TRPC Channels: Dysregulation and Ca2+ Mishandling in Ischemic Heart Disease

 , ,
, ,  , and
, and 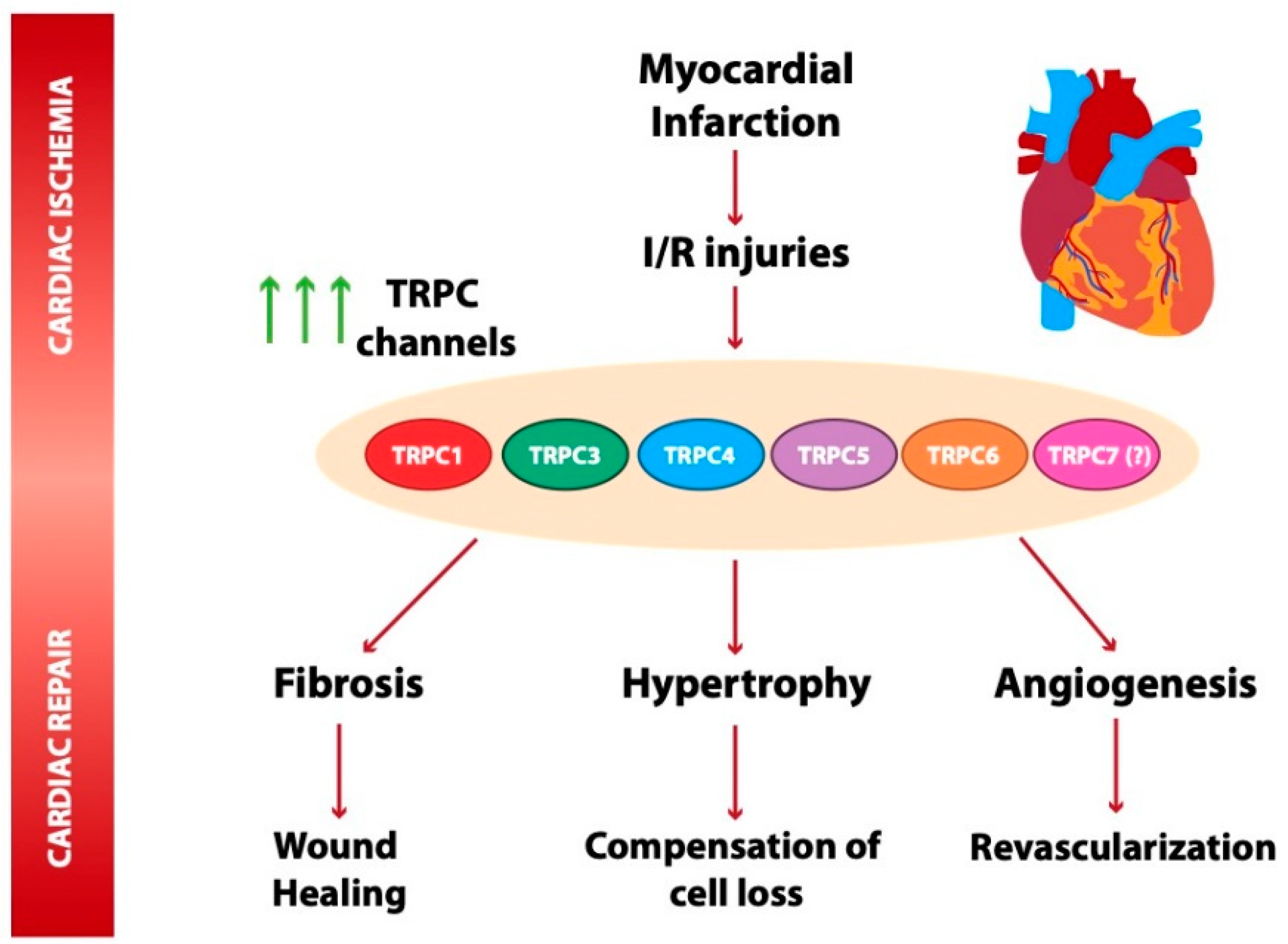{kind=link}
Abstract
1. Introduction
2. TRPC Channels in the Cardiovascular System
3. TRPC Channels Mediate Ca2+ Influx in Cardiac Myocytes
4. Role of TRPC Channels in Cardiac Pathophysiology
5. Role of TRPC Channels in Cardiac Ischemia
5.1. TRPC Channels in Myocardial Infarction
5.2. TRPC Channel Role in Ischemia and Reperfusion Injuries and Cardioprotection
6. TRPC Channels in Post-Ischemia Cardiac Repair
6.1. TRPC Channels in Post-Ischemia Angiogenesis
6.2. TRPC Channels in Early Adaptative Cardiac Remodeling
7. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
References
- Fabiato, A.; Fabiato, F. Excitation-contraction coupling of isolated cardiac fibers with disrupted or closed sarcolemmas. Calcium-dependent cyclic and tonic contractions. Circ. Res. 1972, 31, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Rueda, A.; de Alba-Aguayo, D.R.; Valdivia, H.H. Ryanodine receptor, calcium leak and arrhythmias. Arch. Cardiol. Mex. 2014, 84, 191–201. [Google Scholar] [PubMed]
- Falcón, D.; Galeano-Otero, I.; Calderón-Sánchez, E.; Del Toro, R.; Martín-Bórnez, M.; Rosado, J.A.; Hmadcha, A.; Smani, T. TRP Channels: Current Perspectives in the Adverse Cardiac Remodeling. Front. Physiol. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Eder, P.; Chang, B.; Molkentin, J.D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 7000–7005. [Google Scholar] [CrossRef]
- Tsvilovskyy, V.V.; Zholos, A.V.; Aberle, T.; Philipp, S.E.; Dietrich, A.; Zhu, M.X.; Birnbaumer, L.; Freichel, M.; Flockerzi, V. Deletion of TRPC4 and TRPC6 in Mice Impairs Smooth Muscle Contraction and Intestinal Motility In Vivo. Gastroenterology 2009, 137, 1415–1424. [Google Scholar] [CrossRef]
- Noorani, M.M.Z.; Noel, R.C.; Marrelli, S.P. Upregulated TRPC3 and Downregulated TRPC1 Channel Expression during Hypertension is Associated with Increased Vascular Contractility in Rat. Front. Physiol. 2011, 2, 42. [Google Scholar] [CrossRef]
- Guinamard, R.; Chatelier, A.; Demion, M.; Potreau, D.; Patri, S.; Rahmati, M.; Bois, P. Functional characterization of a Ca 2+-activated non-selective cation channel in human atrial cardiomyocytes. J. Physiol. 2004, 558, 75–83. [Google Scholar] [CrossRef]
- Simard, C.; Sallé, L.; Rouet, R.; Guinamard, R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br. J. Pharmacol. 2012, 165, 2354–2364. [Google Scholar] [CrossRef]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006, 25, 5305–5316. [Google Scholar] [CrossRef]
- Ohba, T.; Watanabe, H.; Murakami, M.; Takahashi, Y.; Iino, K.; Kuromitsu, S.; Mori, Y.; Ono, K.; Iijima, T.; Ito, H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Rodríguez, A.; Ruiz-Hurtado, G.; Sabourin, J.; Gómez, A.M.; Alvarez, J.L.; Benitah, J.P. Proarrhythmic effect of sustained EPAC activation on TRPC3/4 in rat ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 87, 74–78. [Google Scholar] [CrossRef]
- Domínguez-Rodríguez, A.; Mayoral-Gonzalez, I.; Avila-Medina, J.; de Rojas-de Pedro, E.S.; Calderón-Sánchez, E.; Díaz, I.; Hmadcha, A.; Castellano, A.; Rosado, J.A.; Benitah, J.-P.; et al. Urocortin-2 Prevents Dysregulation of Ca2+ Homeostasis and Improves Early Cardiac Remodeling After Ischemia and Reperfusion. Front. Physiol. 2018, 9, 813. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Kiselyov, K.; Shin, D.M.; Chen, J.; Shcheynikov, N.; Kang, S.H.; Dehoff, M.H.; Schwarz, M.K.; Seeburg, P.H.; Muallem, S.; et al. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 2003, 114, 777–789. [Google Scholar] [CrossRef]
