LRP1-Mediated AggLDL Endocytosis Promotes Cholesteryl Ester Accumulation and Impairs Insulin Response in HL-1 Cells

 ,
, 
Abstract
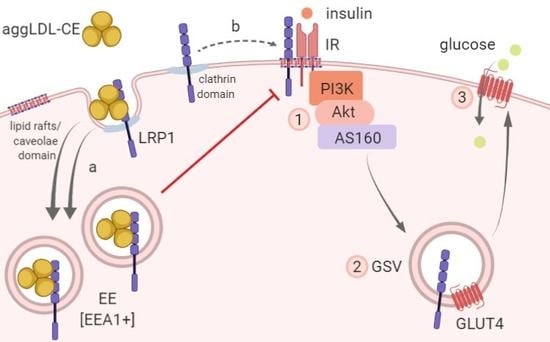
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. HL-1 Cardiomyocyte-Derived Cell Line, Cultures, and Reagents
2.2. LDL Isolation and Modification
2.3. DiI-Labeling of LDL
2.4. Lipid Extraction and Determination of CE, TG, and FC Content
2.5. Western Blot Analysis
2.6. Confocal Microscopy
2.7. Real Time-PCR
2.8. Immunoprecipitation (IP) Assays
2.9. Biotin-Labeling Cell Surface Protein Assay
2.10. 2-NBDG Uptake Assay
2.11. Statistical Treatment of Data
3. Results
3.1. LRP1 Mediates Intracellular CE Accumulation by AggLDL Endocytosis
3.2. Aggregated LDL Endocytosis Leads to AggLDL/LRP1 Complex Accumulation in Endosomal Compartments
3.3. Aggregated LDL Reduces the Molecular Association Between LRP1 and IR
3.4. Aggregated LDL Impairs Insulin-Induced Intracellular Signaling Activation
3.5. Aggregated LDL Affects the GLUT4 Traffic to the Cell Surface and Glucose Uptake
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2-NBDG | (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl) Amino)-2-Deoxyglucose) |
| aggLDL | Aggregated low-density lipoprotein |
| CE | Cholesteryl ester |
| GLUT4 | Glucose transporter type 4 |
| GSV | GLUT4 storage vesicles |
| IR | Insulin receptor |
| LDL | Low-density lipoprotein |
| LRP1 | Low-density lipoprotein receptor-related protein-1 |
References
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin. Sci. Lond. 2008, 114, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Park, T.S.; Goldberg, I.J. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail. Clin. 2012, 8, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Abel, E.D. Insulin Signaling and Heart Failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.I.; Huh, J.Y.; Sohn, J.H.; Choe, S.S.; Lee, Y.S.; Lim, C.Y.; Jo, A.; Park, S.B.; Han, W.; Kim, J.B.; et al. Lipid-overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol. Cell. Biol. 2015, 35, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Taskinen, M.R.; Olofsson, S.O.; Levin, M. Ectopic lipid storage and insulin resistance: A harmful relationship. J. Intern. Med. 2013, 274, 25–40. [Google Scholar] [CrossRef]
- Iqbal, J.; Al Qarni, A.; Hawwari, A.; Alghanem, A.F.; Ahmed, G. Metabolic Syndrome, Dyslipidemia and Regulation of Lipoprotein Metabolism. Curr. Diabetes Rev. 2018, 14, 427–433. [Google Scholar] [CrossRef]
- Hurt-Camejo, E.; Camejo, G.; Sartipy, P. Phospholipase A2 and small, dense low-density lipoprotein. Curr. Opin. Lipidol. 2000, 11, 465–471. [Google Scholar] [CrossRef]
- Martinez-Bujidos, M.; Rull, A.; Gonzalez-Cura, B.; Perez-Cuellar, M.; Montoliu-Gaya, L.; Villegas, S.; Ordonez-Llanos, J.; Sanchez-Quesada, J.L. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. 2015, 29, 1688–1700. [Google Scholar] [CrossRef]
- Cal, R.; Castellano, J.; Revuelta-Lopez, E.; Aledo, R.; Barriga, M.; Farre, J.; Vilahur, G.; Nasarre, L.; Hove-Madsen, L.; Badimon, L.; et al. Low-density lipoprotein receptor-related protein 1 mediates hypoxia-induced very low density lipoprotein-cholesteryl ester uptake and accumulation in cardiomyocytes. Cardiovasc. Res. 2012, 94, 469–479. [Google Scholar] [CrossRef]
- Cal, R.; Garcia-Arguinzonis, M.; Revuelta-Lopez, E.; Castellano, J.; Padro, T.; Badimon, L.; Llorente-Cortes, V. Aggregated low-density lipoprotein induces LRP1 stabilization through E3 ubiquitin ligase CHFR downregulation in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 369–377. [Google Scholar] [CrossRef]
- Dissmore, T.; Seye, C.I.; Medeiros, D.M.; Weisman, G.A.; Bradford, B.; Mamedova, L. The P2Y2 receptor mediates uptake of matrix-retained and aggregated low density lipoprotein in primary vascular smooth muscle cells. Atherosclerosis 2016, 252, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Samouillan, V.; Revuelta-Lopez, E.; Dandurand, J.; Nasarre, L.; Badimon, L.; Lacabanne, C.; Llorente-Cortes, V. Cardiomyocyte intracellular cholesteryl ester accumulation promotes tropoelastin physical alteration and degradation: Role of LRP1 and cathepsin S. Int. J. Biochem. Cell Boil. 2014, 55, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Costales, P.; Castellano, J.; Revuelta-Lopez, E.; Cal, R.; Aledo, R.; Llampayas, O.; Nasarre, L.; Juarez, C.; Badimon, L.; Llorente-Cortes, V.; et al. Lipopolysaccharide downregulates CD91/low-density lipoprotein receptor-related protein 1 expression through SREBP-1 overexpression in human macrophages. Atherosclerosis 2013, 227, 79–88. [Google Scholar] [CrossRef]
