1. Introduction
Diverse pathways that serve to modulate transcript abundance and translation efficiency dynamically regulate gene expression levels. The rates of transcription, maturation, transportation, and degradation mainly determined the abundance of transcripts [
1]. After transported from the nucleus to cytoplasm, the abundance of mature mRNAs is negatively regulated by the decay rate. The bulk of eukaryotic mRNAs are degraded in a deadenylation-dependent pathway, in which deadenylation is the first and rate-limiting step [
2,
3]. The long poly(A) tails at the mRNA 3′-end are removed by deadenylases, a group of 3′-exonucleases with high specificity of poly(A) as the substrate [
1,
2,
3,
4]. Eukaryotic cells generally possess up to a dozen of deadenylases that can be classified into the DEDD and endonuclease-exonuclease-phosphatase (EEP) families [
1,
4]. Various deadenylases have distinct intracellular functions that are specified by their dissimilar enzymatic properties, binding partners, and subcellular localizations. Most deadenylases have multiple cellular localizations and they can shuttle between the nucleus and cytoplasm. Among them, CCR4 and CAF1 has been observed to exist in processing bodies in some cell types [
5,
6], poly(A)-specific ribonuclease (PARN) is abundant in the nucleoli and Cajal bodies [
7,
8], while mammalian PARN-like ribonuclease domain containing 1 (PNLDC1), has been shown to have potential endoplasmic reticulum (ER) distribution [
9]. From the developmental view, PNLDC1 is highly expressed during early development to act as a pre-piRNA trimmer [
9,
10,
11], while the other deadenylases are ubiquitous and expressed with high levels in most cell types [
1,
4]. Abnormality in the expression levels or occurrence of inherited mutations in deadenylases have been linked to human diseases, such as cancer, immune disorder, acute leukemias, developmental delay, bone marrow failure, and dyskeratosis congenital [
12,
13,
14,
15,
16,
17].
Most stable eukaryotic mRNAs have long poly(A) tails of up to ~250 nucleotides [
18], which provide multiple binding sites for the poly(A) binding protein (PABP) [
19]. PABP affects mRNA stability and translation efficiency in a complicate way, probably depending on its binding partners. During active translation, PABP interacts with the 5′-cap-associated eukaryotic initiation factor and thus contributes to the formation of a closed-loop mRNA to facilitate efficient translation [
20,
21,
22]. At the onset of translation repression, PABP stimulates the deadenylase activity of the PAN2-PAN3 complex or CAF1 and it can enhance miRNA-mediated gene silencing [
23,
24]. Very recently, PABP was found to bind CCR4 and regulate the deadenylation activity of the CCR4-NOT complex in human cells and yeasts [
25,
26]. Studies in embryonic cells have suggested that the polyadenylation status of an mRNA is positively coupled to its stability, potency to form a circular structure, assembly of polysomes, and translation efficiency [
27,
28,
29,
30,
31,
32]. This proposal has also been verified in adult mouse heart cells [
33]. However, the genome-wide studies challenge this mechanism and suggest that the abundant mRNAs that are highly expressed in somatic cells may have short poly(A) tails to optimize the translational functions [
34,
35,
36]. ER stress has been found to induce an increase in the poly(A) tail length of the ER stress-induced genes, which positively correlates to their de-repression and stabilization. Meanwhile, the repressed mRNAs in the stress-induced RNA granules generally have shorter poly(A) tails [
37]. It is possible that the cells utilize both tail-length-dependent and -independent mechanisms to reshape translation globally. It is clear that the change of tail-length contributes to translation regulation despite the controversial opinions in the relationship between poly(A) tail length and translation efficiency.
Translation regulation includes not only the control of gene expression levels, but also the spatiotemporal production of proteins. In eukaryotic cells, the concept of translational compartmentalization has been widely recognized since the groundbreaking work by Palade and his coworkers [
38]. In the classical view, ribosomes have two distinct populations: on the ER surface or in the cytosol. The mRNAs encoding membrane, organelle and secreted proteins are sorted to translate on the ER, while the cytosolic and nuclear proteins are produced in the cytosol. The sorting of translation sites is achieved by the recognition of the signaling peptide within nascent proteins by the signal recognition particle (SRP) [
39], which delivers on-translation mRNAs to the SRP receptor that is located at the rough ER membrane [
40]. Recent findings demonstrate that, besides the canonical SRP co-translational delivering pathway, mRNAs can be conveyed to the ER by an alternative pathway independent of SRP or translation [
41,
42]. Furthermore, transcriptome and single-molecule studies reveal that a number of mRNAs encoding cytosolic and nuclear proteins associate with the ER in a translation-dependent manner [
43,
44]. The ER-bound mRNAs have higher ribosome occupancies than those that are retained in the cytosol and they can return back to the cytosol after translation [
43].