- Adebiyi, A.; Thomas-Gatewood, C.M.; Leo, M.D.; Kidd, M.W.; Neeb, Z.P.; Jaggar, J.H. An elevation in physical coupling of type 1 inositol 1,4,5-trisphosphate (IP3) receptors to transient receptor potential 3 (TRPC3) channels constricts mesenteric arteries in genetic hypertension. Hypertension 2012, 60, 1213–1219. [Google Scholar] [CrossRef]
- Adebiyi, A.; Zhao, G.; Narayanan, D.; Thomas-Gatewood, C.M.; Bannister, J.P.; Jaggar, J.H. Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ. Res. 2010, 106, 1603–1612. [Google Scholar] [CrossRef]
- Mery, L.; Magnino, F.; Schmidt, K.; Krause, K.H.; Dufour, J.F. Alternative splice variants of hTrp4 differentially interact with the C-terminal portion of the inositol 1,4,5-trisphosphate receptors. FEBS Lett. 2001, 487, 377–383. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Tikunova, S.; Johnson, J.D.; Chen, Z.; Qin, N.; Dietrich, A.; Stefani, E.; Birnbaumer, L.; Zhu, M.X. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc. Natl. Acad. Sci. USA 2001, 98, 3168–3173. [Google Scholar] [CrossRef]
- Trebak, M.; Lemonnier, L.; Dehaven, W.I.; Wedel, B.J.; Bird, G.S.; Putney, J.W. Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflugers Arch. Eur. J. Physiol. 2009, 457, 757–769. [Google Scholar] [CrossRef]
- Otsuguro, K.I.; Tang, J.; Tang, Y.; Xiao, R.; Freichel, M.; Tsvilovskyy, V.; Ito, S.; Flockerzi, V.; Zhu, M.X.; Zholos, A.V. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2008, 283, 10026–10036. [Google Scholar] [CrossRef]
- Myeong, J.; Ko, J.; Kwak, M.; Kim, J.; Woo, J.; Ha, K.; Hong, C.; Yang, D.; Kim, H.J.; Jeon, J.H.; et al. Dual action of the Gαq-PLCβ-PI(4,5)P2 pathway on TRPC1/4 and TRPC1/5 heterotetramers. Sci. Rep. 2018, 8, 12117. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, J.; Robin, E.; Raddatz, E. A key role of TRPC channels in the regulation of electromechanical activity of the developing heart. Cardiovasc. Res. 2011, 92, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Trebak, M. Transient receptor potential canonical 7: A diacylglycerol-activated non-selective cation channel. Handb. Exp. Pharmacol. 2014, 222, 189–204. [Google Scholar] [PubMed]
- He, X.; Li, S.; Liu, B.; Susperreguy, S.; Formoso, K.; Yao, J.; Kang, J.; Shi, A.; Birnbaumer, L.; Liao, Y. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc. Natl. Acad. Sci. USA 2017, 114, E4582. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Storch, U.; Forst, A.L.; Pardatscher, F.; Erdogmus, S.; Philipp, M.; Gregoritza, M.; Schnitzler, M.M.Y.; Gudermann, T. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc. Natl. Acad. Sci. USA 2017, 114, E37–E46. [Google Scholar] [CrossRef]
- Hof, T.; Chaigne, S.; Récalde, A.; Sallé, L.; Brette, F.; Guinamard, R. Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef]
- Ju, Y.-K.; Lee, B.H.; Trajanovska, S.; Hao, G.; Allen, D.G.; Lei, M.; Cannell, M.B. The involvement of TRPC3 channels in sinoatrial arrhythmias. Front. Physiol. 2015, 6, 86. [Google Scholar] [CrossRef]
- Bush, E.W.; Hood, D.B.; Papst, P.J.; Chapo, J.A.; Minobe, W.; Bristow, M.R.; Olson, E.N.; McKinsey, T.A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 2006, 281, 33487–33496. [Google Scholar] [CrossRef]
