Glutamine Synthetase 1 Increases Autophagy Lysosomal Degradation of Mutant Huntingtin Aggregates in Neurons, Ameliorating Motility in a Drosophila Model for Huntington’s Disease

 , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Fly Husbandry and Lines
2.2. Survival at Eclosure Analysis
2.3. Motility Assays
2.4. Quantification of Fluorescence (GFP) on the Adult Compound Eye
2.5. Western Blot
2.6. Filter Trap Assay
2.7. Patient-Derived Cell Lines
2.8. MS/HPLC for Glutamate and Glutamine Quantification and Analysis of Amino Acids
2.9. Quantitative Real Time PCR
2.10. Quantification of GS1 Activity
2.11. Immunofluorescences and Quantification of the Size of Htt-Q93 Aggregates
2.12. Statistical Analysis
2.13. Autophagy Analysis
3. Results
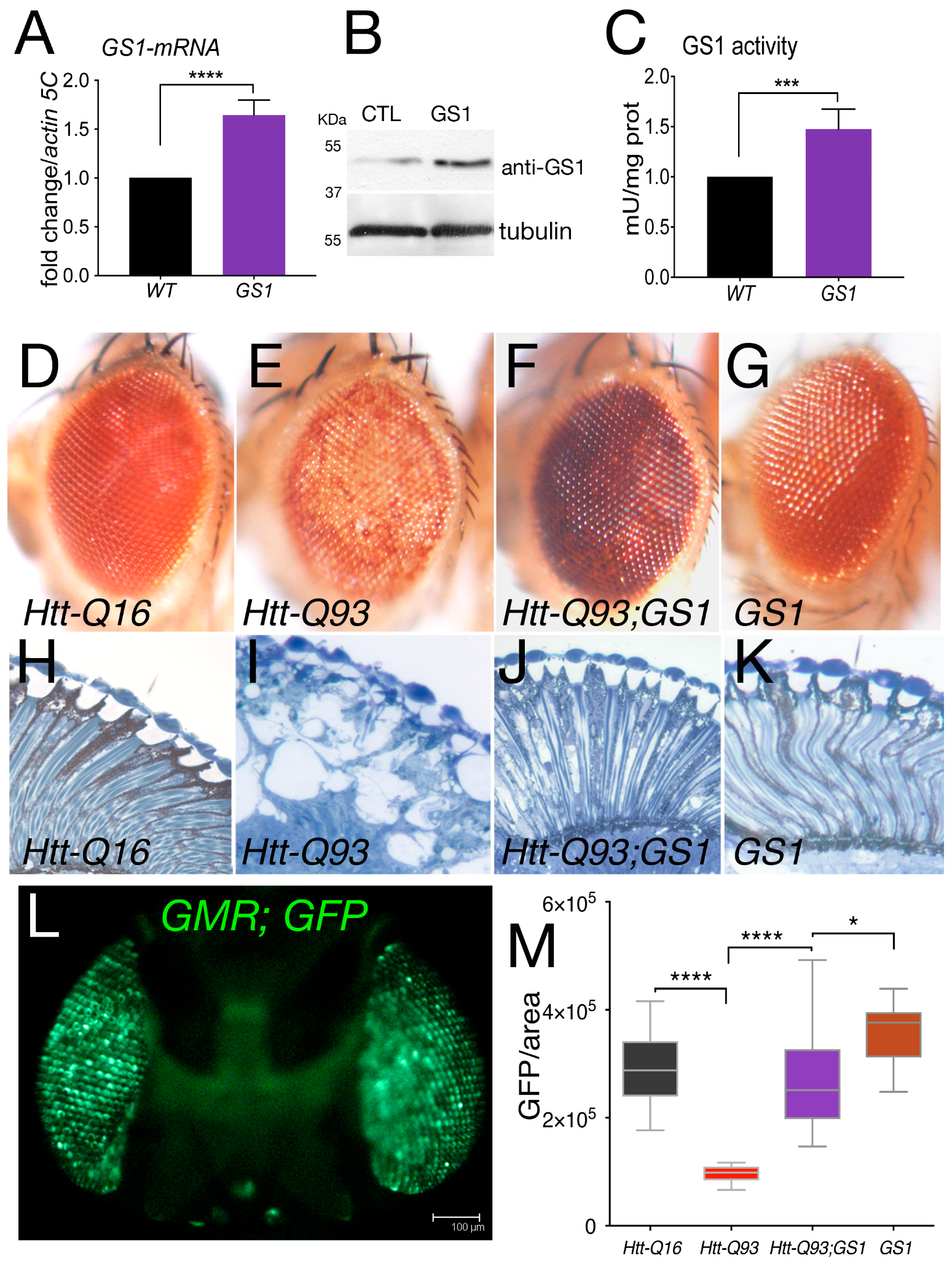
3.1. GS1 Rescues Neuronal Death Induced by Expression of Htt-Q93 in the Retina
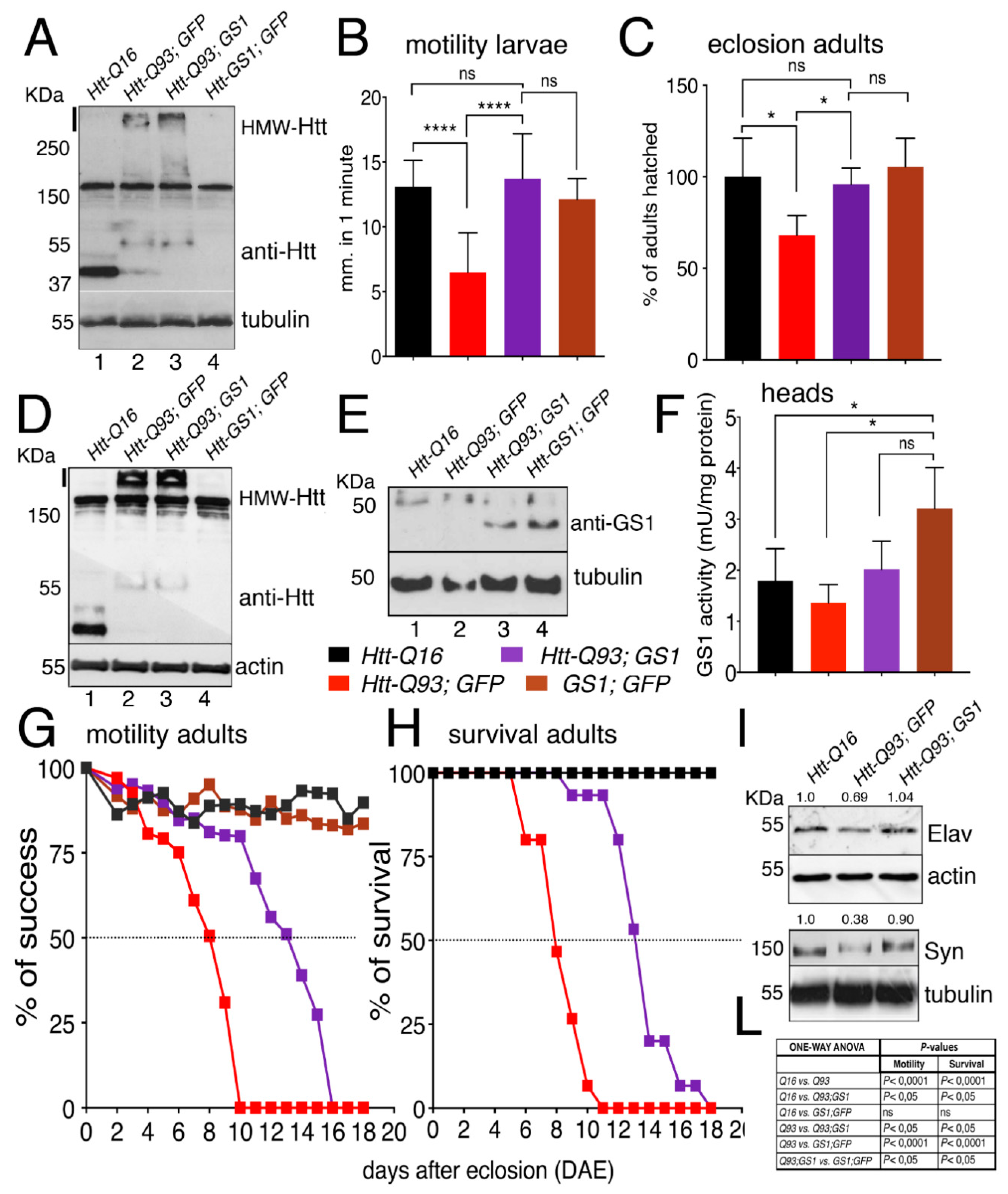
3.2. GS1 Expression in Neurons Improves Locomotion Defects of Htt-Q93 Animals
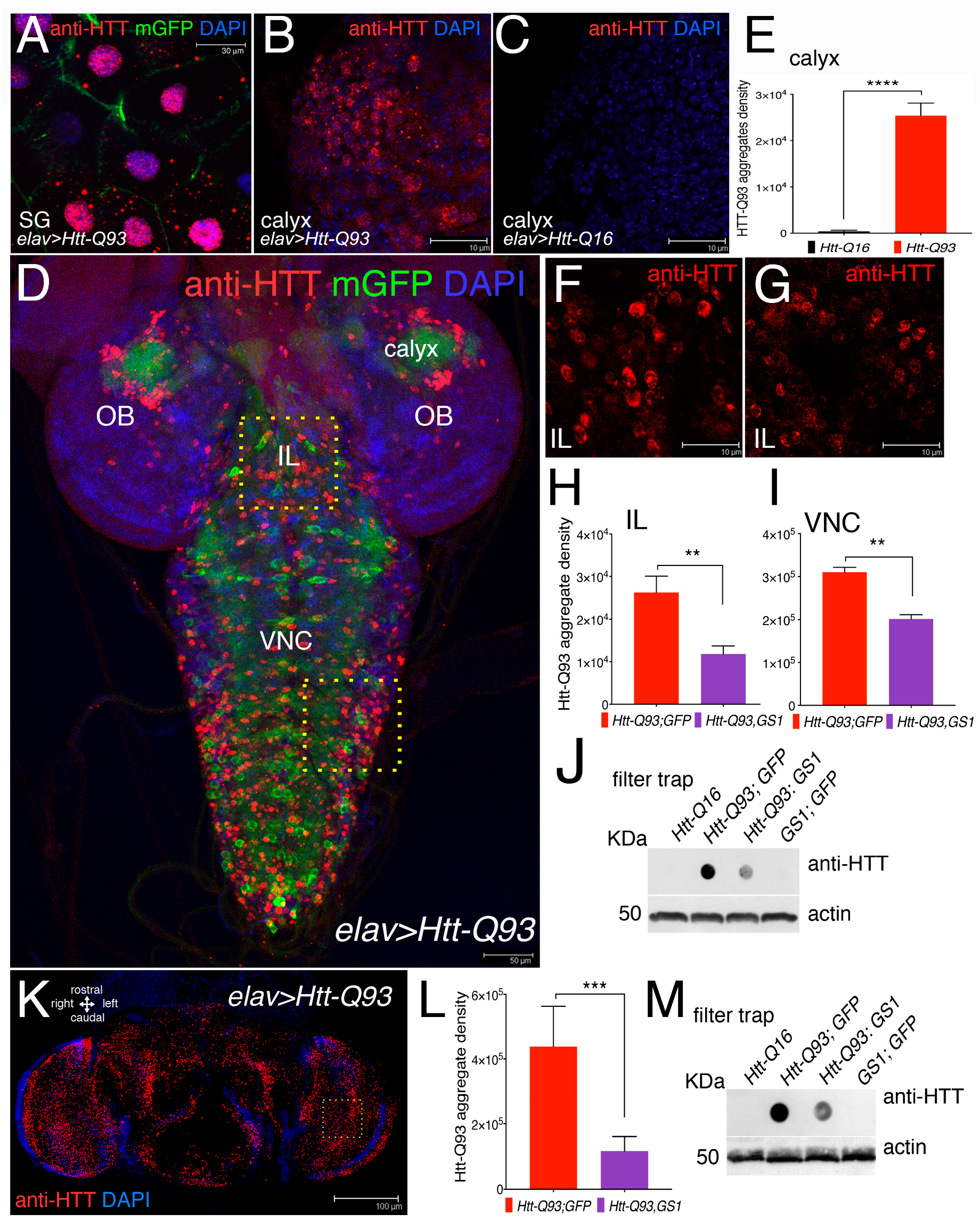
3.3. Expression of GS1 Reduces Htt-Q93 Protein Aggregates in Neurons
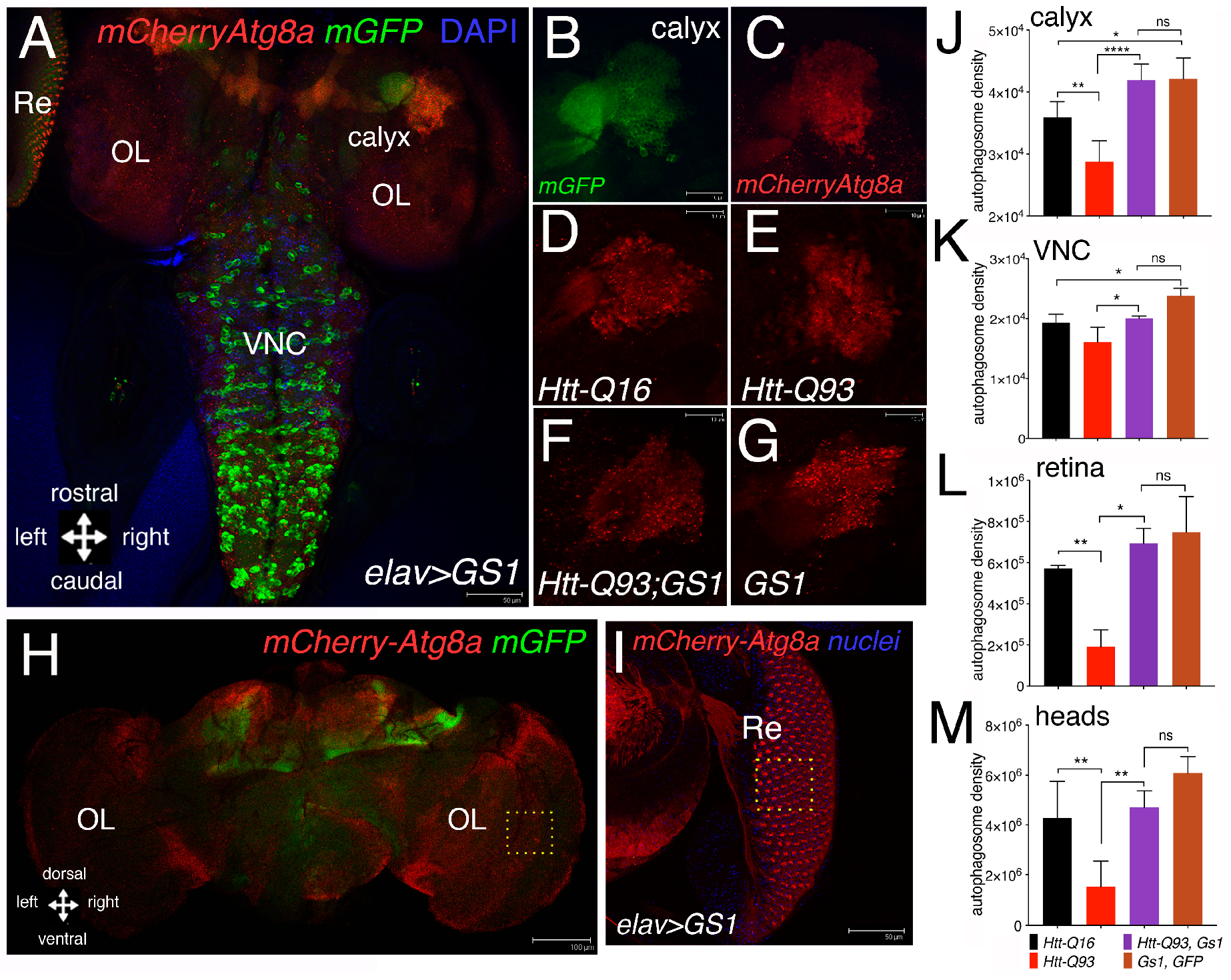
3.4. GS1 Induces Autophagy in Neurons
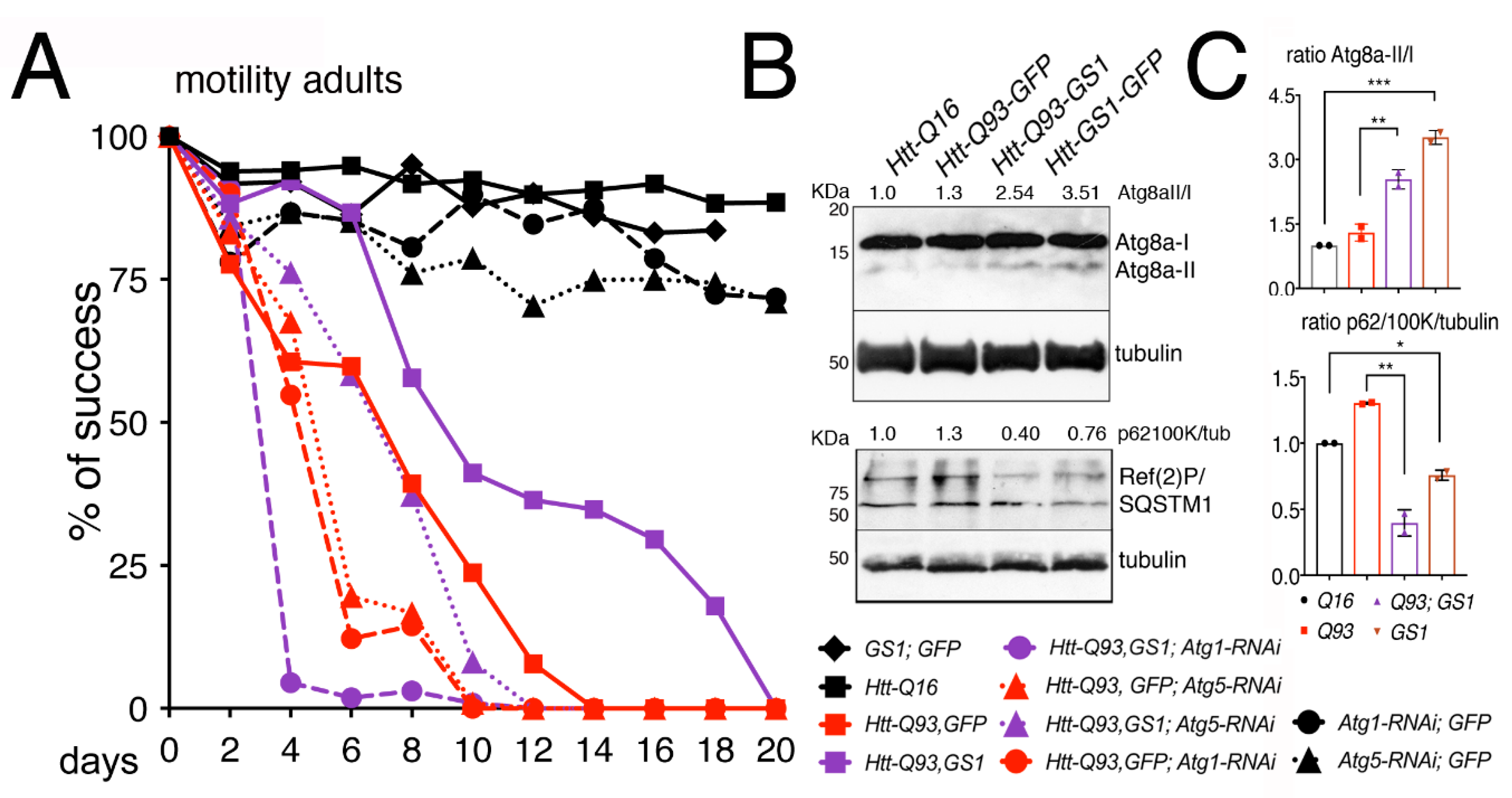
3.5. GS1 Rescue of Htt-Q93 Motility-Defect Depends on the Full Expression of Atg1 and Atg5
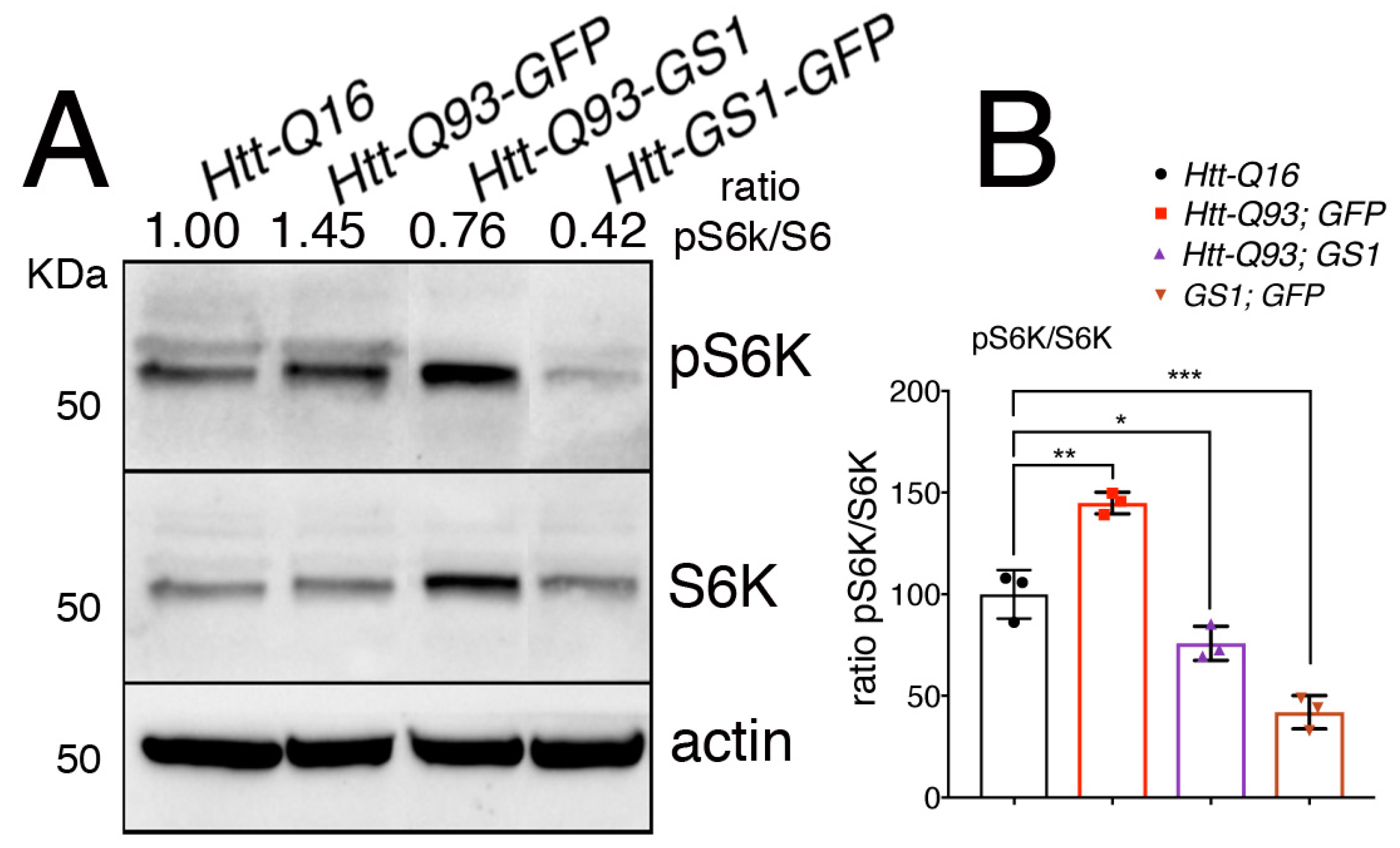
3.6. Expression of GS1 Reduces TOR Activation Visible by Phosphorylation of Its Substrate S6K
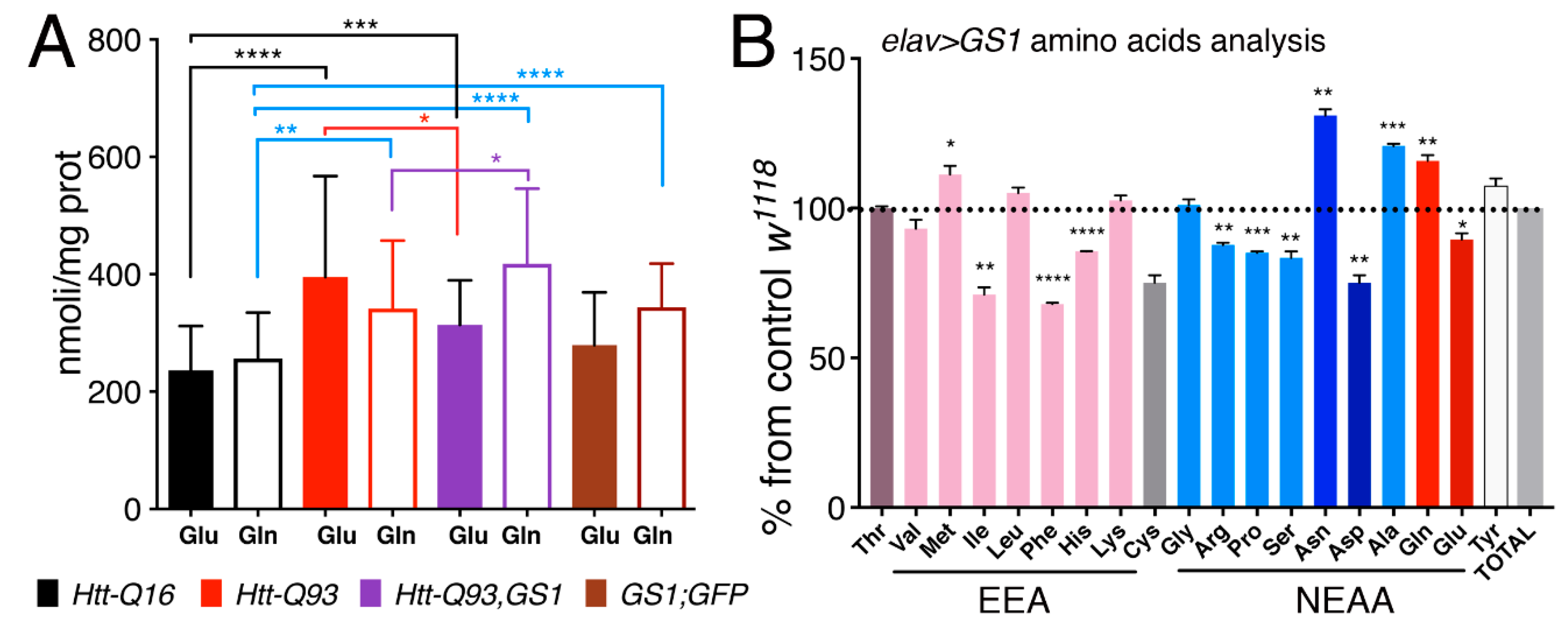
3.7. Expression of GS1 in Neurons affects Glutamate and Glutamine Levels and Reduces Essential Amino Acids
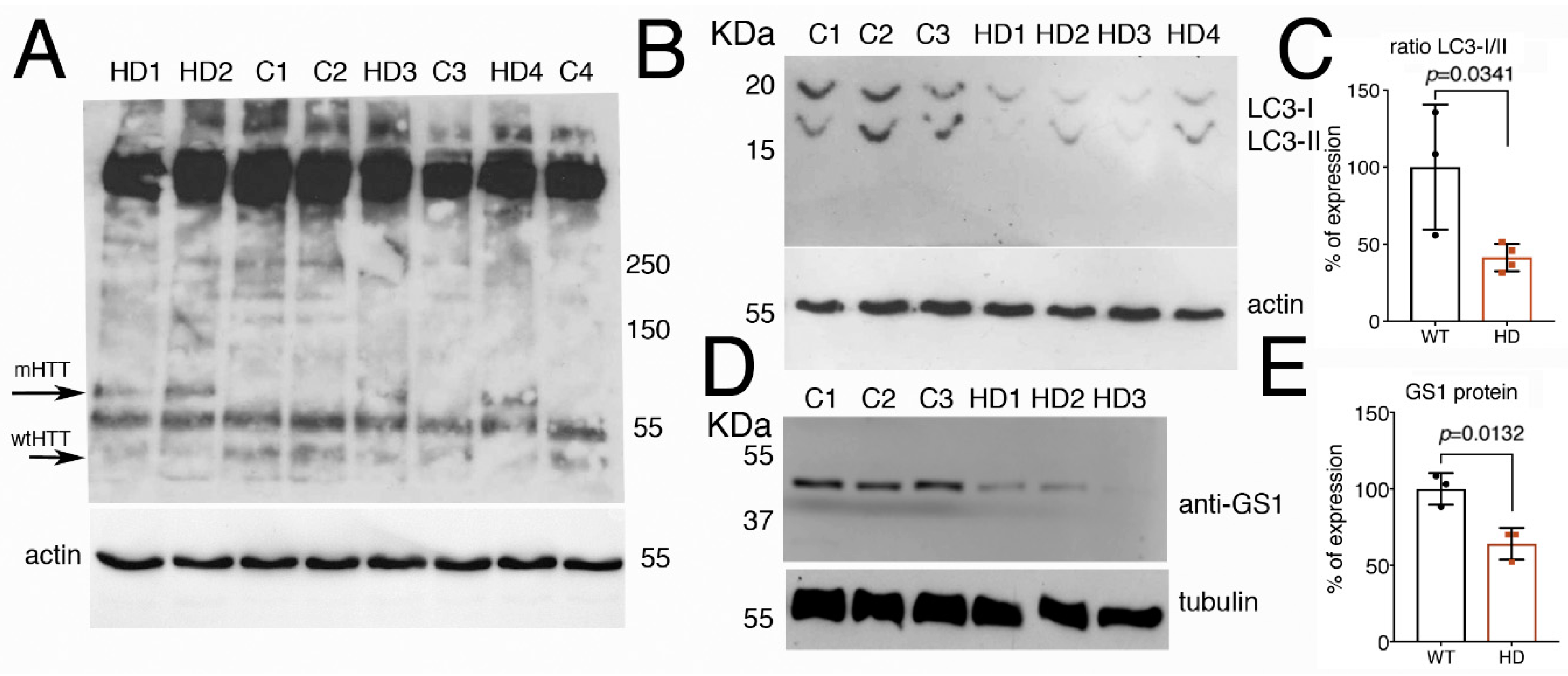
3.8. Fibroblasts from Patients with Huntington’s Disease Have Impaired Autophagy and Reduced Level of GS1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Genetic Modifiers of Huntington’s Disease Consortium. Cag repeat not polyglutamine length determines timing of huntington’s disease onset. Cell 2019, 178, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Spodenkiewicz, M.; Diez-Fernandez, C.; Rufenacht, V.; Gemperle-Britschgi, C.; Haberle, J. Minireview on glutamine synthetase deficiency, an