Crosstalk between PTEN/PI3K/Akt Signalling and DNA Damage in the Oocyte: Implications for Primordial Follicle Activation, Oocyte Quality and Ageing


Abstract
1. Introduction
2. Methods
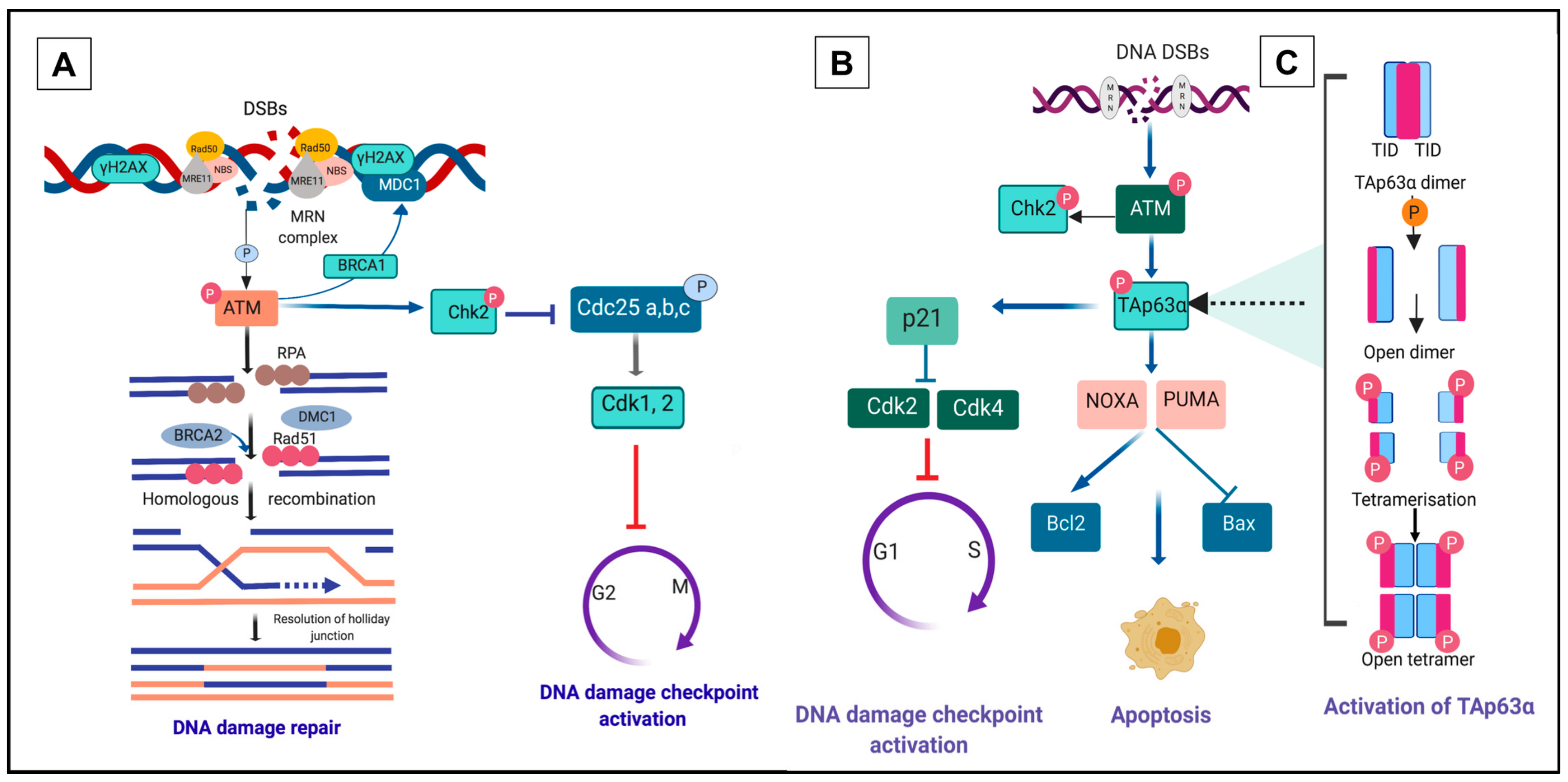
3. DNA Damage Repair Pathway within Primordial Follicles
4. A Unique p63 Pathway Links DNA Damage and Apoptosis in Oocytes within Primordial Follicles
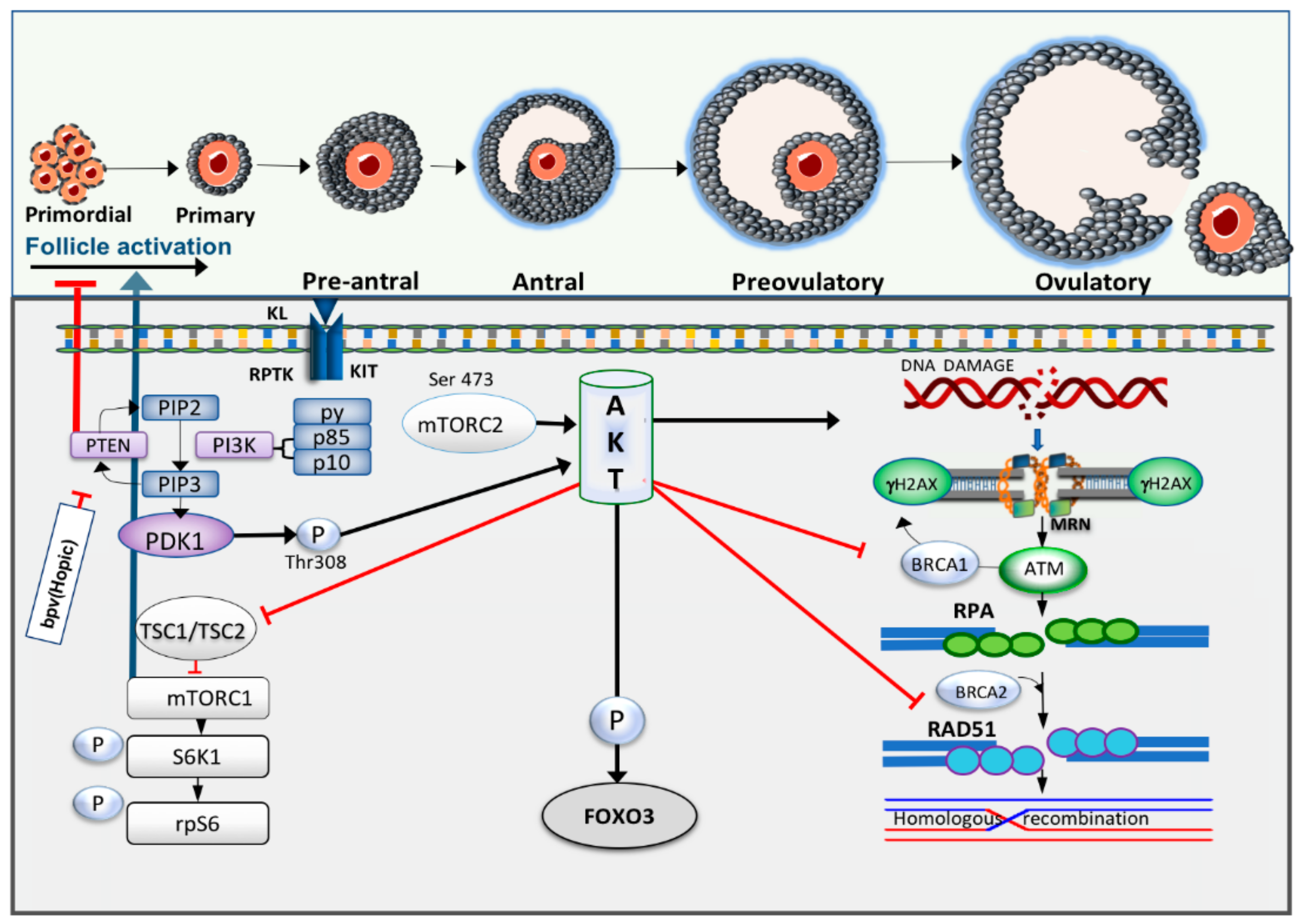
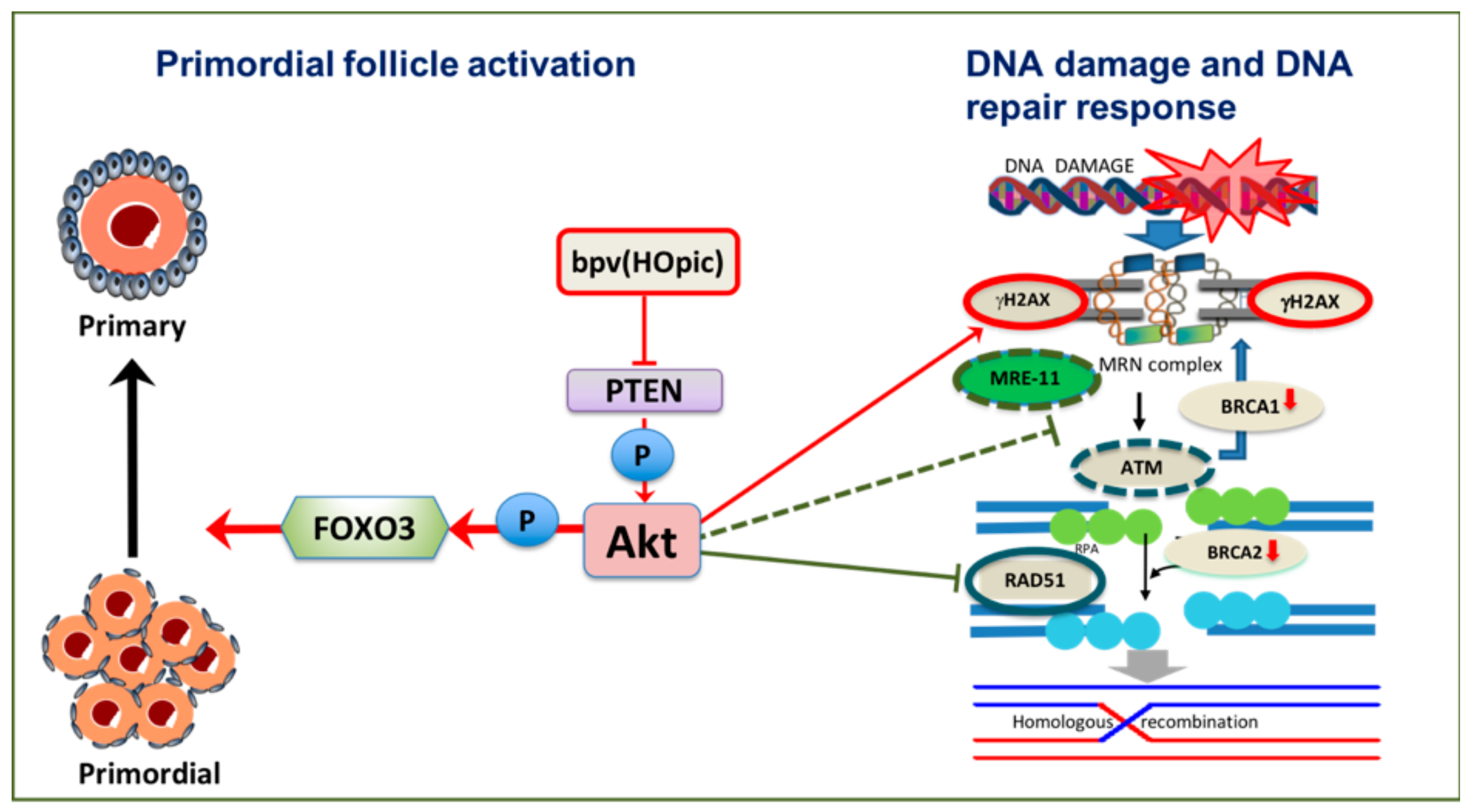
5. The PI3K/Akt Pathway Links Primordial Follicle Growth and the DDR
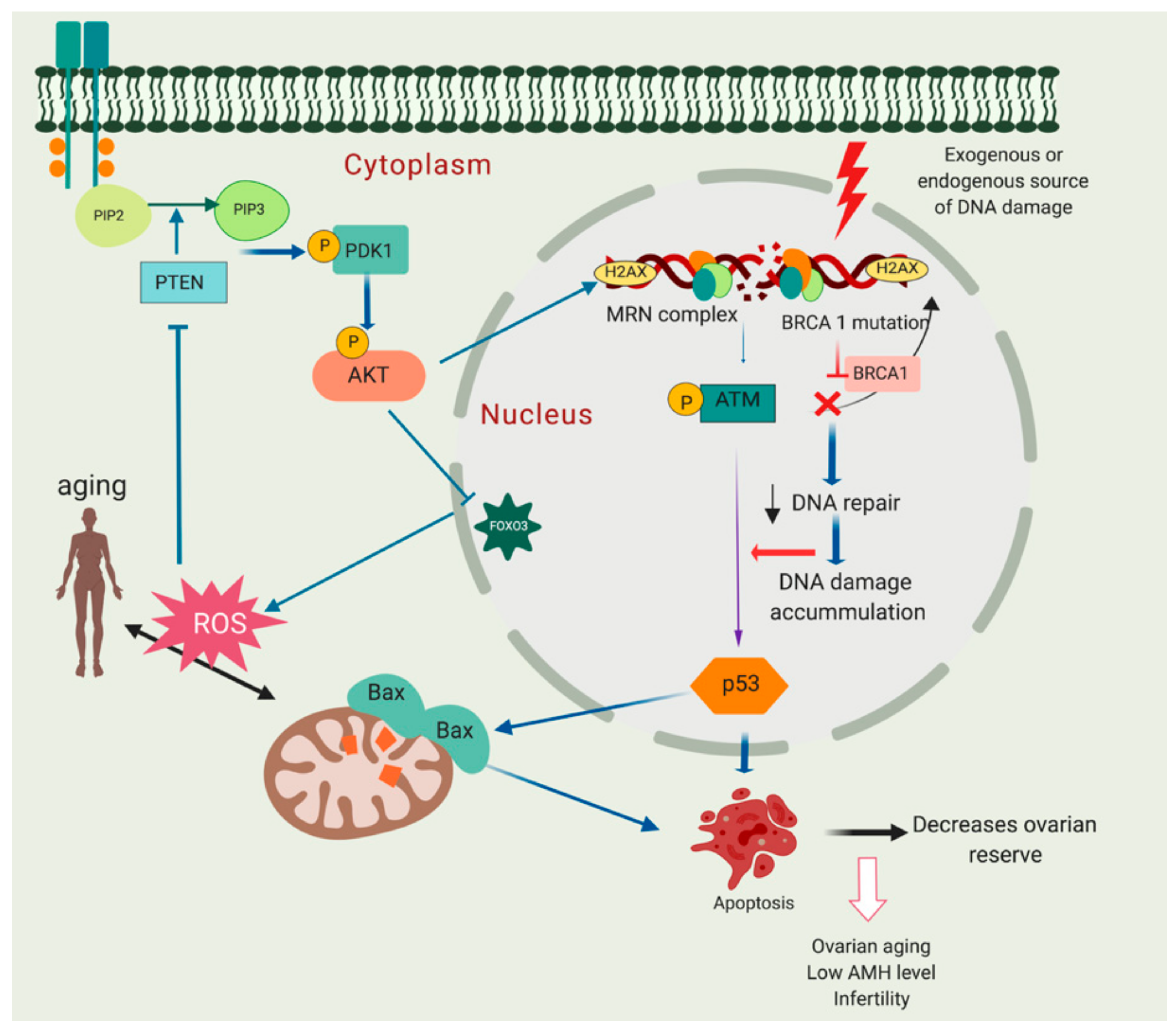
6. DNA Damage Associated with Ovarian Ageing, a Crosstalk between PI3K/Akt/PTEN Signalling, Ageing and DNA Damage Response
7. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Telfer, E.E.; Zelinski, M.B. Ovarian follicle culture: Advances and challenges for human and nonhuman primates. Fertil. Steril. 2013, 99, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Faddy, M.J.; Gosden, R.G. A mathematical model of follicle dynamics in the human ovary. Hum. Reprod. 1995, 10, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.R.; Knowlton, N.S.; Thyer, A.C.; Charleston, J.S.; Soules, M.R.; Klein, N.A. A new model of reproductive aging: The decline in ovarian non-growing follicle number from birth to menopause. Hum. Reprod. 2008, 23, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Broekmans, F.J.; Knauff, E.A.; te Velde, E.R.; Macklon, N.S.; Fauser, B.C. Female reproductive ageing: Current knowledge and future trends. Trends Endocrinol. Metab. 2007, 18, 58–65. [Google Scholar] [CrossRef]
- McGee, E.A.; Hsueh, A.J. Initial and cyclic recruitment of ovarian follicles. Endocr. Rev. 2000, 21, 200–214. [Google Scholar] [CrossRef]
- Adhikari, D.; Liu, K. Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocr. Rev. 2009, 30, 438–464. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Q.Y. Evaluation of oocyte quality: Morphological, cellular and molecular predictors. Reprod. Fertil. Dev. 2007, 19, 1–12. [Google Scholar] [CrossRef]
- Ashwood-Smith, M.J.; Edwards, R.G. DNA repair by oocytes. Mol. Hum. Reprod. 1996, 2, 46–51. [Google Scholar] [CrossRef]
- Tilly, J.L. Commuting the death sentence: How oocytes strive to survive. Nat. Rev. Mol. Cell Biol. 2001, 2, 838–848. [Google Scholar] [CrossRef]
- Winship, A.L.; Stringer, J.M.; Liew, S.H.; Hutt, K.J. The importance of DNA repair for maintaining oocyte quality in response to anti-cancer treatments, environmental toxins and maternal ageing. Hum. Reprod. Update 2018, 24, 119–134. [Google Scholar] [CrossRef]
- Dupont, J.; Scaramuzzi, R.J. Insulin signalling and glucose transport in the ovary and ovarian function during the ovarian cycle. Biochem. J. 2016, 473, 1483–1501. [Google Scholar] [CrossRef]
- Stokoe, D. The phosphoinositide 3-kinase pathway and cancer. Expert. Rev. Mol. Med. 2005, 7, 1–22. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Karimian, A.; Mir, S.M.; Parsian, H.; Refieyan, S.; Mirza-Aghazadeh-Attari, M.; Yousefi, B.; Majidinia, M. Crosstalk between phosphoinositide 3-kinase/akt signaling pathway with DNA damage response and oxidative stress in cancer. J. Cell. Biochem. 2019, 120, 10248–10272. [Google Scholar] [CrossRef]
- Plo, I.; Laulier, C.; Gauthier, L.; Lebrun, F.; Calvo, F.; Lopez, B.S. Akt1 inhibits homologous recombination by inducing cytoplasmic retention of brca1 and rad51. Cancer Res. 2008, 68, 9404–9412. [Google Scholar] [CrossRef]
- Puc, J.; Keniry, M.; Li, H.S.; Pandita, T.K.; Choudhury, A.D.; Memeo, L.; Mansukhani, M.; Murty, V.V.; Gaciong, Z.; Meek, S.E.; et al. Lack of pten sequesters chk1 and initiates genetic instability. Cancer Cell. 2005, 7, 193–204. [Google Scholar] [CrossRef]
- Reddy, P.; Adhikari, D.; Zheng, W.; Liang, S.; Hamalainen, T.; Tohonen, V.; Ogawa, W.; Noda, T.; Volarevic, S.; Huhtaniemi, I.; et al. Pdk1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum. Mol. Genet. 2009, 18, 2813–2824. [Google Scholar] [CrossRef]
- Reddy, P.; Liu, L.; Adhikari, D.; Jagarlamudi, K.; Rajareddy, S.; Shen, Y.; Du, C.; Tang, W.; Hamalainen, T.; Peng, S.L.; et al. Oocyte-specific deletion of pten causes premature activation of the primordial follicle pool. Science 2008, 319, 611–613. [Google Scholar] [CrossRef]
- Govindaraj, V.; Keralapura Basavaraju, R.; Rao, A.J. Changes in the expression of DNA double strand break repair genes in primordial follicles from immature and aged rats. Reprod. Biomed. Online 2015, 30, 303–310. [Google Scholar] [CrossRef]
- Oktay, K.; Turan, V.; Titus, S.; Stobezki, R.; Liu, L. Brca mutations, DNA repair deficiency, and ovarian aging. Biol. Reprod. 2015, 93, 67. [Google Scholar] [CrossRef]
- Titus, S.; Li, F.; Stobezki, R.; Akula, K.; Unsal, E.; Jeong, K.; Dickler, M.; Robson, M.; Moy, F.; Goswami, S.; et al. Impairment of brca1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci. Transl. Med. 2013, 5, 172ra21. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, X.; Zeng, M.; Yuan, J.; Liu, M.; Yin, Y.; Wu, X.; Keefe, D.L.; Liu, L. Increased DNA damage and repair deficiency in granulosa cells are associated with ovarian aging in rhesus monkey. J. Assist. Reprod. Genet. 2015, 32, 1069–1078. [Google Scholar] [CrossRef]
- Chang, E.M.; Lim, E.; Yoon, S.; Jeong, K.; Bae, S.; Lee, D.R.; Yoon, T.K.; Choi, Y.; Lee, W.S. Cisplatin induces overactivation of the dormant primordial follicle through pten/akt/foxo3a pathway which leads to loss of ovarian reserve in mice. PLoS ONE 2015, 10, e0144245. [Google Scholar] [CrossRef]
- Kerr, J.B.; Hutt, K.J.; Michalak, E.M.; Cook, M.; Vandenberg, C.J.; Liew, S.H.; Bouillet, P.; Mills, A.; Scott, C.L.; Findlay, J.K.; et al. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require tap63-mediated induction of puma and noxa. Mol. Cell. 2012, 48, 343–352. [Google Scholar] [CrossRef]
- Lin, W.; Titus, S.; Moy, F.; Ginsburg, E.S.; Oktay, K. Ovarian aging in women with brca germline mutations. J. Clin. Endocrinol. Metab. 2017, 102, 3839–3847. [Google Scholar] [CrossRef]
- Nguyen, Q.N.; Zerafa, N.; Liew, S.H.; Morgan, F.H.; Strasser, A.; Scott, C.L.; Findlay, J.K.; Hickey, M.; Hutt, K.J. Loss of puma protects the ovarian reserve during DNA-damaging chemotherapy and preserves fertility. Cell Death Dis. 2018, 9, 618. [Google Scholar] [CrossRef]