- Sunggip, C.; Shimoda, K.; Oda, S.; Tanaka, T.; Nishiyama, K.; Mangmool, S.; Nishimura, A.; Numaga-Tomita, T.; Nishida, M. TRPC5-eNOS axis negatively regulates ATP-induced cardiomyocyte hypertrophy. Front. Pharmacol. 2018, 9, 523. [Google Scholar] [CrossRef]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, W.; Liu, P.; Jiang, Y.; Zhao, Y.; Wei, H.; Niu, W. TRPC1 expression and distribution in rat hearts. Eur. J. Histochem. 2009, 53, 26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sabourin, J.; Antigny, F.; Robin, E.; Frieden, M.; Raddatz, E. Activation of transient receptor potential canonical 3 (TRPC3)-mediated Ca2+ entry by A1 adenosine receptor in cardiomyocytes disturbs atrioventricular conduction. J. Biol. Chem. 2012, 287, 26688–26701. [Google Scholar] [CrossRef]
- Jiang, Y.; Huang, H.; Liu, P.; Wei, H.; Zhao, H.; Feng, Y.; Wang, W.; Niu, W. Expression and localization of TRPC proteins in rat ventricular myocytes at various developmental stages. Cell Tissue Res. 2014, 355, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.K.; Chu, Y.; Chaulet, H.; Lai, D.; Gervasio, O.L.; Graham, R.M.; Cannell, M.B.; Allen, D.G. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ. Res. 2007, 100, 1605–1614. [Google Scholar] [CrossRef]
- Numaga-Tomita, T.; Kitajima, N.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; Birnbaumer, L.; et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 2016, 6, 39383. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef]
- Rose, R.A.; Hatano, N.; Ohya, S.; Imaizumi, Y.; Giles, W.R. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide C receptor-mediated signalling. J. Physiol. 2007, 580, 255–274. [Google Scholar] [CrossRef]
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ordog, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N.; et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012, 126, 2051–2064. [Google Scholar] [CrossRef]
- Ikeda, K.; Nakajima, T.; Yamamoto, Y.; Takano, N.; Tanaka, T.; Kikuchi, H.; Oguri, G.; Morita, T.; Nakamura, F.; Komuro, I. Roles of transient receptor potential canonical (TRPC) channels and reverse-mode Na+/Ca2+ exchanger on cell proliferation in human cardiac fibroblasts: Effects of transforming growth factor β1. Cell Calcium 2013, 54, 213–225. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Wu, H.-J.; Che, H.; Sun, H.-Y.; Cheng, L.-C.; Li, X.; Au, W.-K.; Tse, H.-F.; Li, G.-R. Functional transient receptor potential canonical type 1 channels in human atrial myocytes. Pflugers Arch. 2013, 465, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Numaga-Tomita, T.; Kitajima, N.; Toyama, T.; Harada, E.; Shimauchi, T.; Nishimura, A.; Ishikawa, T.; Kumagai, Y.; Birnbaumer, L.; et al. TRPC6 counteracts TRPC3-Nox2 protein complex leading to attenuation of hyperglycemia-induced heart failure in mice. Sci. Rep. 2017, 7, 7511. [Google Scholar] [CrossRef] [PubMed]
- Doleschal, B.; Primessnig, U.; Wölkart, G.; Wolf, S.; Schernthaner, M.; Lichtenegger, M.; Glasnov, T.N.; Kappe, C.O.; Mayer, B.; Antoons, G.; et al. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc. Res. 2015, 106, 163–173. [Google Scholar] [CrossRef]
- Goel, M.; Zuo, C.D.; Sinkins, W.G.; Schilling, W.P. TRPC3 channels co-localize with the Na+, Ca2+ exchanger and the Na+ pump in the axial component of the transverse-axial-tubular system (TATS) of rat ventricle. Am. J. Physiol. Heart Circ. Physiol. 2006, 292, H874–H883. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, L.; Trebak, M.; Lievremont, J.P.; Bird, G.S.; Putney, J.W. Protection of TRPC7 cation channels from calcium inhibition by closely associated SERCA pumps. FASEB J. 2006, 20, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef]