ultra-rare inborn error of amino acid biosynthesis. Biology 2016, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin lowering strategies for disease modification in huntington’s disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Fullgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and neurodegeneration: Pathogenic mechanisms and therapeutic opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Bento, C.F.; Ricketts, T.; Vicinanza, M.; Siddiqi, F.; Pavel, M.; Squitieri, F.; Hardenberg, M.C.; Imarisio, S.; Menzies, F.M.; et al. Polyglutamine tracts regulate autophagy. Autophagy 2017, 13, 1613–1614. [Google Scholar] [CrossRef]
- Wang, T.; Lao, U.; Edgar, B.A. Tor-mediated autophagy regulates cell death in drosophila neurodegenerative disease. J. Cell Biol. 2009, 186, 703–711. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mtor induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. Mtor signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C. The glutamate-glutamine cycle is not stoichiometric: Fates of glutamate in brain. J. Neurol. Res. 2007, 85, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, A.R.; Norenberg, M.D. Glutamine synthetase: Role in neurological disorders. Adv. Neurobiol. 2016, 13, 327–350. [Google Scholar] [PubMed]
- Butterworth, J. Changes in nine enzyme markers for neurons, glia, and endothelial cells in agonal state and huntington’s disease caudate nucleus. J. Neurochem. 1986, 47, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.J. Glutamine synthetase and fructose-1, 6-diphosphatase activity in the putamen of control and huntington’s disease brain post mortem. Life Sci. 1983, 32, 1949–1955. [Google Scholar] [CrossRef]
- Carter, C.J. Glutamine synthetase activity in huntington’s disease. Life Sci. 1982, 31, 1151–1159. [Google Scholar] [CrossRef]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in drosophila. Nature 2001, 413, 739–743. [Google Scholar] [CrossRef]
- Caizzi, R.; Bozzetti, M.P.; Caggese, C.; Ritossa, F. Homologous nuclear genes encode cytoplasmic and mitochondrial glutamine synthetase in drosophila melanogaster. J. Mol. Biol. 1990, 212, 17–26. [Google Scholar] [CrossRef]
