas H. Sanderson, and Karin Przyklenk. 2020. "Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury" Cells 9, no. 1: 214. https://doi.org/10.3390/cells9010214
APA StyleKulek, A. R., Anzell, A., Wider, J. M., Sanderson, T. H., & Przyklenk, K. (2020). Mitochondrial Quality Control: Role in Cardiac Models of Lethal Ischemia-Reperfusion Injury. Cells, 9(1), 214. https://doi.org/10.3390/cells9010214