- Qu, Y.; Boutjdir, M. TRPC channels, an overarching Ca(2+) paradigm in the developing heart. Cardiovasc. Res. 2011, 92, 189–190. [Google Scholar] [CrossRef][Green Version]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009, 23, 297–328. [Google Scholar] [CrossRef]
- Chen, J.; Crossland, R.F.; Noorani, M.M.Z.; Marrelli, S.P.; Marrelli, S.P. Inhibition of TRPC1/TRPC3 by PKG contributes to NO-mediated vasorelaxation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, 417–424. [Google Scholar] [CrossRef]
- Jardin, I.; Gómez, L.J.; Salido, G.M.; Rosado, J.A. Dynamic interaction of hTRPC6 with the Orai1-STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem. J. 2009, 420, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Bröker-Lai, J.; Kollewe, A.; Schindeldecker, B.; Pohle, J.; Nguyen Chi, V.; Mathar, I.; Guzman, R.; Schwarz, Y.; Lai, A.; Weißgerber, P.; et al. Heteromeric channels formed by TRPC 1, TRPC 4 and TRPC 5 define hippocampal synaptic transmission and working memory. EMBO J. 2017, 36, 2770–2789. [Google Scholar] [CrossRef] [PubMed]
- Goel, M.; Sinkins, W.G.; Schilling, W.P. Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 2002, 277, 48303–48310. [Google Scholar] [CrossRef] [PubMed]
- Flockerzi, V.; Nilius, B. TRPs: Truly remarkable proteins. Handb. Exp. Pharmacol. 2014, 222, 1–12. [Google Scholar]
- Eder, P. Cardiac Remodeling and Disease: SOCE and TRPC Signaling in Cardiac Pathology. Adv. Exp. Med. Biol. 2017, 993, 505–521. [Google Scholar]
- Abramowitz, J.; Yildirim, E.; Birnbaumer, L. The TRPC Family of Ion Channels: Relation to the TRP Superfamily and Role in Receptor- and Store-Operated Calcium Entry. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2007; pp. 1–18. [Google Scholar]
- Fauconnier, J.; Lanner, J.; Sultan, A.; Zhang, S.; Katz, A.; Bruton, J.; Westerblad, H. Insulin potentiates TRPC3-mediated cation currents in normal but not in insulin-resistant mouse cardiomyocytes. Cardiovasc. Res. 2007, 73, 376–385. [Google Scholar] [CrossRef]
- Wen, H.; Zhao, Z.; Fefelova, N.; Xie, L.-H. Potential Arrhythmogenic Role of TRPC Channels and Store-Operated Calcium Entry Mechanism in Mouse Ventricular Myocytes. Front. Physiol. 2018, 9, 1785. [Google Scholar] [CrossRef]
- Sabourin, J.; Bartoli, F.; Antigny, F.; Gomez, A.M.; Benitah, J.P. Transient receptor potential canonical (trpc)/orai1-dependent store-operated Ca2+ channels; new targets of aldosterone in cardiomyocytes. J. Biol. Chem. 2016, 291, 13394–13409. [Google Scholar] [CrossRef]
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca2+ handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224. [Google Scholar] [CrossRef]
- Bartoli, F.; Moradi Bachiller, S.; Antigny, F.; Bedouet, K.; Gerbaud, P.; Sabourin, J.; Benitah, J.P. Specific Upregulation of TRPC1 and TRPC5 Channels by Mineralocorticoid Pathway in Adult Rat Ventricular Cardiomyocytes. Cells 2019, 9, 47. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Streiff, M.; Hunter, C.; Hu, Q.; Sachse, F.B. Physiological and pathophysiological role of transient receptor potential canonical channels in cardiac myocytes. Prog. Biophys. Mol. Biol. 2017, 130, 254–263. [Google Scholar] [CrossRef]
