Abstract
Transient receptor potential canonical (TRPC) channels are ubiquitously expressed in excitable and non-excitable cardiac cells where they sense and respond to a wide variety of physical and chemical stimuli. As other TRP channels, TRPC channels may form homo or heterotetrameric ion channels, and they can associate with other membrane receptors and ion channels to regulate intracellular calcium concentration. Dysfunctions of TRPC channels are involved in many types of cardiovascular diseases. Significant increase in the expression of different TRPC isoforms was observed in different animal models of heart infarcts and in vitro experimental models of ischemia and reperfusion. TRPC channel-mediated increase of the intracellular Ca2+ concentration seems to be required for the activation of the signaling pathway that plays minor roles in the healthy heart, but they are more relevant for cardiac responses to ischemia, such as the activation of different factors of transcription and cardiac hypertrophy, fibrosis, and angiogenesis. In this review, we highlight the current knowledge regarding TRPC implication in different cellular processes related to ischemia and reperfusion and to heart infarction.
1. Introduction
The heart rate of a healthy adult ranges between 60 and 100 beats/min, which is mainly achieved by adequate function of the cardiac contraction/relaxation cycle. Adequate ventricular contraction is strongly dependent on effective excitation–contraction (EC) coupling in cardiac cells. Electrical stimuli travel across conducting cardiac tissues to the cardiomyocytes, inducing a cell-membrane depolarization activating ion channel and finally activating the cell contractile machinery (reviewed elsewhere [1,2]). EC coupling and cell contraction are critically dependent on Ca2+ influx and Ca2+ channel trafficking. The initial cell-membrane depolarization stimulates sarcolemma L-type Ca2+ channels, prompting a small influx of Ca2+ from the extracellular medium. Ca2+ entry triggers a large release of Ca2+ from the sarcoplasmic reticulum via ryanodine receptors (RyR), resulting in an increase in the intracellular Ca2+ concentration ([Ca2+]i). The rise in [Ca2+]i boosts Ca2+ binding to troponin C, which activates the contractile machinery. After contraction, [Ca2+]i must decrease to allow cell relaxation, which is achieved mainly via two mechanisms: Ca2+ re-uptake by the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) pump and Ca2+ efflux by the sarcoplasmic Na+/Ca2+ exchanger (NCX) [2,3]. Dysregulation of any of these Ca2+ handling processes is commonly associated with cardiac dysfunction.
Recently, other players emerged as key partners in the regulation of cardiac Ca2+ handling. Among these partners are the transient receptor potential (TRP) channels that are classified in a superfamily, including 28 mammalian TRP proteins divided according their genetic and functional homology into six families: TRPP (polycystin), TRPV (vanilloid), TRPM (melastatin), TRPA (ankyrin), TRPML (mucolipin), and TRPC (canonical). TRP channels are composed of six transmembrane domains (TM1–TM6), with a preserved sequence called the “TRP domain” adjacent to the C-terminus of TM6 and a cation-permeable pore region formed by a loop between TM5 and TM6 (reviewed in Reference [4]). TRP channels are located in the plasma membrane, and their activation allows the entry of Ca2+ and/or Na+, with higher permeability for Ca2+. Although most TRP channels lack a voltage sensor, they can be activated by physical or biochemical changes, regulating Ca2+ dynamics by directly conducting Ca2+ or prompting Ca2+ entry secondary to membrane depolarization and modulation of voltage-gated Ca2+ channels [5]. The activation of different isoforms of TRP is associated with cell-membrane depolarization, for example, in smooth muscle cells [6,7] and in cardiac cells [8,9,10].
There is substantial evidence that TRP channels have important roles in mediating cardiac pathological processes, including cardiac hypertrophy and fibrosis [11,12,13], which all lead to deleterious cardiac remodeling and subsequent heart failure (HF). This review focuses on the role of TRPC channels and provides an overview of the most relevant and recent findings related to these channels and ischemia-related disease in the heart. Nevertheless, the activation mechanism of TRPC channels is not yet completely clarified, and even less so in cardiac cells. Previous studies using different cell types suggest that TRPCs can interact physically with different splice variants of the inositol triphosphate receptors (IP3R). For instance, TRPC1 [14], TRPC3 [15,16], and a splice variant of human TRPC4 [17] interact physically with the IP3R. Actually, it appears that IP3R and Ca2+/calmodulin compete for a common binding site on TRPC3 since the displacement of calmodulin by IP3R from the binding domain activates TRPC3 [18]. Others researchers proved that phosphatidylinositol 4,5-bisphosphate (PIP2) participates in the regulation of TRPC4 and TRPC5 [19,20]. Gαq protein also activates TRPC1/4 and TRPC1/5 through direct interaction [21]. Meanwhile, independent studies demonstrated that TRPC3, 6, and 7 are activated by diacylglycerol (DAG) [22,23,24,25]. Interestingly, TRPC4 and 5 channels also become sensitive to DAG when their interactions with other regulators are inhibited, such as protein kinase C (PKC) and Na+/H+ exchanger regulatory factor (NHERF) [26].
2. TRPC Channels in the Cardiovascular System
TRPC channels are classified into seven members (TRPC1–7) that are distributed based on biochemical and functional similarities into TRPC1/4/5, TRPC3/6/7, and TRPC2, which is a pseudogene in humans. The expression of TRPC isoforms in the heart was examined in different stages of animal development, animal models, and areas of the heart. They are expressed at very low levels in normal adult cardiac myocytes but their expression and activity might increase in pathological processes [12,13,27]. However, they likely display different patterns of expression in cardiac cells isolated from the sinoatrial node and in myocytes isolated from atrial or ventricular heart [22,28]. In human cardiac tissues and/or neonatal rat cardiomyocytes, messenger RNA (mRNA) of TRPC5 [29,30] and TRPC6 [31] was detected. In animal models, the expression of TRPC1/3–7 was confirmed in adult rat and mouse ventricle and atrial cardiac myocytes either at mRNA or protein levels [13,32,33]. Other reports showed that TRPC1/3–6 are expressed in rat ventricular myocytes of fetal and neonatal ventricular myocytes [28,34]. In sinoatrial node cells, TRPC1, 2, 3, 4, 6, and 7 mRNA expression levels are detected using RT-qPCR, whereas TRPC5 expression is not observed. Furthermore, experiments using immunohistochemistry confirmed protein expression of TRPC1, 3, 4, and 6, but not TRPC7 in mouse sinoatrial node and in isolated pacemaker cells [35]. In the case of cardiac fibroblasts, all TRPC isoforms were described. In particular, the mRNAs of TRPC1, 3, 4, and 6 are detected in mouse cardiac fibroblasts. Meanwhile, isolated rat ventricular fibroblasts have significant mRNA expression of TRPC2, 3, and 5 [36,37,38]. Experiments using immunocytochemistry and Western blot also revealed the expression of TRPC1, 3, 4, and 6 proteins in rat and human cardiac fibroblasts [39,40,41].
A functional TRPC channel is composed of four proteins, allowing it to form homo or heterotetramers [42]. However, the concept of TRPC multimerization was barely addressed in cardiac myocytes. A previous study from Molkentin’s group suggested multimerization of TRPC3 and homotypic TRPC6 in adult mouse cardiac myocytes since they demonstrated, using an immunoprecipitation approach, that TRPC3 can associate with TRPC4 protein [5]. More recently, TRPC6 was suggested to form a heteromeric complex with TRPC3 and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase 2 (NOX2) protein in diabetic mouse heart. Nonetheless, this study used HEK293 cells to confirm the interaction between TRPC3 and TRPC6 by immunoprecipitation [43]. It should be noted that other studies indicated that TRPC channels can form a macromolecule complex with the NCX [44], Na+/K+ pump [45], and SERCA pump [46]. Therefore, they might create a microenvironment facilitating the fine-tuning of Ca2+ homeostasis and excitation–contraction coupling (reviewed elsewhere [47,48,49]). In fact, recent evidence confirmed that TRPC3 mediates Ca2+ and Na+ entry in proximity of NCX, elevating Ca2+ levels and cardiac contractility [44]. Certainly, more precise investigations about TRPC heteromerization will be welcome to reveal whether this concept is similar to that observed in other cells such as smooth muscle cell [50], platelets [51], hippocampus [52], or rat brain [53]. Actually, Bröker-Lai J et al. [52] combined quantitative high-resolution mass spectrometry with affinity purifications using isoform-specific antibodies on membrane fractions prepared from wild-type (WT) and target-knockout (KO) brains to demonstrate that TRPC1, 4, and 5 form heteromeric complexes in the brain, particularly in the hippocampus.