Although translation is highly localized, the degradation of mRNAs seems likely to occur in the cytosol, since the ER-associated mRNAs are generally occupied by ribosomes and in efficient translation. This leads to an unproved hypothesis that the ER-associated mRNAs need to be released to the cytosol for further deadenylation or degradation purpose. Is it possible for the ER-associated mRNAs to be deadenylated or degraded on the ER surface in situ? Furthermore, can the translation efficiency of the ER-bound mRNAs be re-modulated by the change of tail length but without dissociation from ER? To address these problems, the key is to identify whether there are ER-anchored active deadenylases. Presently, PNLDC1 is the only known deadenylase with potential ER localization [
9]. However, PNLDC1 is a pre-piRNA trimmer during early development and it has low abundance in somatic cells [
9,
10,
11]. In this research, we showed that PARN, the homologue of PNLDC1, has obvious ER distribution in the HeLa and HEK-293T cells, as well as in mouse tissues. We found that PARN overexpression reshapes the poly(A) tail length profile of the ER-associated mRNAs, but not the cytosolic mRNAs. Although the ER-anchored PARN has no effect on the global translation efficiency, it can modulate the transcript levels and translation efficiency of a small subset of mRNAs, particularly of genes that are involved in DNA-damage response and cell cycle control. Strikingly, we observed that PARN overexpression significantly reduces the number of transcripts of several mRNAs, but greatly enhances their translation rates by remodeling the polysome profile. These findings shed new lights on the mechanistic understanding of the regulated communications between mRNA decay and translation on the ER surface.
2. Materials and Methods
2.1. Materials
Rabbit anti-PARN antibody (ab125185 and ab188333) and mouse anti-PDI antibody were purchased from Abcam (Cambridge, MA, USA). Rabbit anti-PARN antibody (A6941) was obtained from Abclonal (Cambridge, MA, USA). Rabbit anti-MAPKAPK-2 antibody (#3042) was from Cell Signaling Technology (Beverly, MA, USA). Mouse antibodies against calnexin, CNOT6, and CNOT7 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Mouse anti-GAPDH antibody was from Bioworld (Louis Park, MN, USA). Rabbit antibodies against LaminA/C, CNOT7, CNOT8, PAN2, MDM2, and RPS3 were from Proteintech (Chicago, IL, USA). The anti-Flag mouse antibody and anti-PNLDC1 rabbit antibody were from Sigma-Aldrich (St Louis, MO, USA). Rabbit antibodies against CDKL1, CCNT2, CLOCK, and MATR3 were purchased from BBI Life Sciences Corp. (Shanghai, China). Horseradish peroxidase (HRP)-conjugated secondary antibodies for Western blot were obtained from Yeasen (Shanghai, China). HRP-labeled sreptavidin for biotin-labeled RNA detection was from Beyotime (Beijing, China). Dylight 488/549/594/649 goat-anti-rabbit/mouse secondary antibodies for immunofluorescence observations were from Bioworld. The transfection reagent Lipofectamine™ and the RNAiMAX Reagent were from Invitrogen (Carlsbad, CA, USA) and Vigofect was from Vigorous (Beijing, China). Doxorubicin hydrochloride (DOX) was from Solarbio (Beijing, China). Cycloheximide (CHX) was from AMRESCO (Solon, OH, USA) and was dissolved in DMSO (Sigma-Aldrich, St Louis, MO, USA). Digitonin, hydroxyurea (HU), urea, and protease inhibitor cocktail used for mammalian cellular extraction were from Sigma. CMPD1 was purchased from Santa Cruz. Recombinant RNasin
® ribonuclease inhibitor and sequencing grade modified trypsin were purchased from Promega (Madison, WI, USA). RNase A/T1 Mix and yeast total RNA were from Thermo Scientific (Waltham, MA, USA). NP-40, DNase/RNase-free sucrose and sodium deoxycholate (DOC) were from AMRESCO. Oligo (dT)
25 cellulose beads for mRNA isolation and the low range ssRNA ladder were purchased from New England Biolabs (Ipswich, MA, USA). Sodium dodecyl sulfate (SDS) and paraformaldehyde were from Merck (Darmstadt, Germany). 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DPPS) was from Avanti Polar Lipids (Alabaster, AL, USA). The kit for cytosolic and membrane proteins extraction was from KeyGen biotech (Nanjing, China). The kits for RNA 3′end biotin labeling and chemiluminescent nucleic acid detection were from Thermo Scientific. Viewsolid synthesized RNA oligos for PARN knockdown [
7]. Takara synthesized Polyadenylate A
20 and A
200 was from Sigma. All other chemicals were of analytical grade.