- Seo, K.; Rainer, P.P.; Shalkey Hahn, V.; Lee, D.I.; Jo, S.H.; Andersen, A.; Liu, T.; Xu, X.; Willette, R.N.; Lepore, J.J.; et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Rainer, P.P.; Lee, D.-I.; Hao, S.; Bedja, D.; Birnbaumer, L.; Cingolani, O.H.; Kass, D.A. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ. Res. 2014, 114, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Camacho Londoño, J.E.; Tian, Q.; Hammer, K.; Schröder, L.; Camacho Londoño, J.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Heart J. 2015, 36, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Freichel, M.; Berlin, M.; Schürger, A.; Mathar, I.; Bacmeister, L.; Medert, R.; Frede, W.; Marx, A.; Segin, S.; Londoño, J.E.C. TRP Channels in the Heart. In Neurobiology of TRP Channels; CRC Press: Boca Raton, FL, USA, 2017; pp. 149–185. [Google Scholar]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordoñez, A.; Rosado, J.A.; Smani, T. The complex role of store operated calcium entry pathways and related proteins in the function of cardiac, skeletal and vascular smooth muscle cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Kirschmer, N.; Bandleon, S.; von Ehrlich-Treuenstätt, V.; Hartmann, S.; Schaaf, A.; Lamprecht, A.-K.; Miranda-Laferte, E.; Langsenlehner, T.; Ritter, O.; Eder, P. TRPC4α and TRPC4β Similarly Affect Neonatal Cardiomyocyte Survival during Chronic GPCR Stimulation. PLoS ONE 2016, 11, e0168446. [Google Scholar] [CrossRef]
- Zou, G.; Hong, H.; Lin, X.; Shi, X.; Wu, Y.; Chen, L. TRPC1, CaN and NFATC3 signaling pathway in the pathogenesis and progression of left ventricular hypertrophy in spontaneously hypertensive rats. Clin. Exp. Hypertens. 2015, 37, 223–234. [Google Scholar] [CrossRef]
- Zhou, R.; Hang, P.; Zhu, W.; Su, Z.; Liang, H.; Du, Z. Whole Genome Network Analysis of Ion Channels and Connexins in Myocardial Infarction. Cell. Physiol. Biochem. 2011, 27, 299–304. [Google Scholar] [CrossRef]
- Makarewich, C.A.; Zhang, H.; Davis, J.; Correll, R.N.; Trappanese, D.M.; Hoffman, N.E.; Troupes, C.D.; Berretta, R.M.; Kubo, H.; Madesh, M.; et al. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ. Res. 2014, 115, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Gené, G.G.; Tomás, M.; Plata, C.; Selent, J.; Pastor, M.; Fandos, C.; Senti, M.; Lucas, G.; Elosua, R.; et al. A gain-of-function SNP in TRPC4 cation channel protects against myocardial infarction. Cardiovasc. Res. 2011, 91, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Marchase, R.B.; Chatham, J.C. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am. J. Physiol. Cell Physiol. 2008, 294, C833–C841. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Fukushima, Y.; Ito, Y.; Ding, W.-G.; Kitagawa, H.; Matsuura, H. Transient Receptor Potential Canonical Channel Blockers Improve Ventricular Contractile Functions After Ischemia/Reperfusion in a Langendorff-perfused Mouse Heart Model. J. Cardiovasc. Pharmacol. 2018, 71, 248–255. [Google Scholar] [PubMed]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, L.; Chen, K.-H.; Sun, H.-Y.; Jin, M.-W.; Xiao, G.-S.; Wang, Y.; Li, G.-R. SKF-96365 blocks human ether-à-go-go-related gene potassium channels stably expressed in HEK 293 cells. Pharmacol. Res. 2016, 104, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, W.-Z.; Shi, Y.-W.; Zhou, B.-F.; Ma, R.; Li, W.