3. TRPC Channels Mediate Ca2+ Influx in Cardiac Myocytes
There are considerable indications that, in cardiac myocytes isolated from the atrium, the ventricle, or from neonatal rat ventricular myocytes (NRVM), TRPC channels participate both in store-operated Ca2+ entry (SOCE) and receptor-operated Ca2+ entry (ROCE) pathways, and their activation and/or upregulation is essential for cardiac Ca2+ signaling, particularly under pathological situations (reviewed elsewhere [54,55,56]). Independent studies showed that DAG, which works as an important mediator of the G-protein coupled receptor (GPCR)-stimulated Ca2+ signaling pathway, activates TRPC3 and 6. For instance, Onohara et al. [10] demonstrated that stimulation of NRVM with angiotensin-II and 1-oleoyl-2-acetyl-sn-glycerol (OAG), a membrane-permeable DAG analogue, activates TRPC3 and 6 channels, causing membrane depolarization. They further demonstrated that small interfering RNA (siRNA) against TRPC3 and 6 significantly reduces responses to angiotensin-II. OAG also activates a cation current in mouse cardiac myocytes that is significantly reduced by cell dialysis with an anti-TRPC3 antibody [57]. Moreover, the activation of A1 adenosine receptor in atrial and ventricular myocytes activates TRPC3, through DAG, since Ca2+ influx is inhibited by Pyr3, considered a specific inhibitor of TRPC3 [33].
It should be noted that other studies focused on the role of TRPC channels in SOCE activation in cardiac myocytes. For instance, a recent study by Wen et al. [58] demonstrated the presence of SOCE in normal adult mouse ventricular myocytes and the participation of TRPC1, 3, and 6, since antibodies against these TRPC channels reduced store depletion-mediated Ca2+ entry. Previously, Wu et al. [5] characterized the participation of TRPC3, 4, and 6 in the exacerbated SOCE observed in mouse cardiac myocytes from hypertrophic hearts. They demonstrated significant reduction of SOCE mediated by specific inhibition of SERCA with cyclopiazonic acid in cardiac-specific transgenic mice expressing dominant-negative (dn) TRPC3 (dn-TRPC3), dn-TRPC6, or dn-TRPC4. The participation of TRPC3 and 4 in SOCE was also characterized in adult rat ventricular myocytes induced by specific activation of EPAC (exchange protein directly activated by cyclic adenosine monophosphate (cAMP)) with 8-(4-Chlorophenylthio) (8-pCPT) [12]. This study revealed significant upregulation of TRPC3 and 4, which correlates with an SOCE increase in 8-CPT-treated cardiac myocyte. In addition, thapsigargin-induced SOCE is inhibited by Pyr3, a TRPC3 inhibitor [12]. Another study suggested a role of TRPC1, 4, and 5 in SOCE caused by aldosterone stimulation of NRVM. Indeed, thapsigargin-induced SOCE is inhibited in aldosterone-treated NRVM transfected with dn-TRPC1 and dn-TRPC4, and with siRNA against TRPC5, whereas dn-TRPC3 did not alter SOCE [59]. Moreover, TRPC1 and 4 overexpression correlates with calcium release activated channel (CRAC)-like current recorded in isolated hypertrophied right ventricular myocytes treated with monocrotaline [60]. Likewise, we proposed that at least TRPC5 may be critical in SOCE since its downregulation inhibits thapsigargin-induced potentiated SOCE in NRVM under ischemia and reperfusion. We further demonstrated that TRPC5 colocalizes with Orai1, the pore-forming sub-unit of store-operated Ca2+ channel (SOCC) [13]. More recently, Bartoli et al. [61] proposed that TRPC1 and 5 are involved in aldosterone activation of SOCE in adult rat ventricular cardiomyocytes. This study revealed that cardiac myocytes treated for 24 h with aldosterone enhance SOCE through the activation of mineralocorticoid receptor, and increase the store-operated Ca2+ current (ISOC), which correlates with specific overexpression of TRPC1 and 5, as well as stromal interaction molecule 1 (STIM1), but not of TRPC3, 4, or 6, nor of Orai1 and Orai3.
It is important to note that all these reports used agents that selectively deplete sarcoplasmic reticulum Ca2+ stores (e.g., cyclopiazonic acid, thapsigargin) to activate SOCC and avoid contribution of ROCE pathways. The combination of using different TRPC inhibitors, together with functional pore inhibitory antibodies for TRPC proteins and RNA silencing, suggests that TRPC channels account for the prominent SOCE in cardiac myocytes, especially under pathological conditions. Nevertheless and despite the increasing number of studies investigating SOCE in cardiac myocytes, the role of TRPC channels in SOCE is still controversial, which requires further investigation.
4. Role of TRPC Channels in Cardiac Pathophysiology
There is a general consensus that the overexpression and activation of TRPC channels are associated with deleterious cardiac pathology. As reviewed recently, under physiological conditions, the function of TRPC channels in the heart does not seem to be essential [4,62]. It appears that hearts from KO mice of different TRPC channels do not present any significant contractile abnormalities. Echocardiography analysis showed that TRPC3 KO and TRPC6 KO mice have similar resting left-ventricular mass and fractional shortening as compared to their respective littermate controls [63]. However, the induced stress-stimulated contractility, known as the Anrep effect, is diminished in isolated papillary muscles and cardiomyocytes from TRPC6 KO, but not TRPC3 KO mice [64]. In addition, TRPC1/4 double-KO mice have normal basal cardiac contractility, as well as normal systolic and diastolic functions. In contrast, isoproterenol-induced chronotropic responses are reduced in TRPC1/4 double-KO mice [65].
TRPC channels might play a role in some physiological processes. TRPC channels likely regulate cardiac pacemaking, conduction, ventricular activity, and contractility during cardiogenesis, through the interaction with the Cav1.2 channel in isolated hearts obtained from four-day-old chick embryos [22]. TRPC channels also contribute to Ca2+ homeostasis by directly conducting Ca2+ or indirectly via membrane depolarization and voltage-gated Ca2+ channel modulation. The resulting TRPC-mediated Ca2+ influx is required for the activation of signaling pathways that play minor roles in the healthy heart. For instance, they are involved in the activation of transcription factors promoting cardiac hypertrophy, fibrosis, and/or arrythmia [5,13,28,55,66,67,68,69]. Here, we discuss the role of TRPC channels in processes related to cardiac ischemic diseases.
5. Role of TRPC Channels in Cardiac Ischemia
5.1. TRPC Channels in Myocardial Infarction
One of the first pieces of evidence of the participation of TRPC in myocardial infarction (MI) was proposed using bioinformatic analysis combined with experimental approaches. Zhou et al. [70] demonstrated an increase in the expression of TRPC6, which was experimentally validated in a one-month post-MI rat model, suggesting TRPC6 as a potential therapeutic target for MI. Later, other studies highlighted the induction of TRPC proteins under MI and explored the idea that Ca2+ influx through TRPC channels overexpressed after MI contributes to cardiac dysfunction and adverse remodeling. In fact, significant increases in TRPC1, 3, 4, and 6 mRNA levels in mice one, two, and six weeks post MI were observed, as compared with sham [71]. This channel upregulation correlates with the increase in Ca2+ entry when myocytes isolated from MI adult mouse are stimulated with cyclopiazonic acid and OAG. Furthermore, mice expressing dn-TRPC4 have less pathological hypertrophy, better cardiac hemodynamic performance, and increased survival after MI, as compared with WT mice [71]. Therefore, the loss of TRPC4 function likely protects against the progression of cardiac dysfunction after MI. Interestingly, Jung et al. [72] suggested that gain of function of TRPC4 due to a genetic variation (I957V) causes an increase in channel activity, which has a protective effect against MI. The authors identified a single-nucleotide polymorphism (SNP) in TRPC4 that associates with MI risk in a case–control study. They further used multivariate analysis to show a protective effect of the I957V allele against MI risk, but only in diabetic patients. Therefore, the mutated TRPC4-I957V is thought to mediate higher Ca2+ signals, perhaps to facilitate the generation of endothelium and nitric oxide-dependent vasorelaxation. Nevertheless, the authors did not experimentally test this hypothesis. Recently, we observed significant dysregulation in the expression of several TRPC isoforms in a Wistar rat model of MI induced by transient ligation of the left coronary artery. A PCR-based micro-array, qRT-PCR, and Western blotting demonstrated significant upregulation of TRPC1, 3, 4, 5, and 6, whether in at-risk or in remote zones of infarcted hearts, as compared to sham. Specific downregulation of TRPC5 in MI rats infused with urocortin-2 at the onset of reperfusion was observed, offering a role of TRPC5 in cardioprotection [13].