2.2. Cell Culture
The HEK-293T and HeLa cell lines were purchased from the China Center of American Type Culture Collection (ATCC, Wuhan, China) and then cultured in the Dulbecco’s modified Eagle’s medium (DMEM, Gibco) with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA) at 37 °C with 5% CO
2. Plasmids containing the wild-type (WT), the truncated form or site mutated human PARN for cell transfection, were constructed while using pcDNA3.1 (N-Flag), pEGFP-C3, and -N1 vectors. The primers that were used for mutagenesis and plasmid construction were the same as those described previously [
16,
45,
46]. The cells were seeded, transfected using the Lipofectamine™ RNAiMAX Reagent or Vigofect according to the manufacturer’s instructions, cultured for 24 h, and then harvested for further analysis. The knockdown of PARN by siRNA was carried out while using the procedures described previously [
7] and the cells were harvested after transfection and then cultured for 72 h. UV treatment was carried out by exposing the cells to UV irradiation for 20 min., and the cells were then cultured in fresh DMEM cell culture medium for 2 h.
2.3. Cell Fractionation
Differential detergent fractionation was used to separate the cytosolic and membrane fractions by a fractionation kit that was provided by KeyGen (Nanjing, China). The fractionation was performed according to the manufacturer’s instructions.
Cell fractionation by differential centrifugation after syringe homogenization was carried out while using a 10-cm dish of HEK-293T cells. The cells were washed twice with 10 mL ice-cold phosphate buffered saline (PBS) and then scraped in 1 mL ice-cold PBS with 1 mM DTT and 1× protease inhibitor cocktail. Subsequently, the cells were transferred to a 1.5 mLEppendorf tube and homogenized by a 25-gauge syringe on ice. The homogenate (whole cell lysates) was centrifuged at 1000× g for 10 min. to remove unbroken cells, nuclei and cell debris. The supernatant fraction was then centrifuged at 20,000× g for 10 min. to remove the large organelles, followed by centrifugation at 100,000× g for 60 min. at 4 °C in a Beckman TLA 55 rotor to separate cytosol from microsomes.
Cell fractionation by differential centrifugation after Dounce homogenization was performed while using a 15-cm dish of the HeLa cells. The cells were washed twice with 10 mL ice-cold PBS and then scraped in 4 mL ice-cold homogenate buffer containing 10 mM HEPES-KOH (pH 7.5) buffer, 10 mM KCl, 1 mM MgCl2, 1 mM DTT, and 1× protease inhibitor cocktail. The cell suspension was transferred into a pre-cooled 5 mL Dounce homogenizer and homogenized with 15–20 strokes while using the pestle at 4 °C. Subsequently, the homogenates were transferred into a new Eppendorf tube with the addition of 1/10 volume of 2.5 M sucrose to make a 250 mM isotonic solution and then subjected to differential centrifugation. The fractions were obtained by collecting the cell pellets after sequential differential centrifugation of the supernatant fraction, as follows: nucleus, mitochondria, and large membrane fractions were obtained from the pellets after centrifuging at 700× g, heavy mitochondria and membrane debris at 3000× g, mitochondria, lysosome, peroxisome, and the intact Golgi apparatus at 6000× g, mitochondria, lysosome, peroxisome, and the Golgi membrane at 10,000× g, lysosome, peroxisome, the Golgi membrane, and large, high-density vesicle from rough endoplasmic reticulum at 20,000× g, and all of the vesicles from the endoplasmic reticulum (ER), the plasma membrane, the Golgi membrane, and endosomes at 100,000× g for 60 min. All of the fractions were washed with the HM buffer twice and then re-suspended in the RIPA buffer with the addition of 1 × protease inhibitor cocktail.