-P. Danshensu protects against ischemia/reperfusion injury and inhibits the apoptosis of H9c2 cells by reducing the calcium overload through the p-JNK-NF-κB-TRPC6 pathway. Int. J. Mol. Med. 2016, 37, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hang, P.; Zhao, J.; Cai, B.; Tian, S.; Huang, W.; Guo, J.; Sun, C.; Li, Y.; Du, Z. Brain-derived neurotrophic factor regulates TRPC3/6 channels and protects against myocardial infarction in rodents. Int. J. Biol. Sci. 2015, 11, 536–545. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Wu, Q.; Xu, B.; Wang, X.; Wu, J.; Huang, L.; Cheng, J. Suppression of Stim1 reduced intracellular calcium concentration and attenuated hypoxia/reoxygenation induced apoptosis in H9C2 cells. Biosci. Rep. 2017, 37, BSR20171249. [Google Scholar] [CrossRef]
- Al-Awar, A.; Almási, N.; Szabó, R.; Takacs, I.; Murlasits, Z.; Szűcs, G.; Török, S.; Pósa, A.; Varga, C.; Kupai, K. Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model. Int. J. Mol. Sci. 2018, 19, 3226. [Google Scholar] [CrossRef]
- Garza, M.A. Cardiac remodeling and physical training post myocardial infarction. World J. Cardiol. 2015, 7, 52. [Google Scholar] [CrossRef]
- Chiarella-Redfern, H.H.; Rayner, K.J.; Suuronen, E.J. Spatio-temporal expression patterns of microRNAs in remodelling and repair of the infarcted heart. Histol. Histopathol. 2015, 30, 141–149. [Google Scholar]
- Mouton, A.J.; Rivera, O.J.; Lindsey, M.L. Myocardial infarction remodeling that progresses to heart failure: A signaling misunderstanding. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H71–H79. [Google Scholar] [CrossRef] [PubMed]
- Ingason, A.B.; Goldstone, A.B.; Paulsen, M.J.; Thakore, A.D.; Truong, V.N.; Edwards, B.B.; Eskandari, A.; Bollig, T.; Steele, A.N.; Woo, Y.J. Angiogenesis precedes cardiomyocyte migration in regenerating mammalian hearts. J. Thorac. Cardiovasc. Surg. 2018, 155, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Melly, L.; Cerino, G.; Frobert, A.; Cook, S.; Giraud, M.N.; Carrel, T.; Tevaearai Stahel, H.T.; Eckstein, F.; Rondelet, B.; Marsano, A.; et al. Myocardial infarction stabilization by cell-based expression of controlled Vascular Endothelial Growth Factor levels. J. Cell. Mol. Med. 2018, 22, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-González, I.; Galeano-Otero, I.; Redondo, P.C.; Rosado, J.A.; Smani, T. Pathophysiological Significance of Store-Operated Calcium Entry in Cardiovascular and Skeletal Muscle Disorders and Angiogenesis. Adv. Exp. Med. Biol. 2020, 1131, 489–504. [Google Scholar] [PubMed]
- Smani, T.; Gómez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.-M.; Rosado, J.A. TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Robich, M.P.; Matyal, R.; Chu, L.M.; Feng, J.; Xu, S.H.; Laham, R.J.; Hess, P.E.; Bianchi, C.; Sellke, F.W. Effects of neuropeptide Y on collateral development in a swine model of chronic myocardial ischemia. J. Mol. Cell. Cardiol. 2010, 49, 1022–1030. [Google Scholar] [CrossRef]
- Arif, M.; Pandey, R.; Alam, P.; Jiang, S.; Sadayappan, S.; Paul, A.; Ahmed, R.P.H. MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J. Mol. Med. 2017, 95, 1369–1385. [Google Scholar] [CrossRef]
- Chen, Z.; Li, B.; Dong, Q.; Qian, C.; Cheng, J.; Wang, Y. Repetitive Transient Ischemia-Induced Cardiac Angiogenesis is Mediated by Camkii Activation. Cell. Physiol. Biochem. 2018, 47, 914–924. [Google Scholar] [CrossRef]
- Muona, K.; Mäkinen, K.; Hedman, M.; Manninen, H.; Ylä-Herttuala, S. 10-Year safety follow-up in patients with local VEGF gene transfer to ischemic lower limb. Gene Ther. 2012, 19, 392–395. [Google Scholar] [CrossRef]