- Lipton, J.O.; Sahin, M. The neurology of mtor. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of tsc2 is a universal response to cellular stress. Nat. Commu. 2016, 7, 10662. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Phosphorylation and signal transduction pathways in translational control. Cold Spring Harbor Perspect. Biol. 2018, 11, a033050. [Google Scholar] [CrossRef] [PubMed]
- Junger, M.A.; Rintelen, F.; Stocker, H.; Wasserman, J.D.; Vegh, M.; Radimerski, T.; Greenberg, M.E.; Hafen, E. The drosophila forkhead transcription factor foxo mediates the reduction in cell number associated with reduced insulin signaling. J. Biol. 2003, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Feany, M.B.; Saraswati, S.; Littleton, J.T.; Perrimon, N. Inactivation of drosophila huntingtin affects long-term adult functioning and the pathogenesis of a huntington’s disease model. Dis. Models Mech. 2009, 2, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, J.C.; Kuroda, M.I. Dosage compensation in drosophila. Cold Spring Harbor Perspect. Biol. 2015, 7, a019398. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Yamamoto, A.; Oshitani-Okamoto, S.; Ohsumi, Y.; Yoshimori, T. Lc3, gabarap and gate16 localize to autophagosomal membrane depending on form-ii formation. J. Cell Sci. 2004, 117, 2805–2812. [Google Scholar] [CrossRef]
- Bhukel, A.; Beuschel, C.B.; Maglione, M.; Lehmann, M.; Juhasz, G.; Madeo, F.; Sigrist, S.J. Autophagy within the mushroom body protects from synapse aging in a non-cell autonomous manner. Nat. Commu. 2019, 10, 1318. [Google Scholar] [CrossRef]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mole. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef]
- Hulf, T.; Bellosta, P.; Furrer, M.; Steiger, D.; Svensson, D.; Barbour, A.; Gallant, P. Whole-genome analysis reveals a strong positional bias of conserved dmyc-dependent e-boxes. Mol. Cell. Biol. 2005, 25, 3401–3410. [Google Scholar] [CrossRef]