- Rinaldi, V.D.; Hsieh, K.; Munroe, R.; Bolcun-Filas, E.; Schimenti, J.C. Pharmacological inhibition of the DNA damage checkpoint prevents radiation-induced oocyte death. Genetics 2017, 206, 1823–1828. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, M.; Johnson, S.B.; Yuan, G.; Arriba, A.K.; Zubizarreta, M.E.; Chatterjee, S.; Nagarkatti, M.; Nagarkatti, P.; Xiao, S. Doxorubicin obliterates mouse ovarian reserve through both primordial follicle atresia and overactivation. Toxicol. Appl. Pharmacol. 2019, 381, 114714. [Google Scholar] [CrossRef]
- Maidarti, M.; Clarkson, Y.L.; McLaughlin, M.; Anderson, R.A.; Telfer, E.E. Inhibition of pten activates bovine non-growing follicles in vitro but increases DNA damage and reduces DNA repair response. Hum. Reprod. 2019, 34, 297–307. [Google Scholar] [CrossRef]
- Jagarlamudi, K.; Liu, L.; Adhikari, D.; Reddy, P.; Idahl, A.; Ottander, U.; Lundin, E.; Liu, K. Oocyte-specific deletion of pten in mice reveals a stage-specific function of pten/pi3k signaling in oocytes in controlling follicular activation. PLoS ONE 2009, 4, e6186. [Google Scholar] [CrossRef]
- Jagarlamudi, K.; Reddy, P.; Adhikari, D.; Liu, K. Genetically modified mouse models for premature ovarian failure (pof). Mol. Cell. Endocrinol. 2010, 315, 1–10. [Google Scholar] [CrossRef]
- Adhikari, D.; Gorre, N.; Risal, S.; Zhao, Z.; Zhang, H.; Shen, Y.; Liu, K. The safe use of a pten inhibitor for the activation of dormant mouse primordial follicles and generation of fertilizable eggs. PLoS ONE 2012, 7, e39034. [Google Scholar] [CrossRef]
- Novella-Maestre, E.; Herraiz, S.; Rodriguez-Iglesias, B.; Diaz-Garcia, C.; Pellicer, A. Short-term pten inhibition improves in vitro activation of primordial follicles, preserves follicular viability, and restores amh levels in cryopreserved ovarian tissue from cancer patients. PLoS ONE 2015, 10, e0127786. [Google Scholar] [CrossRef]
- Suzuki, N.; Yoshioka, N.; Takae, S.; Sugishita, Y.; Tamura, M.; Hashimoto, S.; Morimoto, Y.; Kawamura, K. Successful fertility preservation following ovarian tissue vitrification in patients with primary ovarian insufficiency. Hum. Reprod. 2015, 30, 608–615. [Google Scholar] [CrossRef]
- Lerer-Serfaty, G.; Samara, N.; Fisch, B.; Shachar, M.; Kossover, O.; Seliktar, D.; Ben-Haroush, A.; Abir, R. Attempted application of bioengineered/biosynthetic supporting matrices with phosphatidylinositol-trisphosphate-enhancing substances to organ culture of human primordial follicles. J. Assist. Reprod. Genet. 2013, 30, 1279–1288. [Google Scholar] [CrossRef]
- McLaughlin, M.; Kinnell, H.L.; Anderson, R.A.; Telfer, E.E. Inhibition of phosphatase and tensin homologue (pten) in human ovary in vitro results in increased activation of primordial follicles but compromises development of growing follicles. Mol. Hum. Reprod. 2014, 20, 736–744. [Google Scholar] [CrossRef]
- Grosbois, J.; Demeestere, I. Dynamics of pi3k and hippo signaling pathways during in vitro human follicle activation. Hum. Reprod. 2018, 33, 1705–1714. [Google Scholar] [CrossRef]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell cycle control by pten. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, W.H. Pten: A new guardian of the genome. Oncogene 2008, 27, 5443–5453. [Google Scholar] [CrossRef]
- Adriaens, I.; Smitz, J.; Jacquet, P. The current knowledge on radiosensitivity of ovarian follicle development stages. Hum. Reprod. Update 2009, 15, 359–377. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Menezo, Y.; Dale, B.; Cohen, M. DNA damage and repair in human oocytes and embryos: A review. Zygote 2010, 18, 357–365. [Google Scholar] [CrossRef]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; Group, P.-P. Preferred reporting items for systematic review and meta-analysis protocols (prisma-p) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell. Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Oktay, K.; Moy, F.; Titus, S.; Stobezki, R.; Turan, V.; Dickler, M.; Goswami, S. Age-related decline in DNA repair function explains diminished ovarian reserve, earlier menopause, and possible oocyte vulnerability to chemotherapy in women with brca mutations. J. Clin. Oncol. 2014, 32, 1093–1094. [Google Scholar] [CrossRef]
- Kujjo, L.L.; Laine, T.; Pereira, R.J.; Kagawa, W.; Kurumizaka, H.; Yokoyama, S.; Perez, G.I. Enhancing survival of mouse oocytes following chemotherapy or aging by targeting bax and rad51. PLoS ONE 2010, 5, e9204. [Google Scholar] [CrossRef]
- Martin, J.H.; Bromfield, E.G.; Aitken, R.J.; Lord, T.; Nixon, B. Double strand break DNA repair occurs via non-homologous end-joining in mouse mii oocytes. Sci. Rep. 2018, 8, 9685. [Google Scholar] [CrossRef]
- Collins, J.K.; Jones, K.T. DNA damage responses in mammalian oocytes. Reproduction 2016, 152, R15–R22. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Rodgers, K.; McVey, M. Error-prone repair of DNA double-strand breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef]
- Heijink, A.M.; Krajewska, M.; van Vugt, M.A. The DNA damage response during mitosis. Mutat. Res. 2013, 750, 45–55. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair 2010, 9, 1219–1228. [Google Scholar] [CrossRef]
- Stringer, J.M.; Winship, A.; Liew, S.H.; Hutt, K. The capacity of oocytes for DNA repair. Cell. Mol. Life Sci. 2018, 75, 2777–2792. [Google Scholar] [CrossRef]
- Menezo, Y., Jr.; Russo, G.; Tosti, E.; El Mouatassim, S.; Benkhalifa, M. Expression profile of genes coding for DNA repair in human oocytes using pangenomic microarrays, with a special focus on ros linked decays. J. Assist. Reprod. Genet. 2007, 24, 513–520. [Google Scholar] [CrossRef]
- Jaroudi, S.; Kakourou, G.; Cawood, S.; Doshi, A.; Ranieri, D.M.; Serhal, P.; Harper, J.C.; SenGupta, S.B. Expression profiling of DNA repair genes in human oocytes and blastocysts using microarrays. Hum. Reprod. 2009, 24, 2649–2655. [Google Scholar] [CrossRef]
- Govindaraj, V.; Krishnagiri, H.; Chauhan, M.S.; Rao, A.J. Brca-1 gene expression and comparative proteomic profile of primordial follicles from young and adult buffalo (bubalus bubalis) ovaries. Anim. Biotechnol. 2017, 28, 94–103. [Google Scholar] [CrossRef]
- Govindaraj, V.; Rao, A.J. Comparative proteomic analysis of primordial follicles from ovaries of immature and aged rats. Syst. Biol. Reprod. Med. 2015, 61, 367–375. [Google Scholar] [CrossRef]
- Fiorenza, M.T.; Bevilacqua, A.; Bevilacqua, S.; Mangia, F. Growing dictyate oocytes, but not early preimplantation embryos, of the mouse display high levels of DNA homologous recombination by single-strand annealing and lack DNA nonhomologous end joining. Dev. Biol. 2001, 233, 214–224. [Google Scholar] [CrossRef][Green Version]