The isolation of the microsomes and mitochondria was performed while using the published protocols [
47]. In brief, a 15-cm dish of the HeLa cells with about 95% consistency was used for the isolation. After homogenization using the pestle to disrupt 80–90% of cells and remove of the nucleus and cell debris by centrifugation at 600×
g for 10 min. at 4 °C twice, the pellets isolated by centrifugation at 7000×
g were re-suspended to obtain the Mt0 fraction, further centrifuged at 7000×
g for 10 min. to obtain the Mt1 fraction, centrifuged at 10,000×
g to obtain the Mt2 fraction (crude mitochondria) from the pellets. The supernatants and pellets were collected for each step of separation and they were used for further western blot analysis with an equal amount of total proteins.
2.4. Extraction of ER-Bound Proteins from Mouse Tissues
ER-bound proteins were extracted from mouse lung, liver, heart, and kidney tissues while using a kit from Bestbio (BB-31454, Shanghai, China). Six to eight-week-old male mice (C57BL/6N) were sacrificed under guidelines and approved by IACUC of Tsinghua University. All of the methods were performed in accordance with the relevant guidelines and regulations. Protease inhibitor cocktail (Sigma) was added to all buffers. 50–100 mg fresh tissues were washed by ice-cold PBS, minced into small pieces, and then washed by ice-cold PBS twice. The tissue cells were lysed with 500 μL buffer A with the addition of PMSF and protease inhibitor cocktail for 10 min. on ice. The cell suspensions were transferred into a clean and pre-cooled 5 mL glass homogenizer and homogenized with 30–40 strokes while using pestle. The tissue homogenates were centrifuged at 1000× g at 4 °C. The pellets (nucleus and cell debris) were resuspended in the RIPA buffer, while the supernatants were transferred to a new pre-cooled tube and then centrifuged at 11,000× g at 4°C, followed by 50,000× g at 4 °C by the TLA-55 rotor (Beckman) for 45 min. to obtain the cytosolic protein enriched fraction from the supernatants. Afterwards, the pellets were washed by 400 μL buffer B, resuspended in 150 μL buffer C on ice for 20 min. to obtain the ER fraction. The BCA kit measured the protein concentration and an equal amount of total proteins was used for Western blot analysis while using rabbit antibody towards PARN (ab188333, Abcam), GAPDH (10494-1-AP, Proteintech), calnexin (10427-2-AP, Proteintech), and laminA/C (10298-1-AP, Proteintech).
2.5. Extraction of Membrane Proteins and Trypsin Digestion Assay
The membrane proteins were isolated from the pellet fraction of the 100,000× g centrifugation samples that were obtained from the cell fractionation assay described above. The pellets were collected, washed twice with PBS (pH 7.4), re-suspended in PBS, and then divided into five aliquots. The five aliquots were treated with 100 μL PBS buffer without any additions (control), with the addition of 1% Triton X-100, 1% SDS, 0.1 M sodium carbonate (Na2CO3, pH 11.5), or 1 M sodium chloride (NaCl) for 60 min. After treatment, the samples were centrifuged at 100,000× g for 60 min. to separate soluble and insoluble proteins. The pellets were subsequently re-suspended in the same volume of PBS buffer. Trypsin digestion assay were performed to detect membrane protein topology. The pellets of the 100,000× g centrifugation samples that were obtained from cell fractionation assay described above were treated by trypsin digestion. The final concentration of trypsin was 25 μg/mL and the mass ratio of protease to total protein was about 1:80. The reaction was conducted at 37 °C for different time intervals ranging from 0 to 60 min. After treatment, the protease inhibitor cocktail was added to each aliquot to terminate the reaction. Subsequently, the samples were centrifuged at 100,000× g for 60 min. to separate the soluble and insoluble proteins. After centrifugation, each sample was added with a 5× SDS-PAGE loading buffer and subsequently separated by SDS-PAGE and analyzed by western blot.