- Costales, P.; Aledo, R.; Vernia, S.; Das, A.; Shah, V.H.; Casado, M.; Badimon, L.; Llorente-Cortes, V. Selective role of sterol regulatory element binding protein isoforms in aggregated LDL-induced vascular low density lipoprotein receptor-related protein-1 expression. Atherosclerosis 2010, 213, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Llorente-Cortes, V.; Costales, P.; Bernues, J.; Camino-Lopez, S.; Badimon, L. Sterol regulatory element-binding protein-2 negatively regulates low density lipoprotein receptor-related protein transcription. J. Mol. Biol. 2006, 359, 950–960. [Google Scholar] [CrossRef]
- Bogan, J.S.; Kandror, K.V. Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr. Opin. Cell Boil. 2010, 22, 506–512. [Google Scholar] [CrossRef]
- Bogan, J.S. Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem. 2012, 81, 507–532. [Google Scholar] [CrossRef]
- Leto, D.; Saltiel, A.R. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Boil. 2012, 13, 383–396. [Google Scholar] [CrossRef]
- Jedrychowski, M.P.; Gartner, C.A.; Gygi, S.P.; Zhou, L.; Herz, J.; Kandror, K.V.; Pilch, P.F. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J. Biol. Chem. 2010, 285, 104–114. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Tsai, C.W.; Yue, M.; Melrose, H.L.; Kanekiyo, T.; Bu, G. Neuronal LRP1 regulates glucose metabolism and insulin signaling in the brain. J. Neurosci. 2015, 35, 5851–5859. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Xian, X.; Holland, W.L.; Tsai, S.; Herz, J. Low-Density Lipoprotein Receptor-Related Protein-1 Protects Against Hepatic Insulin Resistance and Hepatic Steatosis. EBioMedicine 2016, 7, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Actis Dato, V.; Grosso, R.A.; Sanchez, M.C.; Fader, C.M.; Chiabrando, G.A. Insulin-induced exocytosis regulates the cell surface level of low-density lipoprotein-related protein-1 in Muller Glial cells. Biochem. J. 2018, 475, 1669–1685. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef]
- White, S.M.; Constantin, P.E.; Claycomb, W.C. Cardiac physiology at the cellular level: Use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, 823–829. [Google Scholar] [CrossRef]
- Bu, G.; Geuze, H.J.; Strous, G.J.; Schwartz, A.L. 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 1995, 14, 2269–2280. [Google Scholar] [CrossRef]
- Llorente-Cortes, V.; Martinez-Gonzalez, J.; Badimon, L. LDL receptor-related protein mediates uptake of aggregated LDL in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1572–1579. [Google Scholar] [CrossRef]
- Llorente-Cortes, V.; Otero-Vinas, M.; Badimon, L. Differential role of heparan sulfate proteoglycans on aggregated LDL uptake in human vascular smooth muscle cells and mouse embryonic fibroblasts. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1905–1911. [Google Scholar] [CrossRef]
- Llorente-Cortes, V.; Royo, T.; Otero-Vinas, M.; Berrozpe, M.; Badimon, L. Sterol regulatory element binding proteins downregulate LDL receptor-related protein (LRP1) expression and LRP1-mediated aggregated LDL uptake by human macrophages. Cardiovasc. Res. 2007, 74, 526–536. [Google Scholar] [CrossRef]
- Llorente-Cortes, V.; Otero-Vinas, M.; Camino-Lopez, S.; Costales, P.; Badimon, L. Cholesteryl esters of aggregated LDL are internalized by selective uptake in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 117–123. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Chen, Y.C.; Lee, T.I.; Kao, Y.H.; Chazo, T.F.; Chen, S.A.; Chen, Y.J. Glucagon-like peptide-1 regulates calcium homeostasis and electrophysiological activities of HL-1 cardiomyocytes. Peptides 2016, 78, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Rawal, S.; Nagesh, P.T.; Coffey, S.; Van Hout, I.; Galvin, I.F.; Bunton, R.W.; Davis, P.; Williams, M.J.A.; Katare, R. Early dysregulation of cardiac-specific microRNA-208a is linked to maladaptive cardiac remodelling in diabetic myocardium. Cardiovasc. Diabetol. 