- Rimon-Dahari, N.; Yerushalmi-Heinemann, L.; Alyagor, L.; Dekel, N. Ovarian folliculogenesis. Results Probl. Cell. Differ. 2016, 58, 167–190. [Google Scholar]
- Perez, G.I.; Acton, B.M.; Jurisicova, A.; Perkins, G.A.; White, A.; Brown, J.; Trbovich, A.M.; Kim, M.R.; Fissore, R.; Xu, J.; et al. Genetic variance modifies apoptosis susceptibility in mature oocytes via alterations in DNA repair capacity and mitochondrial ultrastructure. Cell. Death Differ. 2007, 14, 524–533. [Google Scholar] [CrossRef]
- Turan, V.; Oktay, K. Brca-related atm-mediated DNA double-strand break repair and ovarian aging. Hum. Reprod. Update 2019, 26, 43–57. [Google Scholar] [CrossRef]
- Goedecke, W.; Vielmetter, W.; Pfeiffer, P. Activation of a system for the joining of nonhomologous DNA ends during xenopus egg maturation. Mol. Cell. Biol. 1992, 12, 811–816. [Google Scholar] [CrossRef]
- So, S.; Davis, A.J.; Chen, D.J. Autophosphorylation at serine 1981 stabilizes atm at DNA damage sites. J. Cell Biol. 2009, 187, 977–990. [Google Scholar] [CrossRef]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. Atm activation and its recruitment to damaged DNA require binding to the c terminus of nbs1. Mol. Cell Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef]
- Inagaki, A.; Roset, R.; Petrini, J.H. Functions of the mre11 complex in the development and maintenance of oocytes. Chromosoma 2016, 125, 151–162. [Google Scholar] [CrossRef]
- Ganesan, S.; Keating, A.F. The ovarian DNA damage repair response is induced prior to phosphoramide mustard-induced follicle depletion, and ataxia telangiectasia mutated inhibition prevents pm-induced follicle depletion. Toxicol. Appl. Pharmacol. 2016, 292, 65–74. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone h2ax phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Tomilin, N.V.; Solovjeva, L.V.; Svetlova, M.P.; Pleskach, N.M.; Zalenskaya, I.A.; Yau, P.M.; Bradbury, E.M. Visualization of focal nuclear sites of DNA repair synthesis induced by bleomycin in human cells. Radiat. Res. 2001, 156, 347–354. [Google Scholar] [CrossRef]
- Nazarov, I.B.; Smirnova, A.N.; Krutilina, R.I.; Svetlova, M.P.; Solovjeva, L.V.; Nikiforov, A.A.; Oei, S.L.; Zalenskaya, I.A.; Yau, P.M.; Bradbury, E.M.; et al. Dephosphorylation of histone gamma-h2ax during repair of DNA double-strand breaks in mammalian cells and its inhibition by calyculin a. Radiat. Res. 2003, 160, 309–317. [Google Scholar] [CrossRef]
- Jungmichel, S.; Stucki, M. Mdc1: The art of keeping things in focus. Chromosoma 2010, 119, 337–349. [Google Scholar] [CrossRef]
- Marangos, P.; Carroll, J. Oocytes progress beyond prophase in the presence of DNA damage. Curr. Biol. 2012, 22, 989–994. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. Atm- and cell cycle-dependent regulation of atr in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef]
- Bolcun-Filas, E.; Rinaldi, V.D.; White, M.E.; Schimenti, J.C. Reversal of female infertility by chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 2014, 343, 533–536. [Google Scholar] [CrossRef]
- Kujjo, L.L.; Ronningen, R.; Ross, P.; Pereira, R.J.; Rodriguez, R.; Beyhan, Z.; Goissis, M.D.; Baumann, T.; Kagawa, W.; Camsari, C.; et al. Rad51 plays a crucial role in halting cell death program induced by ionizing radiation in bovine oocytes. Biol. Reprod. 2012, 86, 76. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. Chk2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef]
- Basu, A.; Haldar, S. The relationship between bci2, bax and p53: Consequences for cell cycle progression and cell death. Mol. Hum. Reprod. 1998, 4, 1099–1109. [Google Scholar] [CrossRef]
- Kim, D.A.; Suh, E.K. Defying DNA double-strand break-induced death during prophase i meiosis by temporal tap63alpha phosphorylation regulation in developing mouse oocytes. Mol. Cell. Biol. 2014, 34, 1460–1473. [Google Scholar] [CrossRef]
- Suh, E.K.; Yang, A.; Kettenbach, A.; Bamberger, C.; Michaelis, A.H.; Zhu, Z.; Elvin, J.A.; Bronson, R.T.; Crum, C.P.; McKeon, F. P63 protects the female germ line during meiotic arrest. Nature 2006, 444, 624–628. [Google Scholar] [CrossRef]
- Livera, G.; Petre-Lazar, B.; Guerquin, M.J.; Trautmann, E.; Coffigny, H.; Habert, R. P63 null mutation protects mouse oocytes from radio-induced apoptosis. Reproduction 2008, 135, 3–12. [Google Scholar] [CrossRef]
- Amelio, I.; Grespi, F.; Annicchiarico-Petruzzelli, M.; Melino, G. P63 the guardian of human reproduction. Cell Cycle 2012, 11, 4545–4551. [Google Scholar] [CrossRef]
- Levine, A.J.; Tomasini, R.; McKeon, F.D.; Mak, T.W.; Melino, G. The p53 family: Guardians of maternal reproduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 259–265. [Google Scholar] [CrossRef]
- Gebel, J.; Tuppi, M.; Krauskopf, K.; Coutandin, D.; Pitzius, S.; Kehrloesser, S.; Osterburg, C.; Dotsch, V. Control mechanisms in germ cells mediated by p53 family proteins. J. Cell Sci. 2017. [CrossRef]
- Deutsch, G.B.; Zielonka, E.M.; Coutandin, D.; Dotsch, V. Quality control in oocytes: Domain-domain interactions regulate the activity of p63. Cell Cycle 2011, 10, 1884–1885. [Google Scholar] [CrossRef]
- Straub, W.E.; Weber, T.A.; Schafer, B.; Candi, E.; Durst, F.; Ou, H.D.; Rajalingam, K.; Melino, G.; Dotsch, V. The c-terminus of p63 contains multiple regulatory elements with different functions. Cell Death Dis. 2010, 1, e5. [Google Scholar] [CrossRef]
- Deutsch, G.B.; Zielonka, E.M.; Coutandin, D.; Weber, T.A.; Schafer, B.; Hannewald, J.; Luh, L.M.; Durst, F.G.; Ibrahim, M.; Hoffmann, J.; et al. DNA damage in oocytes induces a switch of the quality control factor tap63alpha from dimer to tetramer. Cell 2011, 144, 566–576. [Google Scholar] [CrossRef]
- Coutandin, D.; Osterburg, C.; Srivastav, R.K.; Sumyk, M.; Kehrloesser, S.; Gebel, J.; Tuppi, M.; Hannewald, J.; Schafer, B.; Salah, E.; et al. Quality control in oocytes by p63 is based on a spring-loaded activation mechanism on the molecular and cellular level. Elife 2016, 5, e13909. [Google Scholar] [CrossRef]
- Tuppi, M.; Kehrloesser, S.; Coutandin, D.W.; Rossi, V.; Luh, L.M.; Strubel, A.; Hotte, K.; Hoffmeister, M.; Schafer, B.; De Oliveira, T.; et al. Oocyte DNA damage quality control requires consecutive interplay of chk2 and ck1 to activate p63. Nat. Struct. Mol. Biol. 2018, 25, 261–269. [Google Scholar] [CrossRef]
- Nowsheen, S.; Yang, E. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012, 34, 243–254. [Google Scholar]