2.6. Recombinant Protein Expression and Purification
Professor Anders Virtanen kindly provided the recombinant plasmid containing the WT human PARN (p74) (Uppsala University, Uppsala, Sweden). Details regarding mutagenesis, recombinant proteins overexpression in the
Escherichia coli cells and purification of the recombinant proteins have been described elsewhere [
45,
46,
48]. In brief, the WT and mutated genes were cloned into the vector pET-28a (Novagen, Madison, WI, USA) and verified by sequencing. The recombinant proteins with a His-tag at the N-terminus were overexpressed in
E. coli BL21 (DE3) (Stratagene, La Jolla, CA, USA) and sequentially purified by Ni
2+ affinity chromatography while using a 1 mL Ni
2+ column (GE Healthcare, Madison, WI, USA) and size-exclusion chromatography (SEC) using a Superdex 200 16/60 GL column that was equipped on an ÄKTA purifier (GE Healthcare). The purity of the final products was above 95%, as estimated by SDS-PAGE and SEC analysis. The protein concentration was determined according to the extinction coefficient and
A280 reads. The proteins that were used for biophysical experiments were prepared in 20 mM Tris-HCl buffer, pH 8.0, containing 100 mM KCl, 0.5 mM DTT, 0.2 mM EDTA, and 20% (
v/
v) glycerol. Recombinant αB-crystallin and γD-crystallin were purified from the
E. coli BL21 cell extracts while using the same protocols as those described previously [
49,
50].
2.7. Enzyme Assay
The deadenylase activity was measured by the methylene blue assay or SEC method, as described previously [
51,
52]. In brief, methylene blue stock solution was prepared by dissolving 1.2 mg methylene blue in 100 mL MOPS buffer (100 mM MOPS-KOH, 2 mM EDTA, pH 7.5), while the stock solution of substrate poly(A) was prepared by dissolving commercial A
200 or synthesized A
20 with a concentration of 100 μg/mL. The reaction was initiated by mixing 10 μL enzyme and 40 μL poly(A) stock solution in the standard reaction buffer containing 20 mM Tris-HCl, pH 7.0, 100 mM KCl, 0.5 mM DTT, 0.2 mM EDTA, and 10% (
v/
v) glycerol. After 8 min. reaction at 30 °C, methylene blue buffer was added to terminate the reaction and the absorbance at 662 nm was measured while using an Ultraspec 4300 pro UV/Visible spectrophotometer. The SEC assay was performed on a ÄKTA purifier that was equipped with a Superdex 200 10/30 GL column (GE Healthcare). The column was pre-equilibrated for two-column volumes until the UV absorbance and conductance lines were at the same level as that of the control (the standard reaction buffer). The RNA substrate (A
20 or commercial A
200) was dissolved in the standard reaction buffer with the addition of 1.5 mM MgCl
2 and then quantified by measuring the standard curve. The reaction was initiated by mixing 20 μL cell lysate containing 60 μg total proteins and 100 μL substrate stock solutions. After being incubated at 37 °C for a given time, the reaction was quenched on ice. Subsequently, SEC analyzed 100 μL samples and the absorbance at 280 nm, 254 nm, and 215 nm were simultaneously monitored.
2.8. Monolayer Surface Pressure Measurements
The classical Langmuir–Blodgett apparatus was used to measure the membrane-binding ability of the purified PARN in vitro [
53]. The monolayer surface pressure experiments were performed on an NIMA 9000 (England) Microbalance at 25 °C, as described previously [
49]. The monolayer was prepared by spreading phospholipids, phosphatidylethanolamine (PE), phosphatidylserine (PS), or cardiolipin (CL) on the water-air interface of the trough containing 6 mL buffer (20 mM Tris-HCl, pH 8.0, 100 mM KCl, 0.5 mM DTT, and 0.2 mM EDTA). After equilibration, 10–50 μL purified proteins with a final concentration of 20, 50, or 100 nM was injected into the sample loading hole of the trough by the Hamilton syringe. Time-course change in surface pressure was recorded for 6000 s. The critical pressure (π
c) was calculated by the linear fitting of the changes in surface pressure (∆π) at various initial surface pressures (π
i) [
53]. Besides PARN, the monolayer surface pressure experiments of αB- and γD-crystallins were also performed to take peripheral membrane proteins and cytosolic proteins as the examples of ER.