- Parisi, F.; Riccardo, S.; Zola, S.; Lora, C.; Grifoni, D.; Brown, L.M.; Bellosta, P. Dmyc expression in the fat body affects dilp2 release and increases the expression of the fat desaturase desat1 resulting in organismal growth. Dev. Biol. 2013, 379, 64–75. [Google Scholar] [CrossRef]
- Vorhaben, J.E.; Wong, L.; Campbell, J.W. Assay for glutamine synthetase activity. Biochem. J. 1973, 135, 893–896. [Google Scholar] [CrossRef] [PubMed]
- Caizzi, R.; Ritossa, F. The enzyme glutamine synthetase i of drosophila melanogaster is associated with a modified rna. Biochem. Genet. 1983, 21, 267–285. [Google Scholar] [CrossRef] [PubMed]
- De Pinto, V.; Caggese, C.; Prezioso, G.; Ritossa, F. Purification of the glutamine synthetase ii isozyme of drosophila melanogaster and structural and functional comparison of glutamine synthetases i and ii. Biochem. Genet. 1987, 25, 821–836. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.L.; Walker, H.; Theisen, H.; Zhu, Y.Z.; Fielder, T.; Purcell, J.; Thompson, L.M. Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in drosophila. Hum. Mol. Genet. 2000, 9, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Binari, R.; Zhou, R.; Perrimon, N. A genomewide rna interference screen for modifiers of aggregates formation by mutant huntingtin in drosophila. Genetics 2010, 184, 1165–1179. [Google Scholar] [CrossRef]
- Krench, M.; Littleton, J.T. Neurotoxicity pathways in drosophila models of the polyglutamine disorders. Curr. Top. Dev. Biol. 2017, 121, 201–223. [Google Scholar]
- Slobodkin, M.R.; Elazar, Z. The atg8 family: Multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013, 55, 51–64. [Google Scholar]
- Nezis, I.P.; Lamark, T.; Velentzas, A.D.; Rusten, T.E.; Bjorkoy, G.; Johansen, T.; Papassideri, I.S.; Stravopodis, D.J.; Margaritis, L.H.; Stenmark, H.; et al. Cell death during drosophila melanogaster early oogenesis is mediated through autophagy. Autophagy 2009, 5, 298–302. [Google Scholar] [CrossRef]
- Neufeld, T.P. Contribution of atg1-dependent autophagy to tor-mediated cell growth and survival. Autophagy 2007, 3, 477–479. [Google Scholar] [CrossRef]
- Nagy, P.; Varga, A.; Kovacs, A.L.; Takats, S.; Juhasz, G. How and why to study autophagy in drosophila: It’s more than just a garbage chute. Methods 2015, 75, 151–161. [Google Scholar] [CrossRef]