- Guo, X.; Keyes, W.M.; Papazoglu, C.; Zuber, J.; Li, W.; Lowe, S.W.; Vogel, H.; Mills, A.A. Tap63 induces senescence and suppresses tumorigenesis in vivo. Nat. Cell Biol. 2009, 11, 1451–1457. [Google Scholar] [CrossRef] [PubMed]
- Tavana, O.; Benjamin, C.L.; Puebla-Osorio, N.; Sang, M.; Ullrich, S.E.; Ananthaswamy, H.N.; Zhu, C. Absence of p53-dependent apoptosis leads to uv radiation hypersensitivity, enhanced immunosuppression and cellular senescence. Cell Cycle 2010, 9, 3328–3336. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, D.; Flohr, G.; Gorre, N.; Shen, Y.; Yang, H.; Lundin, E.; Lan, Z.; Gambello, M.J.; Liu, K. Disruption of tsc2 in oocytes leads to overactivation of the entire pool of primordial follicles. Mol. Hum. Reprod. 2009, 15, 765–770. [Google Scholar] [CrossRef]
- Dole, G.; Nilsson, E.E.; Skinner, M.K. Glial-derived neurotrophic factor promotes ovarian primordial follicle development and cell-cell interactions during folliculogenesis. Reproduction 2008, 135, 671–682. [Google Scholar] [CrossRef]
- Nilsson, E.; Parrott, J.A.; Skinner, M.K. Basic fibroblast growth factor induces primordial follicle development and initiates folliculogenesis. Mol. Cell. Endocrinol. 2001, 175, 123–130. [Google Scholar] [CrossRef]
- Ojeda, S.R.; Romero, C.; Tapia, V.; Dissen, G.A. Neurotrophic and cell-cell dependent control of early follicular development. Mol. Cell. Endocrinol. 2000, 163, 67–71. [Google Scholar] [CrossRef]
- Nilsson, E.E.; Skinner, M.K. Kit ligand and basic fibroblast growth factor interactions in the induction of ovarian primordial to primary follicle transition. Mol. Cell. Endocrinol. 2004, 214, 19–25. [Google Scholar] [CrossRef]
- Nilsson, E.E.; Kezele, P.; Skinner, M.K. Leukemia inhibitory factor (lif) promotes the primordial to primary follicle transition in rat ovaries. Mol. Cell. Endocrinol. 2002, 188, 65–73. [Google Scholar] [CrossRef]
- McLaughlin, E.A.; McIver, S.C. Awakening the oocyte: Controlling primordial follicle development. Reproduction 2009, 137, 1–11. [Google Scholar] [CrossRef]
- Adhikari, D.; Zheng, W.; Shen, Y.; Gorre, N.; Hamalainen, T.; Cooney, A.J.; Huhtaniemi, I.; Lan, Z.J.; Liu, K. Tsc/mtorc1 signaling in oocytes governs the quiescence and activation of primordial follicles. Hum. Mol. Genet. 2010, 19, 397–410. [Google Scholar] [CrossRef]
- Adhikari, D.; Liu, K. Mtor signaling in the control of activation of primordial follicles. Cell Cycle 2010, 9, 1673–1674. [Google Scholar] [CrossRef]
- Castrillon, D.H.; Miao, L.; Kollipara, R.; Horner, J.W.; DePinho, R.A. Suppression of ovarian follicle activation in mice by the transcription factor foxo3a. Science 2003, 301, 215–218. [Google Scholar] [CrossRef]
- John, G.B.; Shirley, L.J.; Gallardo, T.D.; Castrillon, D.H. Specificity of the requirement for foxo3 in primordial follicle activation. Reproduction 2007, 133, 855–863. [Google Scholar] [CrossRef]
- Pelosi, E.; Omari, S.; Michel, M.; Ding, J.; Amano, T.; Forabosco, A.; Schlessinger, D.; Ottolenghi, C. Constitutively active foxo3 in oocytes preserves ovarian reserve in mice. Nat. Commun. 2013, 4, 1843. [Google Scholar] [CrossRef]
- Hunt, C.R.; Gupta, A.; Horikoshi, N.; Pandita, T.K. Does pten loss impair DNA double-strand break repair by homologous recombination? Clin. Cancer Res. 2012, 18, 920–922. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Guo, C.; Xie, A.; Gao, D.; Guo, J.; Zhang, J.; Willis, N.; Su, A.; Asara, J.M.; et al. Akt-mediated phosphorylation of xlf impairs non-homologous end-joining DNA repair. Mol. Cell. 2015, 57, 648–661. [Google Scholar] [CrossRef]
- Xu, N.; Hegarat, N.; Black, E.J.; Scott, M.T.; Hochegger, H.; Gillespie, D.A. Akt/pkb suppresses DNA damage processing and checkpoint activation in late g2. J. Cell Biol. 2010, 190, 297–305. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Evinger, A.J.; Lee, E.; Levin, E.R. Estrogen inhibits atr signaling to cell cycle checkpoints and DNA repair. Mol. Biol. Cell 2009, 20, 3374–3389. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential role for nuclear pten in maintaining chromosomal integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef]
- Thacker, J. The rad51 gene family, genetic instability and cancer. Cancer Lett. 2005, 219, 125–135. [Google Scholar] [CrossRef]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. Akt induces senescence in human cells via mtorc1 and p53 in the absence of DNA damage: Implications for targeting mtor during malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef]
- Kawamura, K.; Cheng, Y.; Suzuki, N.; Deguchi, M.; Sato, Y.; Takae, S.; Ho, C.H.; Kawamura, N.; Tamura, M.; Hashimoto, S.; et al. Hippo signaling disruption and akt stimulation of ovarian follicles for infertility treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 17474–17479. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ebbert, K.; Cordeiro, M.H.; Romero, M.; Zhu, J.; Serna, V.A.; Whelan, K.A.; Woodruff, T.K.; Kurita, T. Cell autonomous phosphoinositide 3-kinase activation in oocytes disrupts normal ovarian function through promoting survival and overgrowth of ovarian follicles. Endocrinology 2015, 156, 1464–1476. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ebbert, K.; Cordeiro, M.H.; Romero, M.M.; Whelan, K.A.; Suarez, A.A.; Woodruff, T.K.; Kurita, T. Constitutive activation of pi3k in oocyte induces ovarian granulosa cell tumors. Cancer Res. 2016, 76, 3851–3861. [Google Scholar] [CrossRef]
- Smitz, J.E.; Cortvrindt, R.G. The earliest stages of folliculogenesis in vitro. Reproduction 2002, 123, 185–202. [Google Scholar] [CrossRef]
- Nguyen, Q.N.; Zerafa, N.; Liew, S.H.; Findlay, J.K.; Hickey, M.; Hutt, K.J.; Bezerra, M.E.S.; Gouveia, B.B.; Barberino, R.S.; Menezes, V.G.; et al. Cisplatin- and cyclophosphamide-induced primordial follicle depletion is caused by direct damage to oocytes resveratrol promotes in vitro activation of ovine primordial follicles by reducing DNA damage and enhancing granulosa cell proliferation via phosphatidylinositol 3-kinase pathway. Mol. Hum. Reprod. 2019, 53, 1298–1305. [Google Scholar]
- Soleimani, R.; Heytens, E.; Darzynkiewicz, Z.; Oktay, K. Mechanisms of chemotherapy-induced human ovarian aging: Double strand DNA breaks and microvascular compromise. Aging 2011, 3, 782–793. [Google Scholar] [CrossRef]
- Roness, H.; Gavish, Z.; Cohen, Y.; Meirow, D. Ovarian follicle burnout: A universal phenomenon? Cell Cycle 2013, 12, 3245–3246. [Google Scholar] [CrossRef]