2.9. Liposome Binding Assay
Liposomes were prepared while using the standard method by dissolving DPPS in the chloroform and methanol (3:1) mixture. Subsequently, the organic solvent was removed by rotary evaporation, followed by vacuum pump for about half an hour. The dried lipid film was re-suspended in 20 mM Tris-HCl buffer, pH 7.0, containing 100 mM KCl, 0.5 mM DTT, and 0.2 mM EDTA, and then sonicated to produce small unilaminar vesicles with diameters that ranged from 15 to 50 nm. The quality of the final products was checked by negative-staining electron microscopy. The freshly prepared DPPS liposome was used for the PARN binding assay with a protin:DPPS molar ratio that ranged from 1:50 to 1:200. The final concentration of liposome was 200 μM, while that of PARN ranged from 1 μM to 4 μM. After 1.5 h incubation at room temperature, the mixture was centrifuged at 15,000× g for 15 min. at 4 °C. The supernatant and pellet fractions were both analyzed by SDS-PAGE electrophoresis and western blot.
2.10. Polysome Profiling
Polysome fractionation was achieved by the standard continuous 10–50% sucrose density gradient centrifugation [
54,
55]. All solutions, tips, and tubes used for polysome fractionation were RNase-free to avoid RNA degradation. The buffer for the sucrose solutions was 10 mM HEPES-KOH buffer, pH 7.4, containing 5 mM MgCl
2, 150 mM KCl, 100 μg/mL CHX, 1 mM DTT, RNase inhibitor (40 U/mL), 1× protease inhibitor cocktail, and the detergent NP-40 (
v/
v 1%). Layering was conducted while using the polyallomer ultracentrifugation tube (14 × 89 mm, 331372, Beckman).
The whole cell lysate polysome samples were prepared while using a 15-cm dish culturing the HEK-293T cells with 80–90% confluence. The cells were treated with 100 μg/mL CHX for 10 min. at 37 °C, washed with ice-cold PBS containing CHX three times, and lysed in 500 μL cell lysis buffer. The lysates were then transferred to an ice cold, RNase-free Eppendorf tube, incubated for 15 min. on ice, and then centrifuged at 13,000× g for 10 min. at 4 °C. The supernatants were collected for further fractionation.
The cytosolic and ER-associated polysome samples were prepared by sequential detergent extraction [
56]. After CHX treatment, 1 mL permeabilization buffer (110 mM KCl, 25 mM K-HEPES, pH 7.2, 2.5 mM MgCl
2, 1 mM EGTA, 0.015% digitonin, 1 mM DTT, 50 μg/mL CHX, 1× Complete Protease Inhibitor Cocktail, and 40 U/mL RNase inhibitor) treated the cells on ice for 5 min., and the soluble fraction (cytosol fraction) was collected. The remaining cells were then washed gently with 5 mL wash buffer (110 mM KCl, 25 mM K-HEPES, pH 7.2, 2.5 mM MgCl
2, 1 mM EGTA, 0.004% digitonin, 1 mM DTT, and 50 μg/mL CHX) and lysed with 1mL lysis buffer containing 400 mM KCl, 25 mM K-HEPES pH 7.2, 15 mM MgCl
2, 1% (
v/
v) NP-40, 0.1% (
w/
v) DOC, 1 mM DTT, 50 μg/mL CHX, 1× Complete Protease Inhibitor Cocktail and 40 U/mL RNase inhibitor on ice for 5 min. The NP-40/DOC soluble fraction (membrane fraction) was collected. The cell debris in the crude cytosolic and membrane fractions were removed by centrifuged at 7500×
g for 10 min. Nanodrop measured the RNA concentration of the supernatants.
The extracted samples were layered on the top of the prepared sucrose gradients for fractionation by centrifugation at 40,000× g for 2 h at 4 °C (Rotor SW41Ti, Beckman, München, Germany). After centrifugation, Fractionator (BIOCOMP) collected the gradient fractions and the UV absorbance at 254 nm was measured simultaneously. Translational efficiency was obtained dividing the sum of the peak area of polysomes with above three ribosomes by that of the monosomes (the 80S subunit). RNAs and proteins in the collected gradient fractions were extracted for further analysis by qRT-PCR, RNA electrophoresis, and Western blot. RNA eletrophoresis was performed by the extraction of RNAs while using the TRIzol reagent, separation using an 8% polyacrylamide gel (in TBE buffer), staining with GelSafe (YPH), and visualization using the ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA, USA).