In the case of TRPC3 and 6, a previous study determined that TRPC6 KO mice had significantly higher rates of mortality due to ventricular wall rupture throughout 3–7 days post MI [37]. In contrast, TRPC3/6/7 triple-KO mice subjected to transient MI (30 min of ischemia followed by 24 h reperfusion) exhibit reduced infarct size, better cardiac performance, and less cardiac tissue damage post MI, as compared with WT animals. In addition, they have reduced apoptosis through the inhibition of the calcineurin–nuclear factor of activated T cells (NFAT) signaling pathway [24]. These results suggest that TRPC3, 6, and 7 contribute significantly to worsening MI impacts on cardiac function. Further investigations will be welcome to clarify the discrepancy between these KO studies. It will be interesting to examine whether the cardioprotective effects observed in the triple-KO mice affect the transformation of myofibroblasts required during wound healing and scar formation.
5.2. TRPC Channel Role in Ischemia and Reperfusion Injuries and Cardioprotection
Ischemia and reperfusion (I/R) injury is the main cause of cell apoptosis and necrosis observed after an MI. Several studies demonstrated evidence linking cytosolic Ca2+ increase through TRPC and apoptosis after I/R [24,73]. Studies using TRPC inhibitors examined their role in I/R injuries. For instance, Kojima et al. [74] showed, in a Langendorff-perfused mouse heart under I/R, that left-ventricular functions are significantly improved by the administration of ion channel blockers (2-aminoethoxydiphenyl borate (APB) and La3+) during the initial 5 min of reperfusion, suggesting a TRPC channel role in contractile dysfunction in reperfused ischemic myocardium. In an atrial cardiac cell line, H9C2, the addition of SKF96365, another widely used inhibitor of TRPC, ameliorates injuries induced by hypoxia–reoxygenation (H/R) [24]. However, it is well known that 2-APB and La3+, as reviewed previously [75,76], as well as SKF96365 [75,76], are not specific to TRPC channels and may block other cationic channels. Therefore, these results should be supported by experiments using siRNA and/or TRPC-deficient mice. Actually, other reports used different molecular approaches to identify TRPC isoforms responsible for Ca2+ entry and its relationship with cardiac myocyte death under I/R. For example, Shan et al. [73] observed that transgenic mice overexpressing TRPC3 in myocardial cells are highly sensitive to injuries after I/R as they enhance apoptosis through increased TRPC3-mediated Ca2+ influx and calpain cleavage. They also demonstrated significant improvement in the viability of cardiomyocytes after SKF96365 treatment. Moreover, Meng et al. [77] observed that in vitro I/R increases TRPC6 protein expression, [Ca2+]i levels, and cell apoptotic rate in a time-dependent manner in H9C2 cell line. In addition, they suggested TRPC6 as a possible target for cardioprotection in H9C2 cells since the administration of danshensu, an active component of Salvia miltiorrhiza, protects against I/R injury by reducing TRPC6 expression via the c-Jun N-terminal kinases (JNK) signaling pathway [77]. Hang et al. [78] also demonstrated that brain-derived neurotrophic factor (BDNF) protects against MI and inhibits H/R-mediated cardiomyocyte apoptosis through TRPC3 and TRPC6 regulation.
On the other hand, the role of TRPC1 in I/R is still unclear. A recent study suggested that it is implicated in I/R injury, as the expressions of mRNA and protein of TRPC1, Orai1, and STIM1 are significantly increased in vivo in mice subjected to myocardial I/R injury and in vitro in H9C2 cells after H/R [79]. Interestingly, the suppression of STIM1 by siRNA decreases the expression of TRPC1 and Orai1, leading to decreased intracellular Ca2+ accumulation and apoptosis produced by H/R in H9C2 cells [79]. Therefore, STIM1 likely regulates the expression of TRPC1 and Orai1 in the context of apoptosis and myocardial I/R injury. In contrast, Al-Awar et al. [80] speculated that TRPC1 plays a cardioprotective role against I/R injury. They showed that sitagliptin, an inhibitor of dipeptidyl peptidase-4 (DPP-4), decreases the infarct size in a rat model of I/R which correlates with the increase in protein levels of TRPC1, TRPV1, and calcitonin gene-related peptide in heart tissue. Nevertheless, a specific experiment targeting TRPC1 was not shown. Our recent study, through Western blot, confirms that TRPC1 and 6 are upregulated in a rat model of I/R although they are not inhibited by urocortin-2-mediated cardioprotection. In contrast, urocortin-2 administration in NRVM undergoing in vitro I/R inhibits SOCE and prevents I/R-induced protein overexpression of TRPC5 and Orai1 [13]. Taking into consideration these results, further investigations are necessary to clarify the functional role of TRPC channel increase after I/R.
6. TRPC Channels in Post-Ischemia Cardiac Repair
After MI, the heart undergoes extensive adaptative processes and myocardial remodeling, involving angiogenesis, cardiac cell hypertrophy, and accumulation of fibrous tissue in both the infarcted and the non-infarcted myocardium, as reviewed previously [81,82,83]. Nonetheless, the role of the TRPC protein in cardiac repair still remains poorly studied.
6.1. TRPC Channels in Post-Ischemia Angiogenesis
Angiogenesis relies on new blood vessels forming from pre-existing vessels and the subsequent expansion of the vascular network in the body. Post-ischemic angiogenesis is considered a protective mechanism motivated by the lack of oxygen and blood supply necessary for physiological heart repair after a MI [84,85]. Angiogenesis involves sprouting, proliferation, migration, and tube formation thanks to the stimulation of endothelial cells (ECs) by growth factors such as vascular endothelial growth factor (VEGF), considered as the most potent pro-angiogenic factor specific for ECs (reviewed elsewhere [86,87]). Compelling evidence demonstrated that chronic and transient ischemia significantly increase the expression of VEGF [88,89,90]. Nevertheless, pre-clinical and clinical trials using solely pro-angiogenic factors, such as VEGF, were not shown to be effective in patients with stable angina or critical lower limb ischemia [91,92]. VEGF stimulates two tyrosine-kinase receptors, VEGFR-1 and VEGFR-2 [84,93], to increase [Ca2+]i in ECs involving Ca2+ release from intracellular stores and extracellular Ca2+ flux through cation channels, such as TRP channels [87,94]. There is increasing interest in the role of TRPC channels in angiogenesis, especially in studies related to cancer and diabetes [95,96,97]. ECs express different TRPC proteins involved in vascular function (TRPC1, 4, and 6), vascular tone remodeling (TRPC4), and oxidative stress-induced responses (TRPC3 and 4) [98]. It is apparent that TRPC3 and 6 are implicated in VEGF-mediated [Ca2+]i increase in ECs and angiogenesis. Indeed, VEGF- and OAG-induced extracellular signal-regulated kinases (ERK) 1/2 activation and tubulogenesis are significantly suppressed by TRPC3 inhibitor and siRNA in human umbilical vein ECs (HUVEC) [99]. Meanwhile, the overexpression of dn-TRPC6 in human microvascular ECs inhibits the VEGF-mediated [Ca2+]i increase, migration, sprouting, and proliferation, well-known hallmarks of angiogenesis [100]. In addition, TRPC4 siRNA attenuates oxidized low-density lipoprotein (oxLDL)-induced human coronary EC proliferation, migration, and angiogenesis tube formation [101].