- Henry, T.D.; Annex, B.H.; McKendall, G.R.; Azrin, M.A.; Lopez, J.J.; Giordano, F.J.; Shah, P.K.; Willerson, J.T.; Benza, R.L.; Berman, D.S.; et al. The VIVA trial: Vascular endothelial growth factor in ischemia for vascular angiogenesis. Circulation 2003, 107, 1359–1365. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular Endothelial Growth Factor Stimulates Endothelial Colony Forming Cells Proliferation and Tubulogenesis by Inducing Oscillations in Intracellular Ca2+ Concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC Channel Dependence of VEGF-Activated Ca2+ Entry and Endothelial Tube Formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Schlotterer, A.; Schumacher, D.; Matka, C.; Mathar, I.; Dietrich, N.; Medert, R.; Kriebs, U.; Lin, J.; Nawroth, P.; et al. TRPC proteins contribute to development of diabetic retinopathy and regulate glyoxalase 1 activity and methylglyoxal accumulation. Mol. Metab. 2018, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Song, H.B.; Jun, H.O.; Kim, J.H.; Fruttiger, M.; Kim, J.H. Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell Calcium 2015, 57, 101–108. [Google Scholar] [CrossRef]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. Trpc6 channels are required for proliferation, migration and invasion of breast cancer cell lines by modulation of orai1 and orai3 surface exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef]
- Yip, H.; Chan, W.Y.; Leung, P.C.; Kwan, H.Y.; Liu, C.; Huang, Y.; Michel, V.; Yew, D.T.W.; Yao, X. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem. Cell Biol. 2004, 122, 553–561. [Google Scholar] [CrossRef]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell. Signal. 2017, 37, 12–30. [Google Scholar] [CrossRef]
- Hamdollah Zadeh, M.A.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [CrossRef]
- Qin, W.; Xie, W.; Xia, N.; He, Q.; Sun, T. Silencing of Transient Receptor Potential Channel 4 Alleviates oxLDL-induced Angiogenesis in Human Coronary Artery Endothelial Cells by Inhibition of VEGF and NF-κB. Med. Sci. Monit. 2016, 22, 930–936. [Google Scholar] [CrossRef]
- Fantozzi, I.; Zhang, S.; Platoshyn, O.; Remillard, C.V.; Cowling, R.T.; Yuan, J.X.J. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1233–L1245. [Google Scholar] [CrossRef]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca2+ signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gao, M.; Zhou, T.; Xie, M.; Mao, A.; Feng, L.; Yao, X.; Wong, W.T.; Ma, X. The TRPC5 channel regulates angiogenesis and promotes recovery from ischemic injury in mice. J. Biol. Chem. 2019, 294, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Hanif, W.; Alex, L.; Su, Y.; Shinde, A.V.; Russo, I.; Li, N.; Frangogiannis, N.G. Left atrial remodeling, hypertrophy, and fibrosis in mouse models of heart failure. Cardiovasc. Pathol. 2017, 30, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Niizeki, T.; Takeishi, Y.; Kitahara, T.; Arimoto, T.; Ishino, M.; Bilim, O.; Suzuki, S.; Sasaki, T.; Nakajima, O.; Walsh, R.A.; et al. Diacylglycerol kinase-ε restores cardiac dysfunction under chronic pressure overload: A new specific regulator of Gαq signaling cascade. Am. J. Physiol. Circ. Physiol. 2008, 295, H245–H255. [Google Scholar] [CrossRef]
- Seth, M.; Zhang, Z.-S.; Mao, L.; Graham, V.; Burch, J.; Stiber, J.; Tsiokas, L.; Winn, M.; Abramowitz, J.; Rockman, H.A.; et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 2009, 105, 1023–1030. [Google Scholar] [CrossRef]