- Nezis, I.P.; Simonsen, A.; Sagona, A.P.; Finley, K.; Gaumer, S.; Contamine, D.; Rusten, T.E.; Stenmark, H.; Brech, A. Ref(2)p, the drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J. Cell Biol. 2008, 180, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mtorc1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Radimerski, T.; Montagne, J.; Rintelen, F.; Stocker, H.; van der Kaay, J.; Downes, C.P.; Hafen, E.; Thomas, G. Ds6k-regulated cell growth is dpkb/dpi(3)k-independent, but requires dpdk1. Nat. Cell Biol. 2002, 4, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Vandekeere, S.; Kalucka, J.; Bierhansl, L.; Zecchin, A.; Bruning, U.; Visnagri, A.; Yuldasheva, N.; Goveia, J.; Cruys, B.; et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017, 36, 2334–2352. [Google Scholar] [CrossRef]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in huntington’s disease: From mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef]
- Persichetti, F.; Carlee, L.; Faber, P.W.; McNeil, S.M.; Ambrose, C.M.; Srinidhi, J.; Anderson, M.; Barnes, G.T.; Gusella, J.F.; MacDonald, M.E. Differential expression of normal and mutant huntington’s disease gene alleles. Neurobiol. Dis. 1996, 3, 183–190. [Google Scholar] [CrossRef]
- Gafni, J.; Ellerby, L.M. Calpain activation in huntington’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 4842–4849. [Google Scholar] [CrossRef]
- Lievens, J.C.; Rival, T.; Iche, M.; Chneiweiss, H.; Birman, S. Expanded polyglutamine peptides disrupt egf receptor signaling and glutamate transporter expression in drosophila. Hum. Mol. Genet. 2005, 14, 713–724. [Google Scholar] [CrossRef]
- Bradford, J.; Shin, J.Y.; Roberts, M.; Wang, C.E.; Li, X.J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc. Natl. Acad. Sci. USA 2009, 106, 22480–22485. [Google Scholar] [CrossRef]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the huntington mutation. J. Neurol. 2002, 125, 1908–1922. [Google Scholar] [CrossRef] [PubMed]