Unfortunately, little is known regarding the role of TRPC channels during post-ischemic angiogenesis. In contrast, TRPC channels appear involved in hypoxia-induced angiogenesis [96,102]. For instance, the expression of TRPC4 protein is significantly upregulated in the retina under hypoxic condition. TRPC4 siRNA inhibits VEGF-induced migration and tube formation of retinal microvascular ECs, which suggests a role of TRPC4 in initiating neovascularization in response to VEGF in retina under hypoxia [96]. Moccia et al. [103] hypothesized and debated whether transfecting TRPC3 into autologous endothelial progenitor cells (EPCs) might enhance revascularization and functional recovery of ischemic hearts. However, functional experiments that tested this hypothesis were not performed. Recently, Zhu et al. [104] demonstrated that TRPC5 activation is necessary for EC sprouting, angiogenesis, and blood perfusion in a hind-limb ischemia model. TRPC5 downregulation prevents NFAT activation and EC tube formation under hypoxia. Moreover, TRPC5 KO mice have worse vascular recovery than WT mice after an ischemic injury. Finally, activation of TRPC5 by riluzole stimulates ECs sprouting and significantly improves limb recovery from ischemia injuries [104]. Therefore, it will be interesting to confirm the beneficial role of other TRPC channels in post-ischemic heart angiogenesis.
6.2. TRPC Channels in Early Adaptative Cardiac Remodeling
An early cardiac hypertrophy and fibrosis are considered compensatory events to the loss of cardiac myocytes, necessary for wound healing and scar formation after heart infarcts. However, prolonged hypertrophy could lead to the development of HF, arrhythmias, and even sudden cardiac death [105]. Since it is known that the activation of TRPC channels mediates the Ca2+ influx, which activates Ca2+ intracellular signaling pathways, such as calcineurin/NFAT, TRPC channels are suggested as Ca2+ effectors and transducers of hypertrophic genes in the heart. Little is known regarding TRPC channel implication in I/R-induced cardiac hypertrophy. In contrast, there is general agreement regarding the role of TRPC channels in pathological cardiac hypertrophy as a consequence of aortic constriction or under chronic GPCR stimulation using endothelin-1, phenylephrine, or angiotensin-II [31,106,107]. Similarly, Makarewich et al. [71] revealed an upregulation of TRPC1, 3, 4, and 6 channels in mice six weeks post MI as compared to sham animals, along with the activation of the so-called fetal gene program, commonly used as markers of cardiac hypertrophy. They also demonstrated that mice expressing dn-TRPC4 have less pathological hypertrophy, better cardiac hemodynamics performance, and increased survival after MI, as compared with wild-type (WT) mice, which all suggest a critical role of TRPC4 in post-MI heart damage. Cardiac hypertrophy is also observed in rat heart tissue as early as one week post I/R, which correlates with the upregulation of the expression of TRPC1, 3, 4, 5, and 6 mRNA [13] and the activation of the fetal gene program (unpublished data). Recently, Dragún et al. [108] examined the expression of TRP channels in 43 patients with end-stage HF. They discovered, among other TRP channels, a significant increase in TRPC1 and 5 gene expression, while TRPC4 expression was decreased in HF patients compared to a healthy donor. Also, they detected a significant correlation of the gene expression of TRPC1 and MEF2c (myocyte enhancer factor 2c), considered a key transcription factor for cardiac hypertrophy [109]. Interestingly, this pilot study did not detect any significant differences in TRP expression between male and female HF patients, nor between HF patients based on ischemic or non-ischemic background. Another recent study observed a similar increase in the expression of TRPC1 in hearts of patients with hypertrophic cardiomyopathy (HCM) or HF. This study further used human pluripotent stem cell lines of TRPC1 KO generated using clustered regularly interspaced short palindromic repeats (CRISPR)/ CRISPR-associated protein 9 (Cas9) to confirm the role of TRPC1 in regulating cardiac myocyte hypertrophy induced by phorbol 12-myristate 13-acetate (PMA), which was associated with abnormal activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [110]. Altogether, this indicates that TRPC channels might play a similar role in cardiac hypertrophy and HF, independently of patient background. Once TRPC expression is triggered, they perhaps activate several Ca2+-dependent factors of transcription and cardiac hypertrophy genes, leading to the same outcome, i.e., HF.
In the case of fibrosis, multiple well-known markers of fibrosis, hypertrophy, and Ca2+ handling protein were identified recently using genome-wide transcriptome analysis of infarcted hearts [111]. TRPC6 is considered a regulator of myofibroblast differentiation, a hallmark of fibrosis, since its silencing in human cardiac fibroblasts attenuates the transforming growth factor beta 1 (TGF-β1)-induced upregulation of alpha smooth muscle actin (α-SMA), a marker of myofibroblast transformation [112]. A recent study confirmed that the serum level of TGF-β1 is increased 28 days after MI in mice, accelerating cardiac fibrosis [113]. On the other hand, Saliba et al. [114] described that polyphenol extracted from grape pomace decreases angiotensin-II-induced Ca2+ entry through a direct regulation of TRPC3 and subsequent activation of NFATc3 in human ventricular cardiac fibroblasts, which abrogates myofibroblast differentiation and fibrosis by decreasing collagen secretion. However, the direct contribution of TRPC channels in cardiac fibrosis mediated by ischemia was barely addressed. Different isoforms of TRPC proteins are upregulated in rats showing fibrosis one week post I/R, although their direct role in promoting fibrosis was not examined [13]. Interestingly, TRPC6, through calcineurin–NFAT signaling, seems to be required for myofibroblast transformation after MI, a critical step during which collagen deposition and scar formation happen to maintain ventricular wall structural integrity in the early days post MI. In fact, TRPC6 KO mice show poor wound healing and fewer myofibroblasts, stained with α-SMA antibody, in the infarcted area [37]. Moreover and independently of studies related to ischemia and heart infarct, several reports proposed the participation of TRPC channels in the cardiac interstitial fibrosis caused by pressure overload by thoracic aortic constriction (TAC) in animal models or using vasoactive agonists, such as phenylephrine [36,106,115]. For instance, experiments performed in TRPC1/4 double-KO mice revealed significant amelioration of pressure overload-induced hypertrophy and interstitial fibrosis, which is explained by a reduced activity of TRPC1- and 4-dependent basal Ca2+ entry in adult ventricular myocytes [65]. At the same time, TRPC3 knockdown, using a small hairpin RNA lentivirus through the tail vein of mice, efficiently suppresses the extent of atrial fibrosis induced by TAC [116].
7. Concluding Remarks
In light of the reviewed studies, TRPC proteins stand out as key ion channels critical for cardiac cell responses under ischemic stress. A clearly defined role for specific TRPC isoforms in cellular events related to ischemic heart diseases still remains elusive, perhaps reflecting the complexity of these channels, the limitations of pharmacological tools, and the lack of specific inhibitors and antibodies. Nevertheless, TRPC channels were extensively studied since they sense and respond to a plethora of endogenous and exogenous stimuli by Ca2+ signaling in cardiac cells. Increasing evidence indicates that TRPC channels contribute to pathophysiological consequences of heart infarction, such as cardiac hypertrophy, fibrosis, and post-ischemic angiogenesis, as summarized in Figure 1. The potential to influence these outcomes by specifically modulating the expression and/or function of TRPC channels requires major efforts and more investigation. Further progress in the mechanistic understanding of TRPC channels will certainly help to identify new therapeutic targets for drug development to mitigate the impact of ischemia on cardiac function and to prevent cardiac transition from adaptive responses to harmful heart failure.
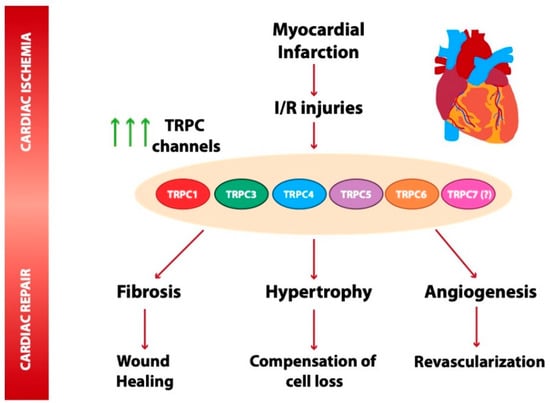
Figure 1.
Scheme summarizing transient receptor potential canonical (TRPC) channel isoforms dysregulated under myocardial infarction (MI) and ischemia and reperfusion. TRPC1, 3, 4, 5, and 6 are upregulated in mouse and rat animal models of MI [5,13,71]. Compelling evidence indicates that TRPC channel overexpression contributes to Ca2+ entry, mediating the activation of Ca2+-sensitive signaling pathways, such as calcineurin–NFAT, a critical pathway involved in apoptosis, cardiac hypertrophy, and fibrosis [13,28,55,66,67]. TRPC proteins are likely also involved in cardiac repair-related processes. The protective role played by TRPC6 in wound healing is of note [37]. Other studies suggested a role of TRPC channels, such as TRPC5, in angiogenesis and revascularization triggered post ischemia [104].