- Kalich-Philosoph, L.; Roness, H.; Carmely, A.; Fishel-Bartal, M.; Ligumsky, H.; Paglin, S.; Wolf, I.; Kanety, H.; Sredni, B.; Meirow, D. Cyclophosphamide triggers follicle activation and “burnout”; as101 prevents follicle loss and preserves fertility. Sci. Transl. Med. 2013, 5, 185ra62. [Google Scholar] [CrossRef]
- Spears, N.; Lopes, F.; Stefansdottir, A.; Rossi, V.; De Felici, M.; Anderson, R.A.; Klinger, F.G. Ovarian damage from chemotherapy and current approaches to its protection. Hum. Reprod. Update 2019, 25, 673–693. [Google Scholar] [CrossRef]
- Govatati, S.; Kodati, V.L.; Deenadayal, M.; Chakravarty, B.; Shivaji, S.; Bhanoori, M.; Yin, X.; Pavone, M.E.; Lu, Z.; Wei, J.; et al. Mutations in the pten tumor gene and risk of endometriosis: A case-control study increased activation of the pi3k/akt pathway compromises decidualization of stromal cells from endometriosis. Hum. Reprod. 2014, 29, 324–336. [Google Scholar] [CrossRef]
- Madanes, D.; Bilotas, M.A.; Baston, J.I.; Singla, J.J.; Meresman, G.F.; Baranao, R.I.; Ricci, A.G.; Takeuchi, A.; Koga, K.; Satake, E.; et al. Pi3k/akt pathway is altered in the endometriosis patient’s endometrium and presents differences according to severity stage endometriosis triggers excessive activation of primordial follicles via pi3k-pten-akt-foxo3 pathway inhibition of pi3k/akt/mtor pathway for the treatment of endometriosis. Gynecol. Endocrinol. 2019, 104, 1–5. [Google Scholar]
- Makker, A.; Goel, M.M.; Das, V.; Agarwal, A. Pi3k-akt-mtor and mapk signaling pathways in polycystic ovarian syndrome, uterine leiomyomas and endometriosis: An update. Gynecol. Endocrinol. 2012, 28, 175–181. [Google Scholar] [CrossRef]
- Takeuchi, A.; Koga, K.; Satake, E.; Makabe, T.; Taguchi, A.; Miyashita, M.; Takamura, M.; Harada, M.; Hirata, T.; Hirota, Y.; et al. Endometriosis triggers excessive activation of primordial follicles via pi3k-pten-akt-foxo3 pathway inhibition of pi3k/akt/mtor pathway for the treatment of endometriosis. J. Clin. Endocrinol. Metab. 2019, 104, 5547–5554. [Google Scholar] [CrossRef]
- Yin, X.; Pavone, M.E.; Lu, Z.; Wei, J.; Kim, J.J. Increased activation of the pi3k/akt pathway compromises decidualization of stromal cells from endometriosis. J. Clin. Endocrinol. Metab. 2012, 97, E35–E43. [Google Scholar] [CrossRef]
- Barra, F.; Ferro Desideri, L.; Ferrero, S. Inhibition of pi3k/akt/mtor pathway for the treatment of endometriosis. Br. J. Pharmacol. 2018, 175, 3626–3627. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, X.; Liu, S.; Li, J.; Wen, Z.; Li, M. 17betae2 promotes cell proliferation in endometriosis by decreasing pten via nfkappab-dependent pathway. Mol. Cell. Endocrinol. 2010, 317, 31–43. [Google Scholar] [CrossRef]
- Kitajima, M.; Dolmans, M.M.; Donnez, O.; Masuzaki, H.; Soares, M.; Donnez, J. Enhanced follicular recruitment and atresia in cortex derived from ovaries with endometriomas. Fertil. Steril. 2014, 101, 1031–1037. [Google Scholar] [CrossRef]
- Choi, Y.S.; Park, J.H.; Lee, J.H.; Yoon, J.K.; Yun, B.H.; Park, J.H.; Seo, S.K.; Sung, H.J.; Kim, H.S.; Cho, S.; et al. Association between impairment of DNA double strand break repair and decreased ovarian reserve in patients with endometriosis. Front Endocrinol. 2018, 9, 772. [Google Scholar] [CrossRef]
- Kacan, T.; Yildiz, C.; Baloglu Kacan, S.; Seker, M.; Ozer, H.; Cetin, A. Everolimus as an mtor inhibitor suppresses endometriotic implants: An experimental rat study. Geburtshilfe Frauenheilkd. 2017, 77, 66–72. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, Z.; Liu, Z.; Chen, R.; Peng, H.; Guo, J.; Chen, X.; Zhang, H. Mtor inhibition attenuates DNA damage and apoptosis through autophagy-mediated suppression of creb1. Autophagy 2013, 9, 2069–2086. [Google Scholar] [CrossRef]
- Zhou, L.; Xie, Y.; Li, S.; Liang, Y.; Qiu, Q.; Lin, H.; Zhang, Q. Rapamycin prevents cyclophosphamide-induced over-activation of primordial follicle pool through pi3k/akt/mtor signaling pathway in vivo. J. Ovarian. Res. 2017, 10, 56. [Google Scholar] [CrossRef]
- Adhikari, D.; Risal, S.; Liu, K.; Shen, Y. Pharmacological inhibition of mtorc1 prevents over-activation of the primordial follicle pool in response to elevated pi3k signaling. PLoS ONE 2013, 8, e53810. [Google Scholar] [CrossRef]
- Zhang, X.M.; Li, L.; Xu, J.J.; Wang, N.; Liu, W.J.; Lin, X.H.; Fu, Y.C.; Luo, L.L. Rapamycin preserves the follicle pool reserve and prolongs the ovarian lifespan of female rats via modulating mtor activation and sirtuin expression. Gene 2013, 523, 82–87. [Google Scholar] [CrossRef]
- Goldman, K.N.; Chenette, D.; Arju, R.; Duncan, F.E.; Keefe, D.L.; Grifo, J.A.; Schneider, R.J. Mtorc1/2 inhibition preserves ovarian function and fertility during genotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 2017, 114, 3186–3191. [Google Scholar] [CrossRef]
- Wang, W.; Luo, S.M.; Ma, J.Y.; Shen, W.; Yin, S. Cytotoxicity and DNA damage caused from diazinon exposure by inhibiting the pi3k-akt pathway in porcine ovarian granulosa cells. J. Agric. Food Chem. 2019, 67, 19–31. [Google Scholar] [CrossRef]
- Ganesan, S.; Keating, A.F. Bisphenol a-induced ovotoxicity involves DNA damage induction to which the ovary mounts a protective response indicated by increased expression of proteins involved in DNA repair and xenobiotic biotransformation. Toxicol. Sci. 2016, 152, 169–180. [Google Scholar] [CrossRef][Green Version]
- Sun, X.; Su, Y.; He, Y.; Zhang, J.; Liu, W.; Zhang, H.; Hou, Z.; Liu, J.; Li, J. New strategy for in vitro activation of primordial follicles with mtor and pi3k stimulators. Cell Cycle 2015, 14, 721–731. [Google Scholar] [CrossRef]
- Li, J.; Kawamura, K.; Cheng, Y.; Liu, S.; Klein, C.; Liu, S.; Duan, E.K.; Hsueh, A.J. Activation of dormant ovarian follicles to generate mature eggs. Proc. Natl. Acad. Sci. USA 2010, 107, 10280–10284. [Google Scholar] [CrossRef]
- Faddy, M.J.; Gosden, R.G.; Gougeon, A.; Richardson, S.J.; Nelson, J.F. Accelerated disappearance of ovarian follicles in mid-life: Implications for forecasting menopause. Hum. Reprod. 1992, 7, 1342–1346. [Google Scholar] [CrossRef]
- De Bruin, J.P.; Dorland, M.; Spek, E.R.; Posthuma, G.; van Haaften, M.; Looman, C.W.; te Velde, E.R. Age-related changes in the ultrastructure of the resting follicle pool in human ovaries. Biol. Reprod. 2004, 70, 419–424. [Google Scholar] [CrossRef]
- Li, Q.; Geng, X.; Zheng, W.; Tang, J.; Xu, B.; Shi, Q. Current understanding of ovarian aging. Sci. China Life Sci. 2012, 55, 659–669. [Google Scholar] [CrossRef]
- Gougeon, A. Dynamics of follicular growth in the human: A model from preliminary results. Hum. Reprod. 1986, 1, 81–87. [Google Scholar] [CrossRef]