- Rose, C.F.; Verkhratsky, A.; Parpura, V. Astrocyte glutamine synthetase: Pivotal in health and disease. Biochem. Soc. Trans. 2013, 41, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Dis. 2018, 17, 660–688. [Google Scholar] [CrossRef] [PubMed]
- Metzger, S.; Saukko, M.; Van Che, H.; Tong, L.; Puder, Y.; Riess, O.; Nguyen, H.P. Age at onset in huntington’s disease is modified by the autophagy pathway: Implication of the v471a polymorphism in atg7. Hum. Genet. 2010, 128, 453–459. [Google Scholar] [CrossRef]
- Komatsu, M.; Wang, Q.J.; Holstein, G.R.; Friedrich, V.L., Jr.; Iwata, J.; Kominami, E.; Chait, B.T.; Tanaka, K.; Yue, Z. Essential role for autophagy protein atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc. Natl. Acad. Sci. USA 2007, 104, 14489–14494. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Kim, M.; Sandford, E.; Gatica, D.; Qiu, Y.; Liu, X.; Zheng, Y.; Schulman, B.A.; Xu, J.; Semple, I.; Ro, S.H.; et al. Mutation in atg5 reduces autophagy and leads to ataxia with developmental delay. eLife 2016, 5, e12245. [Google Scholar] [CrossRef]
- Juhasz, G.; Erdi, B.; Sass, M.; Neufeld, T.P. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in drosophila. Genes Dev. 2007, 21, 3061–3066. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Sabatini, D.M. The dawn of the age of amino acid sensors for the mtorc1 pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Differential regulation of mtorc1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mtor and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Hoffman, G.R.; Poulogiannis, G.; Buel, G.R.; Jang, Y.J.; Lee, K.W.; Kim, B.Y.; Erikson, R.L.; Cantley, L.C.; Choo, A.Y.; et al. Metabolic stress controls mtorc1 lysosomal localization and dimerization by regulating the ttt-ruvbl1/2 complex. Mol. Cell 2013, 49, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mtorc1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Lysosomal amino acid transporter slc38a9 signals arginine sufficiency to mtorc1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef]
- van der Vos, K.E.; Eliasson, P.; Proikas-Cezanne, T.; Vervoort, S.J.; van Boxtel, R.; Putker, M.; van Zutphen, I.J.; Mauthe, M.; Zellmer, S.; Pals, C.; et al. Modulation of glutamine metabolism by the pi(3)k-pkb-foxo network regulates autophagy. Nat. Cell Biol. 2012, 14, 829–837. [Google Scholar] [CrossRef]