Funding
This research was funded by FEDER funds, by the Spanish Ministry of Economy and Competitiveness “BFU2016-74932-C2-1-P and BFU2016-74932-C2-2-P”, by the Institute of Carlos III “PI18/01197”, and by the Andalusia Government “PI-0193-2018 and PI-0313-2016”.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
Diacylglycerol (DAG); dominant-negative (dn); endothelial cells (ECs); hypoxia–reoxygenation (H/R); intracellular Ca2+ concentration ([Ca2+]i); inositol triphosphate receptors (IP3R); ischemia and reperfusion (I/R); heart failure (HF); knockout (KO); myocardial infarction (MI); neonatal rat ventricular myocytes (NRVM); 1-oleoyl-2-acetyl-sn-glycerol (OAG); sarco-endoplasmic reticulum Ca2+ ATPase (SERCA); sarcoplasmic Na+/Ca2+ exchanger (NCX); transient receptor potential (TRP); thoracic aortic constriction (TAC); vascular endothelial growth factor (VEGF); wild-type (WT).
References
- Fabiato, A.; Fabiato, F. Excitation-contraction coupling of isolated cardiac fibers with disrupted or closed sarcolemmas. Calcium-dependent cyclic and tonic contractions. Circ. Res. 1972, 31, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Rueda, A.; de Alba-Aguayo, D.R.; Valdivia, H.H. Ryanodine receptor, calcium leak and arrhythmias. Arch. Cardiol. Mex. 2014, 84, 191–201. [Google Scholar] [PubMed]
- Falcón, D.; Galeano-Otero, I.; Calderón-Sánchez, E.; Del Toro, R.; Martín-Bórnez, M.; Rosado, J.A.; Hmadcha, A.; Smani, T. TRP Channels: Current Perspectives in the Adverse Cardiac Remodeling. Front. Physiol. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Eder, P.; Chang, B.; Molkentin, J.D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 7000–7005. [Google Scholar] [CrossRef]
- Tsvilovskyy, V.V.; Zholos, A.V.; Aberle, T.; Philipp, S.E.; Dietrich, A.; Zhu, M.X.; Birnbaumer, L.; Freichel, M.; Flockerzi, V. Deletion of TRPC4 and TRPC6 in Mice Impairs Smooth Muscle Contraction and Intestinal Motility In Vivo. Gastroenterology 2009, 137, 1415–1424. [Google Scholar] [CrossRef]
- Noorani, M.M.Z.; Noel, R.C.; Marrelli, S.P. Upregulated TRPC3 and Downregulated TRPC1 Channel Expression during Hypertension is Associated with Increased Vascular Contractility in Rat. Front. Physiol. 2011, 2, 42. [Google Scholar] [CrossRef]
- Guinamard, R.; Chatelier, A.; Demion, M.; Potreau, D.; Patri, S.; Rahmati, M.; Bois, P. Functional characterization of a Ca 2+-activated non-selective cation channel in human atrial cardiomyocytes. J. Physiol. 2004, 558, 75–83. [Google Scholar] [CrossRef]
- Simard, C.; Sallé, L.; Rouet, R.; Guinamard, R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br. J. Pharmacol. 2012, 165, 2354–2364. [Google Scholar] [CrossRef]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006, 25, 5305–5316. [Google Scholar] [CrossRef]
- Ohba, T.; Watanabe, H.; Murakami, M.; Takahashi, Y.; Iino, K.; Kuromitsu, S.; Mori, Y.; Ono, K.; Iijima, T.; Ito, H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Rodríguez, A.; Ruiz-Hurtado, G.; Sabourin, J.; Gómez, A.M.; Alvarez, J.L.; Benitah, J.P. Proarrhythmic effect of sustained EPAC activation on TRPC3/4 in rat ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 87, 74–78. [Google Scholar] [CrossRef]
- Domínguez-Rodríguez, A.; Mayoral-Gonzalez, I.; Avila-Medina, J.; de Rojas-de Pedro, E.S.; Calderón-Sánchez, E.; Díaz, I.; Hmadcha, A.; Castellano, A.; Rosado, J.A.; Benitah, J.-P.; et al. Urocortin-2 Prevents Dysregulation of Ca2+ Homeostasis and Improves Early Cardiac Remodeling After Ischemia and Reperfusion. Front. Physiol. 2018, 9, 813. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Kiselyov, K.; Shin, D.M.; Chen, J.; Shcheynikov, N.; Kang, S.H.; Dehoff, M.H.; Schwarz, M.K.; Seeburg, P.H.; Muallem, S.; et al. Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 2003, 114, 777–789. [Google Scholar] [CrossRef]
- Adebiyi, A.; Thomas-Gatewood, C.M.; Leo, M.D.; Kidd, M.W.; Neeb, Z.P.; Jaggar, J.H. An elevation in physical coupling of type 1 inositol 1,4,5-trisphosphate (IP3) receptors to transient receptor potential 3 (TRPC3) channels constricts mesenteric arteries in genetic hypertension. Hypertension 2012, 60, 1213–1219. [Google Scholar] [CrossRef]
- Adebiyi, A.; Zhao, G.; Narayanan, D.; Thomas-Gatewood, C.M.; Bannister, J.P.; Jaggar, J.H. Isoform-selective physical coupling of TRPC3 channels to IP3 receptors in smooth muscle cells regulates arterial contractility. Circ. Res. 2010, 106, 1603–1612. [Google Scholar] [CrossRef]
- Mery, L.; Magnino, F.; Schmidt, K.; Krause, K.H.; Dufour, J.F. Alternative splice variants of hTrp4 differentially interact with the C-terminal portion of the inositol 1,4,5-trisphosphate receptors. FEBS Lett. 2001, 487, 377–383. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Tikunova, S.; Johnson, J.D.; Chen, Z.; Qin, N.; Dietrich, A.; Stefani, E.; Birnbaumer, L.; Zhu, M.X. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc. Natl. Acad. Sci. USA 2001, 98, 3168–3173. [Google Scholar] [CrossRef]
- Trebak, M.; Lemonnier, L.; Dehaven, W.I.; Wedel, B.J.; Bird, G.S.; Putney, J.W. Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflugers Arch. Eur. J. Physiol. 2009, 457, 757–769. [Google Scholar] [CrossRef]
- Otsuguro, K.I.; Tang, J.; Tang, Y.; Xiao, R.; Freichel, M.; Tsvilovskyy, V.; Ito, S.; Flockerzi, V.; Zhu, M.X.; Zholos, A.V. Isoform-specific inhibition of TRPC4 channel by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2008, 283, 10026–10036. [Google Scholar] [CrossRef]
- Myeong, J.; Ko, J.; Kwak, M.; Kim, J.; Woo, J.; Ha, K.; Hong, C.; Yang, D.; Kim, H.J.; Jeon, J.H.; et al. Dual action of the Gαq-PLCβ-PI(4,5)P2 pathway on TRPC1/4 and TRPC1/5 heterotetramers. Sci. Rep. 2018, 8, 12117. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, J.; Robin, E.; Raddatz, E. A key role of TRPC channels in the regulation of electromechanical activity of the developing heart. Cardiovasc. Res. 2011, 92, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Trebak, M. Transient receptor potential canonical 7: A diacylglycerol-activated non-selective cation channel. Handb. Exp. Pharmacol. 2014, 222, 189–204. [Google Scholar] [PubMed]
- He, X.; Li, S.; Liu, B.; Susperreguy, S.; Formoso, K.; Yao, J.; Kang, J.; Shi, A.; Birnbaumer, L.; Liao, Y. Major contribution of the 3/6/7 class of TRPC channels to myocardial ischemia/reperfusion and cellular hypoxia/reoxygenation injuries. Proc. Natl. Acad. Sci. USA 2017, 114, E4582. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Storch, U.; Forst, A.L.; Pardatscher, F.; Erdogmus, S.; Philipp, M.; Gregoritza, M.; Schnitzler, M.M.Y.; Gudermann, T. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc. Natl. Acad. Sci. USA 2017, 114, E37–E46. [Google Scholar] [CrossRef]
- Hof, T.; Chaigne, S.; Récalde, A.; Sallé, L.; Brette, F.; Guinamard, R. Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef]
- Ju, Y.-K.; Lee, B.H.; Trajanovska, S.; Hao, G.; Allen, D.G.; Lei, M.; Cannell, M.B. The involvement of TRPC3 channels in sinoatrial arrhythmias. Front. Physiol. 2015, 6, 86. [Google Scholar] [CrossRef]