- Oktay, K.; Kim, J.Y.; Barad, D.; Babayev, S.N. Association of brca1 mutations with occult primary ovarian insufficiency: A possible explanation for the link between infertility and breast/ovarian cancer risks. J. Clin. Oncol. 2010, 28, 240–244. [Google Scholar] [CrossRef]
- Sharan, S.K.; Pyle, A.; Coppola, V.; Babus, J.; Swaminathan, S.; Benedict, J.; Swing, D.; Martin, B.K.; Tessarollo, L.; Evans, J.P.; et al. Brca2 deficiency in mice leads to meiotic impairment and infertility. Development 2004, 131, 131–142. [Google Scholar] [CrossRef]
- Weinberg-Shukron, A.; Rachmiel, M.; Renbaum, P.; Gulsuner, S.; Walsh, T.; Lobel, O.; Dreifuss, A.; Ben-Moshe, A.; Zeligson, S.; Segel, R.; et al. Essential role of brca2 in ovarian development and function. N. Engl. J. Med. 2018, 379, 1042–1049. [Google Scholar] [CrossRef]
- Day, F.R.; Ruth, K.S.; Thompson, D.J.; Lunetta, K.L.; Pervjakova, N.; Chasman, D.I.; Stolk, L.; Finucane, H.K.; Sulem, P.; Bulik-Sullivan, B.; et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and brca1-mediated DNA repair. Nat. Genet. 2015, 47, 1294–1303. [Google Scholar] [CrossRef]
- Phillips, K.A.; Collins, I.M.; Milne, R.L.; McLachlan, S.A.; Friedlander, M.; Hickey, M.; Stern, C.; Hopper, J.L.; Fisher, R.; Kannemeyer, G.; et al. Anti-mullerian hormone serum concentrations of women with germline brca1 or brca2 mutations. Hum. Reprod. 2016, 31, 1126–1132. [Google Scholar] [CrossRef]
- Shi, L.; Zhang, J.; Lai, Z.; Tian, Y.; Fang, L.; Wu, M.; Xiong, J.; Qin, X.; Luo, A.; Wang, S. Long-term moderate oxidative stress decreased ovarian reproductive function by reducing follicle quality and progesterone production. PLoS ONE 2016, 11, e0162194. [Google Scholar] [CrossRef]
- Yang, B.; Oo, T.N.; Rizzo, V. Lipid rafts mediate h2o2 prosurvival effects in cultured endothelial cells. FASEB J. 2006, 20, 1501–1503. [Google Scholar] [CrossRef]
- Das, S.; Chattopadhyay, R.; Ghosh, S.; Ghosh, S.; Goswami, S.K.; Chakravarty, B.N.; Chaudhury, K. Reactive oxygen species level in follicular fluid--embryo quality marker in ivf? Hum. Reprod. 2006, 21, 2403–2407. [Google Scholar] [CrossRef]
- Kitagawa, T.; Suganuma, N.; Nawa, A.; Kikkawa, F.; Tanaka, M.; Ozawa, T.; Tomoda, Y. Rapid accumulation of deleted mitochondrial deoxyribonucleic acid in postmenopausal ovaries. Biol. Reprod. 1993, 49, 730–736. [Google Scholar] [CrossRef]
- Keefe, D.L.; Niven-Fairchild, T.; Powell, S.; Buradagunta, S. Mitochondrial deoxyribonucleic acid deletions in oocytes and reproductive aging in women. Fertil. Steril. 1995, 64, 577–583. [Google Scholar] [CrossRef]
- Nogueira, V.; Hay, N. Molecular pathways: Reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin. Cancer Res. 2013, 19, 4309–4314. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Matsuda, S. Redox regulation of tumor suppressor pten in cancer and aging (review). Int. J. Mol. Med. 2013, 31, 511–515. [Google Scholar] [CrossRef]
- Tait, I.S.; Li, Y.; Lu, J. Pten, longevity and age-related diseases. Biomedicines 2013, 1, 17–48. [Google Scholar] [CrossRef]
- Grynberg, M.; Dagher Hayeck, B.; Papanikolaou, E.G.; Sifer, C.; Sermondade, N.; Sonigo, C. Brca1/2 gene mutations do not affect the capacity of oocytes from breast cancer candidates for fertility preservation to mature in vitro. Hum. Reprod. 2019, 34, 374–379. [Google Scholar] [CrossRef]
- Gunnala, V.; Fields, J.; Irani, M.; D’Angelo, D.; Xu, K.; Schattman, G.; Rosenwaks, Z. Brca carriers have similar reproductive potential at baseline to noncarriers: Comparisons in cancer and cancer-free cohorts undergoing fertility preservation. Fertil. Steril. 2019, 111, 363–371. [Google Scholar] [CrossRef]
- Derks-Smeets, I.A.P.; van Tilborg, T.C.; van Montfoort, A.; Smits, L.; Torrance, H.L.; Meijer-Hoogeveen, M.; Broekmans, F.; Dreesen, J.; Paulussen, A.D.C.; Tjan-Heijnen, V.C.G.; et al. Brca1 mutation carriers have a lower number of mature oocytes after ovarian stimulation for ivf/pgd. J. Assist. Reprod. Genet. 2017, 34, 1475–1482. [Google Scholar] [CrossRef]
- Johnson, L.; Sammel, M.D.; Domchek, S.; Schanne, A.; Prewitt, M.; Gracia, C. Antimullerian hormone levels are lower in brca2 mutation carriers. Fertil. Steril. 2017, 107, 1256–1265. [Google Scholar] [CrossRef]
- Van Tilborg, T.C.; Derks-Smeets, I.A.; Bos, A.M.; Oosterwijk, J.C.; van Golde, R.J.; de Die-Smulders, C.E.; van der Kolk, L.E.; van Zelst-Stams, W.A.; Velthuizen, M.E.; Hoek, A.; et al. Serum amh levels in healthy women from brca1/2 mutated families: Are they reduced? Hum. Reprod. 2016, 31, 2651–2659. [Google Scholar] [CrossRef]
- Wang, E.T.; Pisarska, M.D.; Bresee, C.; Chen, Y.D.; Lester, J.; Afshar, Y.; Alexander, C.; Karlan, B.Y. Brca1 germline mutations may be associated with reduced ovarian reserve. Fertil. Steril. 2014, 102, 1723–1728. [Google Scholar] [CrossRef]
- Finch, A.; Valentini, A.; Greenblatt, E.; Lynch, H.T.; Ghadirian, P.; Armel, S.; Neuhausen, S.L.; Kim-Sing, C.; Tung, N.; Karlan, B.; et al. Frequency of premature menopause in women who carry a brca1 or brca2 mutation. Fertil. Steril. 2013, 99, 1724–1728. [Google Scholar] [CrossRef]
- Guglielmino, M.R.; Santonocito, M.; Vento, M.; Ragusa, M.; Barbagallo, D.; Borzi, P.; Casciano, I.; Banelli, B.; Barbieri, O.; Astigiano, S.; et al. Tap73 is downregulated in oocytes from women of advanced reproductive age. Cell Cycle 2011, 10, 3253–3256. [Google Scholar] [CrossRef]
- Pan, H.; Ma, P.; Zhu, W.; Schultz, R.M. Age-associated increase in aneuploidy and changes in gene expression in mouse eggs. Dev. Biol. 2008, 316, 397–407. [Google Scholar] [CrossRef]
- Jang, H.; Na, Y.; Hong, K.; Lee, S.; Moon, S.; Cho, M.; Park, M.; Lee, O.H.; Chang, E.M.; Lee, D.R.; et al. Synergistic effect of melatonin and ghrelin in preventing cisplatin-induced ovarian damage via regulation of foxo3a phosphorylation and binding to the p27(kip1) promoter in primordial follicles. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- Alexandri, C.; Stratopoulou, C.A.; Demeestere, I. Answer to controversy: Mir-10a replacement approaches do not offer protection against chemotherapy-induced gonadotoxicity in mouse model. Int. J. Mol. Sci. 2019, 20, 4958. [Google Scholar] [CrossRef]
- Wang, Y.; Taniguchi, T. Micrornas and DNA damage response: Implications for cancer therapy. Cell Cycle 2013, 12, 32–42. [Google Scholar] [CrossRef]