2.11. Poly(A) Tail Length Determination
The length distribution of the poly(A) tails were analyzed by electrophoresis of the biotinylated poly(A)+ RNAs that were extracted from the total RNAs. In detail, the total RNAs in the cytosolic or membrane fractions were obtained while using the standard methods. The poly(A)+ RNAs were extracted from 500 μg total RNAs using the Oligo(dT)
25 cellulose beads. After extraction, biotinylation of the 3′-end of poly(A)+ RNAs was performed in a 30 μL reaction solutions containing 8 μL denatured poly(A)+ RNA (~5 μg, 1–50 pmol), 3 μL 10× RNA ligase reaction buffer, 1 μL RNase inhibitor (40U), 1 μL biotinylated cytidine (Bis) phosphate (1 nmol), 2 μL T4 RNA ligase (40 U), and 15 μL 30% PEG at 16 °C overnight. Subsequently, the RNA body of the biotin-labeled RNAs was digested while using the published procedure [
57]. In brief, the RNase A/T1 mixture digested the RNA body. The 200 μL reaction solutions contained 12 μL 5 M NaCl, 2.0 μL 0.5 M EDTA, 2 μL RNase A/T1 mixture (2 mg/mL of RNase A and 5000 U/mL of RNase T1), 1 μL total yeast RNA (10 mg/mL) used as the carrier and 153 μL DEPC-treated double distilled water. After 30 min. reaction at 37 °C, the digested RNAs were extracted by TRIzol, which was precipitated by isopropanol and then dissolved in 5 μL DEPC-treated double distilled water. Finally, the length of the RNA body-digested RNAs, which was dominated by the 3′-end poly(A) tail, was determined by 12% polyacrylamide (29:1 acrylamide:bis-acrylamide) separating gel with the addition of 8 M urea and 1× TBE [
34,
36,
57]. The separated RNAs were transferred to a Hybond N+ nylon membrane (GE Healthcare) at 100 V for 1 h, cross-linked twice by ultraviolet at 120 mJ/cm
2 for 60 s, blocked for 20 min., incubated with the streptavidin-HRP conjugate for 1 h, washed three times, and then used for chemiluminescent detection by the ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA, USA). An alternative method for determining the poly(A) length distribution was the use of an Agilent RNA 6000 Pico Kit analyzed by the Agilent 2100 bioanalyzer (Palo Alto, CA, USA) through capillary electrophoresis in tiny chips. The Agilent RNA 6000 Pico Kit analysis was performed according to the manufacture’s instructions.
2.12. Transcriptome Sequencing
The RNA library was prepared from the total RNAs extracted from the cytosol and membrane fractions of the HEK-293T cell while using the KAPA Stranded RNA-Seq Library Preparation Kit. The samples were sequenced by the Illumina HiSeq X-ten platform at Tsinghua University. The obtained RNA sequences were mapped to the human whole genome (Homo sapiens: HG38) by STAR. The Cuffdiff software analyzed differences in the gene expression profiles. Functional enrichment analysis for the differentially expressed genes was performed while using the PANTHER classification platform of the Gene Ontology Consortium tool (
http://geneontology.org/).