- Bush, E.W.; Hood, D.B.; Papst, P.J.; Chapo, J.A.; Minobe, W.; Bristow, M.R.; Olson, E.N.; McKinsey, T.A. Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J. Biol. Chem. 2006, 281, 33487–33496. [Google Scholar] [CrossRef]
- Sunggip, C.; Shimoda, K.; Oda, S.; Tanaka, T.; Nishiyama, K.; Mangmool, S.; Nishimura, A.; Numaga-Tomita, T.; Nishida, M. TRPC5-eNOS axis negatively regulates ATP-induced cardiomyocyte hypertrophy. Front. Pharmacol. 2018, 9, 523. [Google Scholar] [CrossRef]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, W.; Liu, P.; Jiang, Y.; Zhao, Y.; Wei, H.; Niu, W. TRPC1 expression and distribution in rat hearts. Eur. J. Histochem. 2009, 53, 26. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sabourin, J.; Antigny, F.; Robin, E.; Frieden, M.; Raddatz, E. Activation of transient receptor potential canonical 3 (TRPC3)-mediated Ca2+ entry by A1 adenosine receptor in cardiomyocytes disturbs atrioventricular conduction. J. Biol. Chem. 2012, 287, 26688–26701. [Google Scholar] [CrossRef]
- Jiang, Y.; Huang, H.; Liu, P.; Wei, H.; Zhao, H.; Feng, Y.; Wang, W.; Niu, W. Expression and localization of TRPC proteins in rat ventricular myocytes at various developmental stages. Cell Tissue Res. 2014, 355, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.K.; Chu, Y.; Chaulet, H.; Lai, D.; Gervasio, O.L.; Graham, R.M.; Cannell, M.B.; Allen, D.G. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ. Res. 2007, 100, 1605–1614. [Google Scholar] [CrossRef]
- Numaga-Tomita, T.; Kitajima, N.; Kuroda, T.; Nishimura, A.; Miyano, K.; Yasuda, S.; Kuwahara, K.; Sato, Y.; Ide, T.; Birnbaumer, L.; et al. TRPC3-GEF-H1 axis mediates pressure overload-induced cardiac fibrosis. Sci. Rep. 2016, 6, 39383. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Burr, A.R.; Davis, G.F.; Birnbaumer, L.; Molkentin, J.D. A TRPC6-Dependent Pathway for Myofibroblast Transdifferentiation and Wound Healing In Vivo. Dev. Cell 2012, 23, 705–715. [Google Scholar] [CrossRef]
- Rose, R.A.; Hatano, N.; Ohya, S.; Imaizumi, Y.; Giles, W.R. C-type natriuretic peptide activates a non-selective cation current in acutely isolated rat cardiac fibroblasts via natriuretic peptide C receptor-mediated signalling. J. Physiol. 2007, 580, 255–274. [Google Scholar] [CrossRef]
- Harada, M.; Luo, X.; Qi, X.Y.; Tadevosyan, A.; Maguy, A.; Ordog, B.; Ledoux, J.; Kato, T.; Naud, P.; Voigt, N.; et al. Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 2012, 126, 2051–2064. [Google Scholar] [CrossRef]
- Ikeda, K.; Nakajima, T.; Yamamoto, Y.; Takano, N.; Tanaka, T.; Kikuchi, H.; Oguri, G.; Morita, T.; Nakamura, F.; Komuro, I. Roles of transient receptor potential canonical (TRPC) channels and reverse-mode Na+/Ca2+ exchanger on cell proliferation in human cardiac fibroblasts: Effects of transforming growth factor β1. Cell Calcium 2013, 54, 213–225. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Wu, H.-J.; Che, H.; Sun, H.-Y.; Cheng, L.-C.; Li, X.; Au, W.-K.; Tse, H.-F.; Li, G.-R. Functional transient receptor potential canonical type 1 channels in human atrial myocytes. Pflugers Arch. 2013, 465, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Numaga-Tomita, T.; Kitajima, N.; Toyama, T.; Harada, E.; Shimauchi, T.; Nishimura, A.; Ishikawa, T.; Kumagai, Y.; Birnbaumer, L.; et al. TRPC6 counteracts TRPC3-Nox2 protein complex leading to attenuation of hyperglycemia-induced heart failure in mice. Sci. Rep. 2017, 7, 7511. [Google Scholar] [CrossRef] [PubMed]
- Doleschal, B.; Primessnig, U.; Wölkart, G.; Wolf, S.; Schernthaner, M.; Lichtenegger, M.; Glasnov, T.N.; Kappe, C.O.; Mayer, B.; Antoons, G.; et al. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc. Res. 2015, 106, 163–173. [Google Scholar] [CrossRef]
- Goel, M.; Zuo, C.D.; Sinkins, W.G.; Schilling, W.P. TRPC3 channels co-localize with the Na+, Ca2+ exchanger and the Na+ pump in the axial component of the transverse-axial-tubular system (TATS) of rat ventricle. Am. J. Physiol. Heart Circ. Physiol. 2006, 292, H874–H883. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, L.; Trebak, M.; Lievremont, J.P.; Bird, G.S.; Putney, J.W. Protection of TRPC7 cation channels from calcium inhibition by closely associated SERCA pumps. FASEB J. 2006, 20, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef]
- Qu, Y.; Boutjdir, M. TRPC channels, an overarching Ca(2+) paradigm in the developing heart. Cardiovasc. Res. 2011, 92, 189–190. [Google Scholar] [CrossRef][Green Version]
- Abramowitz, J.; Birnbaumer, L. Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J. 2009, 23, 297–328. [Google Scholar] [CrossRef]
- Chen, J.; Crossland, R.F.; Noorani, M.M.Z.; Marrelli, S.P.; Marrelli, S.P. Inhibition of TRPC1/TRPC3 by PKG contributes to NO-mediated vasorelaxation. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, 417–424. [Google Scholar] [CrossRef]
- Jardin, I.; Gómez, L.J.; Salido, G.M.; Rosado, J.A. Dynamic interaction of hTRPC6 with the Orai1-STIM1 complex or hTRPC3 mediates its role in capacitative or non-capacitative Ca2+ entry pathways. Biochem. J. 2009, 420, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Bröker-Lai, J.; Kollewe, A.; Schindeldecker, B.; Pohle, J.; Nguyen Chi, V.; Mathar, I.; Guzman, R.; Schwarz, Y.; Lai, A.; Weißgerber, P.; et al. Heteromeric channels formed by TRPC 1, TRPC 4 and TRPC 5 define hippocampal synaptic transmission and working memory. EMBO J. 2017, 36, 2770–2789. [Google Scholar] [CrossRef] [PubMed]
- Goel, M.; Sinkins, W.G.; Schilling, W.P. Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 2002, 277, 48303–48310. [Google Scholar] [CrossRef] [PubMed]
- Flockerzi, V.; Nilius, B. TRPs: Truly remarkable proteins. Handb. Exp. Pharmacol. 2014, 222, 1–12. [Google Scholar]
- Eder, P. Cardiac Remodeling and Disease: SOCE and TRPC Signaling in Cardiac Pathology. Adv. Exp. Med. Biol. 2017, 993, 505–521. [Google Scholar]
- Abramowitz, J.; Yildirim, E.; Birnbaumer, L. The TRPC Family of Ion Channels: Relation to the TRP Superfamily and Role in Receptor- and Store-Operated Calcium Entry. In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2007; pp. 1–18. [Google Scholar]
- Fauconnier, J.; Lanner, J.; Sultan, A.; Zhang, S.; Katz, A.; Bruton, J.; Westerblad, H. Insulin potentiates TRPC3-mediated cation currents in normal but not in insulin-resistant mouse cardiomyocytes. Cardiovasc. Res. 2007, 73, 376–385. [Google Scholar] [CrossRef]
- Wen, H.; Zhao, Z.; Fefelova, N.; Xie, L.-H. Potential Arrhythmogenic Role of TRPC Channels and Store-Operated Calcium Entry Mechanism in Mouse Ventricular Myocytes. Front. Physiol. 2018, 9, 1785. [Google Scholar] [CrossRef]
- Sabourin, J.; Bartoli, F.; Antigny, F.; Gomez, A.M.; Benitah, J.P. Transient receptor potential canonical (trpc)/orai1-dependent store-operated Ca2+ channels; new targets of aldosterone in cardiomyocytes. J. Biol. Chem. 2016, 291, 13394–13409. [Google Scholar] [CrossRef]
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca2+ handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224. [Google Scholar] [CrossRef]