- Alexandri, C.; Stamatopoulos, B.; Rothe, F.; Bareche, Y.; Devos, M.; Demeestere, I. Microrna profiling and identification of let-7a as a target to prevent chemotherapy-induced primordial follicles apoptosis in mouse ovaries. Sci. Rep. 2019, 9, 9636. [Google Scholar] [CrossRef]
- Dolmans, M.M.; Martinez-Madrid, B.; Gadisseux, E.; Guiot, Y.; Yuan, W.Y.; Torre, A.; Camboni, A.; Van Langendonckt, A.; Donnez, J. Short-term transplantation of isolated human ovarian follicles and cortical tissue into nude mice. Reproduction 2007, 134, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Gavish, Z.; Spector, I.; Peer, G.; Schlatt, S.; Wistuba, J.; Roness, H.; Meirow, D. Follicle activation is a significant and immediate cause of follicle loss after ovarian tissue transplantation. J. Assist. Reprod. Genet. 2018, 35, 61–69. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agents Used/Compounds/Concentration | Mechanism of Action | Species/Methods | Effects on Follicular Growth/Survival | Specific Effects on Granulosa Cells/Oocyte | Study |
|---|---|---|---|---|---|
| 1 and 10 μM Dipotassium bisperoxo (5-hydroxypyridine-2-carboxyl) oxovanadate (V) (bpv(HOpic)) for 24 h/ PTEN inhibitors. | Increase PI3K/Akt | Bovine/ovarian cortical fragments cultured. | Decreases in higher dose. | Compromises DNA damage response (DDR). | [29] |
| 20, 40, 60, 80, 120 and 140 μM diazinon (DZN) | Inhibit PI3K/Akt | Porcine isolated granulosa cells. | Granulosa cells death | Increase DNA damage, mRNA level of Ataxia telangiectasia mutated (ATM), Rad51 and breast cancer susceptibility gene1 (BRCA1) increase p53 leading to granulosa cell death. | [137] |
| 30 μM bpv (HOpic) + 150 μg/mL 740Y-P for 24 and 48 h or 100 nM everolimus. | Increase PI3K/Akt and inhibit mTOR activation respectively | Cryopreserved human ovarian cortical fragments cultured | Lowering the rate of activation improves follicular growth. | PTEN inhibition compromises granulosa cell estradiol production. | [37] |
| Cyclophosphamide (CP) 75 mg, 100 mg, 150 mg per kg body weight and 5 mg/kg body weight per day 1 week before and after CP administration. | PI3K/Akt activation | Mice, in vivo | CP induces non-growing and growing follicle loss. Rapamycin prevents CP induced primordial follicle activation. | Anti-mullerian hormone (AMH) expression decreases after CP exposure. | [133] |
| Transgenic mouse model | Increase PI3K activation in transgenic mice, Cre+ | Transgenic mice, Cre+ and Cre− | Normal secondary follicles, granulosa cell tumour (GCT) in primordial and primary follicles. | Bilateral GCT due to increased activin A. | [115] |
| 440 μM bisphenol A(BPA). | Increases PI3K/Akt activation | Rat ovarian fragment culture exposed to BPA. | BPA induces DNA damage both in oocytes and granulosa cells. PI3K signalling pathway involved in BPA-induced DNA damage. | Primordial follicle is activated to replace the larger follicle depletion. | [138] |
| Transgenic mouse model | Increase PI3K activation in transgenic mice, Cre+ | Transgenic mice, Cre+ and Cre− | Increases follicles survival | Asynchronous oocytes and granulosa cells growth. | [114] |
| 100 μM bpv(HOpic) for 25 h | Increase PI3K/Akt activation | Human ovarian cortical fragments cultured. | No damage to the follicular growth. | Enhance estradiol production without any damage to follicles compared to control group. | [33] |
| 200 μM phosphatidic acid (PA) and 50 μM propranolol (PRO) for 24 h in mice; bpv(HOpic)(100 μM) /740Y-P (250 μM /mL) for 24 h, 740Y-P (250 μM /mL) only for another 24 h; PA (100 mM)/740Y-P (200 μM)/PRO (50 μM) for 24 h in human. | Increase PI3K/Akt /mTOR activation. | Mice and human ovaries transiently incubated in mTOR activators followed by grafting into female mice. | No damage to the follicular growth. | NA | [139] |
| 30 μM of bpv(HOpic), and 150 μM /mL of 740YP for 24 h followed by incubation with 740YP alone for another 24 h | Increase PI3K/Akt activation. | Human ovarian cortical fragments transplantation following in vitro activation (IVA). | Autografting of ovarian fragments following in vitro activation (IVA) procedure to infertility related primary ovarian insufficiency (POI) patients. | NA | [34] |
| 1 μM bpv(HOpic) and 10 and 100 μM bpv(HOpic) (unpublished) | Increase PI3K/Akt activation | Human ovarian cortical fragments and isolated preantral follicle culture. | Higher dose compromises follicular growth. The lower dose is associated with deleterious effects on subsequent growth of preantral follicles. | NA | [36] |
| Cisplatin, once daily at doses of 0.5, 1.0, 1.5 and 2.0 mg/ kg for 5 to 14 days | Activation of PI3K/Akt | Intraperitoneal injection of cisplatin in mice | Increases the proportion of growing follicles. | Induces ovarian failure. | [23] |
| 100 μM bpv(HOpic) and 500 μM /mL 740Y-P for 24 and 48 h | Increase PI3K/Akt activation. | Human ovarian cortical fragments cultured in polyethylene glycol (PEG)-fibrinogen hydrogels. | Compromises follicle survival. | NA | [35] |
| 30 μM bpv(HOpic) and 150 μg/mL 740YP for 24 h | Increase PI3K/Akt activation. | Mice ovarian transplantation and human ovarian fragments transplantation following IVA. | Promotes primordial follicle activation both in mice and human. | NA | [113] |
| Female mice deficient in PTEN | Increase PI3K/Akt activation | PTEN knockout mice | Rapamycin reduces the primordial follicles activation in PTEN knockout mice. | Rapamycin prevents global primordial follicles activation induced by the absence of PTEN. | [134] |
| 1 μM bpv(HOpic) for 24 h | Increase PI3K/Akt activity. | Mice cortical fragments IVA followed by transplantation and bpv(HOpic) directly injected to female mice. | Does not compromise follicular health. | More mature and fertilised oocytes in PTEN inhibition group. | [32] |
| 100 μM bpv(HOpic) and/or 500 μg/mL 740Y-P for 48 h or bpv(HOpic) plus 740Y-P together with the Akt inhibitor SH-550 μM or the PI3K inhibitor Wortmannin 25 μM. | Increase PI3K/Akt, Akt inhibitor decreases the activation. | Mice and human cortical fragments incubated in Akt activators followed by xenografting. | Increases in the number of secondary and antral stage follicles following xenografting and does not affect follicular health. | No malignancy observed after long term ovarian transplantation. | [140] |
| Mice lacking Tuberous sclerosis complex 1 (TSC1), PTEN; TSC1 and PTEN; Phosphatidylinositol-dependent kinase 1 (PDK1) and PDK1 and TSC1 in oocytes. | Enhances mTOR activation. | Mutant female mice | Degenerated activated primordial follicles (short term), diminished follicular health (long term). | Rapamycin prevents global primordial follicle activation. Activation does not cause tumour development. | [100] |