- Yelamanchi, S.D.; Jayaram, S.; Thomas, J.K.; Gundimeda, S.; Khan, A.A.; Singhal, A.; Keshava Prasad, T.S.; Pandey, A.; Somani, B.L.; Gowda, H. A pathway map of glutamate metabolism. J. Cell Commun. Signal 2016, 10, 69–75. [Google Scholar] [CrossRef]
- Wakabayashi, Y.; Iwashima, A.; Yamada, E.; Yamada, R. Enzymological evidence for the indispensability of small intestine in the synthesis of arginine from glutamate. Ii. N-acetylglutamate synthase. Arch. Biochem. Biophys. 1991, 291, 9–14. [Google Scholar] [CrossRef]
- Murphy, J.M.; Murch, S.J.; Ball, R.O. Proline is synthesized from glutamate during intragastric infusion but not during intravenous infusion in neonatal piglets. J. Nutr. 1996, 126, 878–886. [Google Scholar] [CrossRef]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. Mtorc1 activator slc38a9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 2017, 171, 642–654. [Google Scholar] [CrossRef]
- Featherstone, D.E. Glial solute carrier transporters in drosophila and mice. Glia 2011, 59, 1351–1363. [Google Scholar] [CrossRef]
- Romero-Calderon, R.; Shome, R.M.; Simon, A.F.; Daniels, R.W.; DiAntonio, A.; Krantz, D.E. A screen for neurotransmitter transporters expressed in the visual system of drosophila melanogaster identifies three novel genes. Dev. Neurobiol. 2007, 67, 550–569. [Google Scholar] [CrossRef] [PubMed]
- Limmer, S.; Weiler, A.; Volkenhoff, A.; Babatz, F.; Klambt, C. The drosophila blood-brain barrier: Development and function of a glial endothelium. Front. Neurosci. 2014, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, B.; Erickson, J.D. Sodium-coupled neutral amino acid (system n/a) transporters of the slc38 gene family. Pflugers Arch. 2004, 447, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.L.; Andring, J.T.; McKenna, R.; Kilberg, M.S. Asparagine synthetase: Function, structure, and role in disease. J. Biol. Chem. 2017, 292, 19952–19958. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Morales, I.; Sabate, M.; Rodriguez, M. Striatal glutamate induces retrograde excitotoxicity and neuronal degeneration of intralaminar thalamic nuclei: Their potential relevance for parkinson’s disease. Eur. J. Neurosci. 2013, 38, 2172–2182. [Google Scholar] [CrossRef]