- Bartoli, F.; Moradi Bachiller, S.; Antigny, F.; Bedouet, K.; Gerbaud, P.; Sabourin, J.; Benitah, J.P. Specific Upregulation of TRPC1 and TRPC5 Channels by Mineralocorticoid Pathway in Adult Rat Ventricular Cardiomyocytes. Cells 2019, 9, 47. [Google Scholar] [CrossRef]
- Ahmad, A.A.; Streiff, M.; Hunter, C.; Hu, Q.; Sachse, F.B. Physiological and pathophysiological role of transient receptor potential canonical channels in cardiac myocytes. Prog. Biophys. Mol. Biol. 2017, 130, 254–263. [Google Scholar] [CrossRef]
- Seo, K.; Rainer, P.P.; Shalkey Hahn, V.; Lee, D.I.; Jo, S.H.; Andersen, A.; Liu, T.; Xu, X.; Willette, R.N.; Lepore, J.J.; et al. Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2014, 111, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.; Rainer, P.P.; Lee, D.-I.; Hao, S.; Bedja, D.; Birnbaumer, L.; Cingolani, O.H.; Kass, D.A. Hyperactive adverse mechanical stress responses in dystrophic heart are coupled to transient receptor potential canonical 6 and blocked by cGMP-protein kinase G modulation. Circ. Res. 2014, 114, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Camacho Londoño, J.E.; Tian, Q.; Hammer, K.; Schröder, L.; Camacho Londoño, J.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Heart J. 2015, 36, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Freichel, M.; Berlin, M.; Schürger, A.; Mathar, I.; Bacmeister, L.; Medert, R.; Frede, W.; Marx, A.; Segin, S.; Londoño, J.E.C. TRP Channels in the Heart. In Neurobiology of TRP Channels; CRC Press: Boca Raton, FL, USA, 2017; pp. 149–185. [Google Scholar]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordoñez, A.; Rosado, J.A.; Smani, T. The complex role of store operated calcium entry pathways and related proteins in the function of cardiac, skeletal and vascular smooth muscle cells. Front. Physiol. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed]
- Kirschmer, N.; Bandleon, S.; von Ehrlich-Treuenstätt, V.; Hartmann, S.; Schaaf, A.; Lamprecht, A.-K.; Miranda-Laferte, E.; Langsenlehner, T.; Ritter, O.; Eder, P. TRPC4α and TRPC4β Similarly Affect Neonatal Cardiomyocyte Survival during Chronic GPCR Stimulation. PLoS ONE 2016, 11, e0168446. [Google Scholar] [CrossRef]
- Zou, G.; Hong, H.; Lin, X.; Shi, X.; Wu, Y.; Chen, L. TRPC1, CaN and NFATC3 signaling pathway in the pathogenesis and progression of left ventricular hypertrophy in spontaneously hypertensive rats. Clin. Exp. Hypertens. 2015, 37, 223–234. [Google Scholar] [CrossRef]
- Zhou, R.; Hang, P.; Zhu, W.; Su, Z.; Liang, H.; Du, Z. Whole Genome Network Analysis of Ion Channels and Connexins in Myocardial Infarction. Cell. Physiol. Biochem. 2011, 27, 299–304. [Google Scholar] [CrossRef]
- Makarewich, C.A.; Zhang, H.; Davis, J.; Correll, R.N.; Trappanese, D.M.; Hoffman, N.E.; Troupes, C.D.; Berretta, R.M.; Kubo, H.; Madesh, M.; et al. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ. Res. 2014, 115, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Gené, G.G.; Tomás, M.; Plata, C.; Selent, J.; Pastor, M.; Fandos, C.; Senti, M.; Lucas, G.; Elosua, R.; et al. A gain-of-function SNP in TRPC4 cation channel protects against myocardial infarction. Cardiovasc. Res. 2011, 91, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Marchase, R.B.; Chatham, J.C. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia-reperfusion in adult mouse cardiomyocytes. Am. J. Physiol. Cell Physiol. 2008, 294, C833–C841. [Google Scholar] [CrossRef] [PubMed]
- Kojima, A.; Fukushima, Y.; Ito, Y.; Ding, W.-G.; Kitagawa, H.; Matsuura, H. Transient Receptor Potential Canonical Channel Blockers Improve Ventricular Contractile Functions After Ischemia/Reperfusion in a Langendorff-perfused Mouse Heart Model. J. Cardiovasc. Pharmacol. 2018, 71, 248–255. [Google Scholar] [PubMed]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, L.; Chen, K.-H.; Sun, H.-Y.; Jin, M.-W.; Xiao, G.-S.; Wang, Y.; Li, G.-R. SKF-96365 blocks human ether-à-go-go-related gene potassium channels stably expressed in HEK 293 cells. Pharmacol. Res. 2016, 104, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Li, W.-Z.; Shi, Y.-W.; Zhou, B.-F.; Ma, R.; Li, W.-P. Danshensu protects against ischemia/reperfusion injury and inhibits the apoptosis of H9c2 cells by reducing the calcium overload through the p-JNK-NF-κB-TRPC6 pathway. Int. J. Mol. Med. 2016, 37, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hang, P.; Zhao, J.; Cai, B.; Tian, S.; Huang, W.; Guo, J.; Sun, C.; Li, Y.; Du, Z. Brain-derived neurotrophic factor regulates TRPC3/6 channels and protects against myocardial infarction in rodents. Int. J. Biol. Sci. 2015, 11, 536–545. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Wu, Q.; Xu, B.; Wang, X.; Wu, J.; Huang, L.; Cheng, J. Suppression of Stim1 reduced intracellular calcium concentration and attenuated hypoxia/reoxygenation induced apoptosis in H9C2 cells. Biosci. Rep. 2017, 37, BSR20171249. [Google Scholar] [CrossRef]
- Al-Awar, A.; Almási, N.; Szabó, R.; Takacs, I.; Murlasits, Z.; Szűcs, G.; Török, S.; Pósa, A.; Varga, C.; Kupai, K. Novel Potentials of the DPP-4 Inhibitor Sitagliptin against Ischemia-Reperfusion (I/R) Injury in Rat Ex-Vivo Heart Model. Int. J. Mol. Sci. 2018, 19, 3226. [Google Scholar] [CrossRef]
- Garza, M.A. Cardiac remodeling and physical training post myocardial infarction. World J. Cardiol. 2015, 7, 52. [Google Scholar] [CrossRef]
- Chiarella-Redfern, H.H.; Rayner, K.J.; Suuronen, E.J. Spatio-temporal expression patterns of microRNAs in remodelling and repair of the infarcted heart. Histol. Histopathol. 2015, 30, 141–149. [Google Scholar]
- Mouton, A.J.; Rivera, O.J.; Lindsey, M.L. Myocardial infarction remodeling that progresses to heart failure: A signaling misunderstanding. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H71–H79. [Google Scholar] [CrossRef] [PubMed]
- Ingason, A.B.; Goldstone, A.B.; Paulsen, M.J.; Thakore, A.D.; Truong, V.N.; Edwards, B.B.; Eskandari, A.; Bollig, T.; Steele, A.N.; Woo, Y.J. Angiogenesis precedes cardiomyocyte migration in regenerating mammalian hearts. J. Thorac. Cardiovasc. Surg. 2018, 155, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Melly, L.; Cerino, G.; Frobert, A.; Cook, S.; Giraud, M.N.; Carrel, T.; Tevaearai Stahel, H.T.; Eckstein, F.; Rondelet, B.; Marsano, A.; et al. Myocardial infarction stabilization by cell-based expression of controlled Vascular Endothelial Growth Factor levels. J. Cell. Mol. Med. 2018, 22, 2580–2591. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-González, I.; Galeano-Otero, I.; Redondo, P.C.; Rosado, J.A.; Smani, T. Pathophysiological Significance of Store-Operated Calcium Entry in Cardiovascular and Skeletal Muscle Disorders and Angiogenesis. Adv. Exp. Med. Biol. 2020, 1131, 489–504. [Google Scholar] [PubMed]
- Smani, T.; Gómez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.-M.; Rosado, J.A. TRP Channels in Angiogenesis and Other Endothelial Functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef] [PubMed]