2019, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Lugenbiel, P.; Govorov, K.; Rahm, A.K.; Wieder, T.; Gramlich, D.; Syren, P.; Weiberg, N.; Seyler, C.; Katus, H.A.; Thomas, D.; et al. Inhibition of Histone Deacetylases Induces K+ Channel Remodeling and Action Potential Prolongation in HL-1 Atrial Cardiomyocytes. Cell Physiol. Biochem. 2018, 49, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Willnow, T.E.; Orth, K.; Herz, J. Molecular dissection of ligand binding sites on the low density lipoprotein receptor-related protein. J. Biol. Chem. 1994, 269, 15827–15832. [Google Scholar] [PubMed]
- May, P.; Bock, H.H.; Herz, J. Integration of endocytosis and signal transduction by lipoprotein receptors. Sci. STKE 2003, 2003, PE12. [Google Scholar] [CrossRef] [PubMed]
- Actis Dato, V.; Chiabrando, G.A. The Role of Low-Density Lipoprotein Receptor-Related Protein 1 in Lipid Metabolism, Glucose Homeostasis and Inflammation. Int. J. Mol. Sci. 2018, 19, 1780. [Google Scholar] [CrossRef]
- May, P.; Herz, J.; Bock, H.H. Molecular mechanisms of lipoprotein receptor signalling. Cell Mol. Life Sci. 2005, 62, 2325–2338. [Google Scholar] [CrossRef]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. LDL Receptor-Related Protein 1: Unique Tissue-Specific Functions Revealed by Selective Gene Knockout Studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef] [PubMed]
- Delint-Ramirez, I.; Maldonado Ruiz, R.; Torre-Villalvazo, I.; Fuentes-Mera, L.; Garza Ocanas, L.; Tovar, A.; Camacho, A. Genetic obesity alters recruitment of TANK-binding kinase 1 and AKT into hypothalamic lipid rafts domains. Neurochem. Int. 2015, 80, 23–32. [Google Scholar] [CrossRef]
- Zhang, H.; Links, P.H.; Ngsee, J.K.; Tran, K.; Cui, Z.; Ko, K.W.; Yao, Z. Localization of low density lipoprotein receptor-related protein 1 to caveolae in 3T3-L1 adipocytes in response to insulin treatment. J. Biol. Chem. 2004, 279, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Roura, S.; Cal, R.; Galvez-Monton, C.; Revuelta-Lopez, E.; Nasarre, L.; Badimon, L.; Bayes-Genis, A.; Llorente-Cortes, V. Inverse relationship between raft LRP1 localization and non-raft ERK1,2/MMP9 activation in idiopathic dilated cardiomyopathy: Potential impact in ventricular remodeling. Int. J. Cardiol. 2014, 176, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Laatsch, A.; Merkel, M.; Talmud, P.J.; Grewal, T.; Beisiegel, U.; Heeren, J. Insulin stimulates hepatic low density lipoprotein receptor-related protein 1 (LRP1) to increase postprandial lipoprotein clearance. Atherosclerosis 2009, 204, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Fazakerley, D.J.; Krycer, J.R.; Kearney, A.L.; Hocking, S.L.; James, D.E. Muscle and adipose tissue insulin resistance: Malady without mechanism? J. Lipid Res. 2019, 60, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nunez, S.; Nuno-Lambarri, N.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Actis Dato, V.; Benitez-Amaro, A.; de Gonzalo-Calvo, D.; Vazquez, M.; Bonacci, G.; Llorente-Cortés, V.; Chiabrando, G.A. LRP1-Mediated AggLDL Endocytosis Promotes Cholesteryl Ester Accumulation and Impairs Insulin Response in HL-1 Cells. Cells 2020, 9, 182. https://doi.org/10.3390/cells9010182
Actis Dato V, Benitez-Amaro A, de Gonzalo-Calvo D, Vazquez M, Bonacci G, Llorente-Cortés V, Chiabrando GA. LRP1-Mediated AggLDL Endocytosis Promotes Cholesteryl Ester Accumulation and Impairs Insulin Response in HL-1 Cells. Cells. 2020; 9(1):182. https://doi.org/10.3390/cells9010182
Chicago/Turabian StyleActis Dato, Virginia, Aleyda Benitez-Amaro, David de Gonzalo-Calvo, Maximiliano Vazquez, Gustavo Bonacci, Vicenta Llorente-Cortés, and Gustavo Alberto Chiabrando. 2020. "LRP1-Mediated AggLDL Endocytosis Promotes Cholesteryl Ester Accumulation and Impairs Insulin Response in HL-1 Cells" Cells 9, no. 1: 182. https://doi.org/10.3390/cells9010182
APA StyleActis Dato, V., Benitez-Amaro, A., de Gonzalo-Calvo, D., Vazquez, M., Bonacci, G., Llorente-Cortés, V., & Chiabrando, G. A. (2020). LRP1-Mediated AggLDL Endocytosis Promotes Cholesteryl Ester Accumulation and Impairs Insulin Response in HL-1 Cells. Cells, 9(1), 182. https://doi.org/10.3390/cells9010182