| Homozygous mutant female mice deficient Tuberous sclerosis complex 2 ( TSC2) in oocytes. | Enhances mTOR activation. | Mutant female mice. | Massive primordial follicle activation. | Depletion of follicle reserve. | [93] |
| Female mice lacking PTEN, PDK1 and r ibosomal protein S6 kinase (rpS6) | Increases PI3K/Akt | Mutant female mice. | Follicles with degenerating oocytes in PDK1 deletion and enlarged oocytes in PTEN deletion. | The absence of PTEN causes Primary Ovarian Insufficiency (POI) that can be reversed by PDK1 deletion. | [17] |
| PTEN deletion in mice. | Increases PI3K/Akt | PTEN mutant mice | Tends to be normal follicle morphology but with enlarged oocytes and flattened granulosa cells. | PTEN deletion leads to excessive primordial follicle activation. | [18] |
| Study Focus | Study Type | DDR Pathway Affected | Main Outcomes | References |
|---|---|---|---|---|
| Oocyte maturation rate of breast cancer patient with breast cancer susceptibility gene 1 (BRCA1) and Breast cancer 2 (BRCA2) mutation. | Retrospective cohort study. | BRCA1 and BRCA2 | The number of mature oocytes resulted from in vitro maturation (IVM) procedure is not different between women with BRCA1 and without BRCA mutation. | [158] |
| Ovarian reserve of patients with BRCA mutation carriers and non-carriers with or without malignancy. | Retrospective cohort study. | BRCA1 and BRCA2 | Patients with BRCA mutation carriers and noncarriers show comparable ovarian reserve and number of oocytes yield following ovarian stimulation. | [159] |
| The role of BRCA2 in ovarian development and puberty onset. | A case control study in human. | BRCA2 and Rad51 | Lack of BRCA2 reduces Rad51 recruitment during homologous recombination. | [147] |
| Ovarian reserve in patients with BRCA1 mutation. | Case-control study. | γH2AX, BRCA1 and BRCA2 | DNA double-strand breaks (DSBs) increase in BRCA1 mutation group but not BRCA2. DNA damage increases with age in BRCA1/2 mutation. | [25] |
| Oocyte yield following ovarian stimulation in patients with BRCA1/2 mutation. | Retrospective cohort study. | BRCA1 and BRCA2 | The number of oocytes produced by women with BRCA mutation is lower than without BRCA1 mutation. | [160] |
| DNA damage and repair capacity of aged and young buffalo ovaries. | Experimental study in buffalo ovaries. | BRCA1, γH2AX, MRE11, Rad51 and ATM | mRNA expression of BRCA1, meiotic recombination 11 (MRE11), Rad51 and ataxia telangiectasia mutated (ATM) decline significantly in aged buffalo ovaries. | [58] |
| The effects of BRCA mutation on anti-Mullerian hormone (AMH) serum level. | Prospective cohort study | BRCA1 and BRCA2 | Patients with BRCA2 mutations exhibit a lower AMH level compare to low-risk control patients. | [161] |
| Anti-mullerian hormone (AMH) serum level in patients with BRCA1/2 mutation. | Cross-sectional study | BRCA1 and BRCA2 | AMH serum level of patients with BRCA1/2 mutation carriers does not significantly different from non-carriers. | [162] |
| AMH serum level in women with BRCA1 and BRCA2 mutation. | Cross-sectional study | BRCA1 and BRCA2 | BRCA1 but not in BRCA2 mutation carriers have a lower AMH level. | [149] |
| Ovarian ageing effects on DNA damage repair response in rat ovaries. | Experimental | γH2AX, BRCA1, MRE11, Rad51, ATM, BRCA1 and BRCA2 | DNA repair proteins BRCA1, Rad51, ATM and γH2AX in aged rat primordial follicles declined compared to immature rats. | [19] |
| Comparison of proteins profile of primordial follicles isolated from immature rat and aged rat. | Experimental | Heat shock cognate 71kDa (Hsp71C), calreticulin, Bcl-2-related ovarian killer protein (BOK) | Protein expression for DSBs response decreases significantly in aged rats. | [59] |
| The association between DNA DSBs in granulosa cells and ageing. | Experimental | γH2AX, BRCA1, Telomeric repeat binding factor (TRF2) | Increased γH2AX and decreased BRCA1 expression in all follicle types with age. | [22] |
| The association between AMH serum level and BRCA mutation. | Cross-sectional study | BRCA1 and BRCA2 | AMH serum level of patients with a BRCA1 mutation is lower than without BRCA1 mutation. | [163] |
| The effect of ovarian ageing on DNA DSBs of oocytes and granulosa cells. | Experimental | γH2AX, BRCA1, MRE11, Rad51, ATM, BRCA1 and BRCA2 | Increased DNA damage and decreased DDR capacity with advancing age. | [21] |
| Time to menopause in BRCA1 and 2 mutations carriers. | Case control study. | BRCA1 and BRCA2 | Both BRCA1 and 2 mutation patients experience menopause earlier than control. | [164] |
| Doxorubicin effects on ovarian ageing. | Experimental | γH2AX, ATM and activated caspase 3 | γH2AX expression is higher in ovarian tissue exposed to doxorubicin in vitro. | [118] |
| Transactivation p73 (TAp73) expression in young and aged female oocytes. | Experimental | TAp73 | TAp73 is downregulated in older women’s oocytes. | [165] |
| The effects of age on the occurrence of aneuploidy in mouse oocytes. | Experimental | BRCA1 | BRCA1 expression is decreased in oocytes of aged mice. Aneuploidy increases in aged oocytes. | [166] |
| The role of BRCA2 in male and female gametogenesis. | Experimental | BRCA2 | BRCA2 deficiency in mice leads to infertility. | [146] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maidarti, M.; Anderson, R.A.; Telfer, E.E. Crosstalk between PTEN/PI3K/Akt Signalling and DNA Damage in the Oocyte: Implications for Primordial Follicle Activation, Oocyte Quality and Ageing. Cells 2020, 9, 200. https://doi.org/10.3390/cells9010200
Maidarti M, Anderson RA, Telfer EE. Crosstalk between PTEN/PI3K/Akt Signalling and DNA Damage in the Oocyte: Implications for Primordial Follicle Activation, Oocyte Quality and Ageing. Cells. 2020; 9(1):200. https://doi.org/10.3390/cells9010200
Chicago/Turabian StyleMaidarti, Mila, Richard A. Anderson, and Evelyn E. Telfer. 2020. "Crosstalk between PTEN/PI3K/Akt Signalling and DNA Damage in the Oocyte: Implications for Primordial Follicle Activation, Oocyte Quality and Ageing" Cells 9, no. 1: 200. https://doi.org/10.3390/cells9010200
APA StyleMaidarti, M., Anderson, R. A., & Telfer, E. E. (2020). Crosstalk between PTEN/PI3K/Akt Signalling and DNA Damage in the Oocyte: Implications for Primordial Follicle Activation, Oocyte Quality and Ageing. Cells, 9(1), 200. https://doi.org/10.3390/cells9010200