- Plaitakis, A.; Shashidharan, P. Glutamate transport and metabolism in dopaminergic neurons of substantia nigra: Implications for the pathogenesis of parkinson’s disease. J. Neurol. 2000, 247, II25–II35. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vernizzi, L.; Paiardi, C.; Licata, G.; Vitali, T.; Santarelli, S.; Raneli, M.; Manelli, V.; Rizzetto, M.; Gioria, M.; Pasini, M.E.; et al. Glutamine Synthetase 1 Increases Autophagy Lysosomal Degradation of Mutant Huntingtin Aggregates in Neurons, Ameliorating Motility in a Drosophila Model for Huntington’s Disease. Cells 2020, 9, 196. https://doi.org/10.3390/cells9010196
Vernizzi L, Paiardi C, Licata G, Vitali T, Santarelli S, Raneli M, Manelli V, Rizzetto M, Gioria M, Pasini ME, et al. Glutamine Synthetase 1 Increases Autophagy Lysosomal Degradation of Mutant Huntingtin Aggregates in Neurons, Ameliorating Motility in a Drosophila Model for Huntington’s Disease. Cells. 2020; 9(1):196. https://doi.org/10.3390/cells9010196
Chicago/Turabian StyleVernizzi, Luisa, Chiara Paiardi, Giusimaria Licata, Teresa Vitali, Stefania Santarelli, Martino Raneli, Vera Manelli, Manuela Rizzetto, Mariarosa Gioria, Maria E. Pasini, and et al. 2020. "Glutamine Synthetase 1 Increases Autophagy Lysosomal Degradation of Mutant Huntingtin Aggregates in Neurons, Ameliorating Motility in a Drosophila Model for Huntington’s Disease" Cells 9, no. 1: 196. https://doi.org/10.3390/cells9010196
APA StyleVernizzi, L., Paiardi, C., Licata, G., Vitali, T., Santarelli, S., Raneli, M., Manelli, V., Rizzetto, M., Gioria, M., Pasini, M. E., Grifoni, D., Vanoni, M. A., Gellera, C., Taroni, F., & Bellosta, P. (2020). Glutamine Synthetase 1 Increases Autophagy Lysosomal Degradation of Mutant Huntingtin Aggregates in Neurons, Ameliorating Motility in a Drosophila Model for Huntington’s Disease. Cells, 9(1), 196. https://doi.org/10.3390/cells9010196