- Dragún, M.; Gažová, A.; Kyselovič, J.; Hulman, M.; Máťuš, M. TRP Channels Expression Profile in Human End-Stage Heart Failure. Medicina 2019, 55, 380. [Google Scholar] [CrossRef]
- Duran, J.; Lagos, D.; Pavez, M.; Troncoso, M.F.; Ramos, S.; Barrientos, G.; Ibarra, C.; Lavandero, S.; Estrada, M. Ca2+/calmodulin-dependent protein kinase II and androgen signaling pathways modulate MEF2 activity in testosterone-induced cardiac myocyte hypertrophy. Front. Pharmacol. 2017, 8, 604. [Google Scholar] [CrossRef]
- Tang, L.; Yao, F.; Wang, H.; Wang, X.; Shen, J.; Dai, B.; Wu, H.; Zhou, D.; Guo, F.; Wang, J.; et al. Inhibition of TRPC1 prevents cardiac hypertrophy via NF-κB signaling pathway in human pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2019, 126, 143–154. [Google Scholar] [CrossRef]
- Lacraz, G.P.A.; Junker, J.P.; Gladka, M.M.; Molenaar, B.; Scholman, K.T.; Vigil-Garcia, M.; Versteeg, D.; De Ruiter, H.; Vermunt, M.W.; Creyghton, M.P.; et al. Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis during Ischemic Injury. Circulation 2017, 136, 1396–1409. [Google Scholar] [CrossRef]
- Kapur, N.K.; Qiao, X.; Paruchuri, V.; Mackey, E.E.; Daly, G.H.; Ughreja, K.; Morine, K.J.; Levine, J.; Aronovitz, M.J.; Hill, N.S.; et al. Reducing endoglin activity limits calcineurin and TRPC-6 expression and improves survival in a mouse model of right ventricular pressure overload. J. Am. Heart Assoc. 2014, 3, e000965. [Google Scholar] [CrossRef]
- Gao, S.; Li, L.; Li, L.; Ni, J.; Guo, R.; Mao, J.; Fan, G. Effects of the combination of tanshinone IIA and puerarin on cardiac function and inflammatory response in myocardial ischemia mice. J. Mol. Cell. Cardiol. 2019, 137, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Saliba, Y.; Jebara, V.; Hajal, J.; Maroun, R.; Chacar, S.; Smayra, V.; Abramowitz, J.; Birnbaumer, L.; Farès, N. Transient Receptor Potential Canonical 3 and Nuclear Factor of Activated T Cells C3 Signaling Pathway Critically Regulates Myocardial Fibrosis. Antioxid. Redox Signal. 2019, 30, 1851–1879. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Tang, Y.; Li, S.; Wu, Y.; Chen, X.; Wu, Q.; Hong, K.; Li, J. Protective mechanism of SIRT1 on Hcy-induced atrial fibrosis mediated by TRPC3. J. Cell. Mol. Med. 2020, 24, 488–510. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcón, D.; Galeano-Otero, I.; Martín-Bórnez, M.; Fernández-Velasco, M.; Gallardo-Castillo, I.; Rosado, J.A.; Ordóñez, A.; Smani, T. TRPC Channels: Dysregulation and Ca2+ Mishandling in Ischemic Heart Disease. Cells 2020, 9, 173. https://doi.org/10.3390/cells9010173
Falcón D, Galeano-Otero I, Martín-Bórnez M, Fernández-Velasco M, Gallardo-Castillo I, Rosado JA, Ordóñez A, Smani T. TRPC Channels: Dysregulation and Ca2+ Mishandling in Ischemic Heart Disease. Cells. 2020; 9(1):173. https://doi.org/10.3390/cells9010173
Chicago/Turabian StyleFalcón, Débora, Isabel Galeano-Otero, Marta Martín-Bórnez, María Fernández-Velasco, Isabel Gallardo-Castillo, Juan A. Rosado, Antonio Ordóñez, and Tarik Smani. 2020. "TRPC Channels: Dysregulation and Ca2+ Mishandling in Ischemic Heart Disease" Cells 9, no. 1: 173. https://doi.org/10.3390/cells9010173
APA StyleFalcón, D., Galeano-Otero, I., Martín-Bórnez, M., Fernández-Velasco, M., Gallardo-Castillo, I., Rosado, J. A., Ordóñez, A., & Smani, T. (2020). TRPC Channels: Dysregulation and Ca2+ Mishandling in Ischemic Heart Disease. Cells, 9(1), 173. https://doi.org/10.3390/cells9010173