- Robich, M.P.; Matyal, R.; Chu, L.M.; Feng, J.; Xu, S.H.; Laham, R.J.; Hess, P.E.; Bianchi, C.; Sellke, F.W. Effects of neuropeptide Y on collateral development in a swine model of chronic myocardial ischemia. J. Mol. Cell. Cardiol. 2010, 49, 1022–1030. [Google Scholar] [CrossRef]
- Arif, M.; Pandey, R.; Alam, P.; Jiang, S.; Sadayappan, S.; Paul, A.; Ahmed, R.P.H. MicroRNA-210-mediated proliferation, survival, and angiogenesis promote cardiac repair post myocardial infarction in rodents. J. Mol. Med. 2017, 95, 1369–1385. [Google Scholar] [CrossRef]
- Chen, Z.; Li, B.; Dong, Q.; Qian, C.; Cheng, J.; Wang, Y. Repetitive Transient Ischemia-Induced Cardiac Angiogenesis is Mediated by Camkii Activation. Cell. Physiol. Biochem. 2018, 47, 914–924. [Google Scholar] [CrossRef]
- Muona, K.; Mäkinen, K.; Hedman, M.; Manninen, H.; Ylä-Herttuala, S. 10-Year safety follow-up in patients with local VEGF gene transfer to ischemic lower limb. Gene Ther. 2012, 19, 392–395. [Google Scholar] [CrossRef]
- Henry, T.D.; Annex, B.H.; McKendall, G.R.; Azrin, M.A.; Lopez, J.J.; Giordano, F.J.; Shah, P.K.; Willerson, J.T.; Benza, R.L.; Berman, D.S.; et al. The VIVA trial: Vascular endothelial growth factor in ischemia for vascular angiogenesis. Circulation 2003, 107, 1359–1365. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular Endothelial Growth Factor Stimulates Endothelial Colony Forming Cells Proliferation and Tubulogenesis by Inducing Oscillations in Intracellular Ca2+ Concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC Channel Dependence of VEGF-Activated Ca2+ Entry and Endothelial Tube Formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, R.; Schlotterer, A.; Schumacher, D.; Matka, C.; Mathar, I.; Dietrich, N.; Medert, R.; Kriebs, U.; Lin, J.; Nawroth, P.; et al. TRPC proteins contribute to development of diabetic retinopathy and regulate glyoxalase 1 activity and methylglyoxal accumulation. Mol. Metab. 2018, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Song, H.B.; Jun, H.O.; Kim, J.H.; Fruttiger, M.; Kim, J.H. Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell Calcium 2015, 57, 101–108. [Google Scholar] [CrossRef]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. Trpc6 channels are required for proliferation, migration and invasion of breast cancer cell lines by modulation of orai1 and orai3 surface exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef]
- Yip, H.; Chan, W.Y.; Leung, P.C.; Kwan, H.Y.; Liu, C.; Huang, Y.; Michel, V.; Yew, D.T.W.; Yao, X. Expression of TRPC homologs in endothelial cells and smooth muscle layers of human arteries. Histochem. Cell Biol. 2004, 122, 553–561. [Google Scholar] [CrossRef]
- Andrikopoulos, P.; Eccles, S.A.; Yaqoob, M.M. Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell. Signal. 2017, 37, 12–30. [Google Scholar] [CrossRef]
- Hamdollah Zadeh, M.A.; Glass, C.A.; Magnussen, A.; Hancox, J.C.; Bates, D.O. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 2008, 15, 605–614. [Google Scholar] [CrossRef]
- Qin, W.; Xie, W.; Xia, N.; He, Q.; Sun, T. Silencing of Transient Receptor Potential Channel 4 Alleviates oxLDL-induced Angiogenesis in Human Coronary Artery Endothelial Cells by Inhibition of VEGF and NF-κB. Med. Sci. Monit. 2016, 22, 930–936. [Google Scholar] [CrossRef]
- Fantozzi, I.; Zhang, S.; Platoshyn, O.; Remillard, C.V.; Cowling, R.T.; Yuan, J.X.J. Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1233–L1245. [Google Scholar] [CrossRef]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca2+ signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Gao, M.; Zhou, T.; Xie, M.; Mao, A.; Feng, L.; Yao, X.; Wong, W.T.; Ma, X. The TRPC5 channel regulates angiogenesis and promotes recovery from ischemic injury in mice. J. Biol. Chem. 2019, 294, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Hanif, W.; Alex, L.; Su, Y.; Shinde, A.V.; Russo, I.; Li, N.; Frangogiannis, N.G. Left atrial remodeling, hypertrophy, and fibrosis in mouse models of heart failure. Cardiovasc. Pathol. 2017, 30, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Niizeki, T.; Takeishi, Y.; Kitahara, T.; Arimoto, T.; Ishino, M.; Bilim, O.; Suzuki, S.; Sasaki, T.; Nakajima, O.; Walsh, R.A.; et al. Diacylglycerol kinase-ε restores cardiac dysfunction under chronic pressure overload: A new specific regulator of Gαq signaling cascade. Am. J. Physiol. Circ. Physiol. 2008, 295, H245–H255. [Google Scholar] [CrossRef]
- Seth, M.; Zhang, Z.-S.; Mao, L.; Graham, V.; Burch, J.; Stiber, J.; Tsiokas, L.; Winn, M.; Abramowitz, J.; Rockman, H.A.; et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 2009, 105, 1023–1030. [Google Scholar] [CrossRef]
- Dragún, M.; Gažová, A.; Kyselovič, J.; Hulman, M.; Máťuš, M. TRP Channels Expression Profile in Human End-Stage Heart Failure. Medicina 2019, 55, 380. [Google Scholar] [CrossRef]
- Duran, J.; Lagos, D.; Pavez, M.; Troncoso, M.F.; Ramos, S.; Barrientos, G.; Ibarra, C.; Lavandero, S.; Estrada, M. Ca2+/calmodulin-dependent protein kinase II and androgen signaling pathways modulate MEF2 activity in testosterone-induced cardiac myocyte hypertrophy. Front. Pharmacol. 2017, 8, 604. [Google Scholar] [CrossRef]
- Tang, L.; Yao, F.; Wang, H.; Wang, X.; Shen, J.; Dai, B.; Wu, H.; Zhou, D.; Guo, F.; Wang, J.; et al. Inhibition of TRPC1 prevents cardiac hypertrophy via NF-κB signaling pathway in human pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2019, 126, 143–154. [Google Scholar] [CrossRef]
- Lacraz, G.P.A.; Junker, J.P.; Gladka, M.M.; Molenaar, B.; Scholman, K.T.; Vigil-Garcia, M.; Versteeg, D.; De Ruiter, H.; Vermunt, M.W.; Creyghton, M.P.; et al. Tomo-Seq Identifies SOX9 as a Key Regulator of Cardiac Fibrosis during Ischemic Injury. Circulation 2017, 136, 1396–1409. [Google Scholar] [CrossRef]
- Kapur, N.K.; Qiao, X.; Paruchuri, V.; Mackey, E.E.; Daly, G.H.; Ughreja, K.; Morine, K.J.; Levine, J.; Aronovitz, M.J.; Hill, N.S.; et al. Reducing endoglin activity limits calcineurin and TRPC-6 expression and improves survival in a mouse model of right ventricular pressure overload. J. Am. Heart Assoc. 2014, 3, e000965. [Google Scholar] [CrossRef]
- Gao, S.; Li, L.; Li, L.; Ni, J.; Guo, R.; Mao, J.; Fan, G. Effects of the combination of tanshinone IIA and puerarin on cardiac function and inflammatory response in myocardial ischemia mice. J. Mol. Cell. Cardiol. 2019, 137, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Saliba, Y.; Jebara, V.; Hajal, J.; Maroun, R.; Chacar, S.; Smayra, V.; Abramowitz, J.; Birnbaumer, L.; Farès, N. Transient Receptor Potential Canonical 3 and Nuclear Factor of Activated T Cells C3 Signaling Pathway Critically Regulates Myocardial Fibrosis. Antioxid. Redox Signal. 2019, 30, 1851–1879. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Tang, Y.; Li, S.; Wu, Y.; Chen, X.; Wu, Q.; Hong, K.; Li, J. Protective mechanism of SIRT1 on Hcy-induced atrial fibrosis mediated by TRPC3. J. Cell. Mol. Med. 2020, 24, 488–510. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).