2.13. qRT-PCR
The total RNAs extracted from the cytosol and membrane fractions of the HEK-293T cell were reverse transcribed to construct the cDNA library while using the standard methods. The qPCR reactions were run with 2× RealStar Power SYBR Mixture (Genstar, Beijing, China). The real-time PCR was performed at 95 °C for 5 min. and followed by 40 cycles of 15 s 95 °C, 20 s at 60 °C, and 35 s at 72 °C. The primers used for the qPCR reactions were as follows: MDM2: forward, 5′-TGCCAAGCTTCTCTGTGAAAG-3′, reverse, 5′-TCCTTTTGATCACTCCCACC-3′; GAPDH: forward, 5′-CGCTCTCTGCTCCTCCTGTT-3′, reverse, 5′-CCATGGTGTCTGAGCGATGT-3′; PDIA3: forward, 5′-TGAGGGATAACTACCGATTTGC-3′, reverse, 5′-TGTATATGCCACAGTCTTGTCC-3′; CDKL1: forward, 5′-AATGTAGAAACAGGGACACGG-3′, reverse, 5′-AGGTTGGGATGCTTGAGTTG-3′; CCNT2: forward, 5′-TGAGATCACCATTGAACACCC-3′, reverse, 5′-CACTGTTGGTTTGTACTGAAGAC-3′; CLOCK: forward, 5′-TCAGTTCAGCAACCATCTCAG-3′, reverse, 5′-GATGTGACTGAGGGAAGGTG-3′; and MATR3: forward, 5′-AGTCTACAAATCCAGCACCAG-3′, reverse, 5′-AGTTTCCACTCTGCCTTTCTG-3′. Quantification was achieved by the determination of the standard curve of MDM2 by real time PCR and TAE-agarose gel electrophoresis to obtain the copy number per ng of the PCR product.
2.14. mRNA Stability
mRNA stability was measured in the HEK-293T cells that were cultured in six-well plates with the DMEM medium containing 10% FBS. Transcription inhibition was accomplished by the addition of 5 μg actinomycin D for 0, 2, 4, 8, or 16 h. After treatment, the cells were fractionated into cytosolic and ER-associated fractions, as described above. The total RNA in the ER-associated fraction was extracted by standard procedures. The amount of MDM2 mRNA was determined by real time RT-PCR while using PDIA3 as an internal control. The untreated control group normalized the data.
2.15. Western Blot
The samples with equal amounts of total proteins ranged from 10 to 60 μg were mixed with 5× loading buffer, boiled, separated by 7.5%, 10%, or 12.5% SDS-PAGE gel, and then transferred to a PVDF membrane (GE). Bound primary antibodies were detected by the HRP-conjugated secondary antibodies (1:3000) while using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA, USA). The PVDF membrane was detected by chemiluminescence and then imaged by ChemiDoc Touch Imaging System (Bio-Rad).
2.16. Immunofluorescence
The HeLa cells that were used for immunofluorescence studies were washed twice with PBS (pH 7.4), fixed with 4% paraformaldehyde, penetrated with 0.4% Triton, and then blocked with 10% FBS. The fixed cells were stained with the anti-Flag, anti-PARN, and anti-calnexin antibodies at 4 °C for 14 h. Subsequently, the samples were mounted with Dylight 488 or 594 at room temperature for 1.5 h and then used for the confocal microscopy studies. The nuclei were stained with DAPI. The Imaris software and the Colocalization analysis plugin in Image J were used for colocalization analysis.
2.17. Cell Viability
The cells were seeded in a 96-well plate and cultured for 24 h prior to transfection. After 20 h of transfection, the cells were exposed to different stress conditions, including DMSO (control), CMPD1, DOX, and UV irradiation, with various treating time and/or concentrations. After treatment, the wells were moved to fresh DMEM medium with the addition of 10 μL CCK8 reagent each well and then incubated for 1 h at 37 °C. The OD450 value was read by a microplate spectrophotometer. The DMEM medium with the addition of the same amount of the CCK8 reagent was used as the blank for spectrophotometer analysis.
2.18. Cell Cycle Analysis
The HEK-293T cells were fixed with 70% ethanol in PBS at 4 °C overnight. After washing with PBS three times, the cells were incubated with 100 μg/mL RNase A in PBS for 20 min. at 37 °C and then stained with 50 μg/mL propidium iodide. The percentages of cells in the G0/G1, S, and G2/M phases were determined by FACSCalibur (BD Biosciences, San Jose, CA, USA) and then analyzed with Flowjo 7.6 (Treestar Inc., Ashland, OR, USA).
2.19. Ligase Mediated poly(A) tail (LM-PAT) Assay
The ligase mediated poly(A) tail (LM-PAT) assay was performed while using the published protocols [
58,
59], with some modifications. In brief, the poly(A) tails of 500 ng total RNAs were saturated with 5′-phosphorylated oligo(dT)
16 at 42 °C in the presence of T4 DNA ligase. An excess amount of oligo(dT) anchor primer (5′-AATGCCAGCTCCGCGGCCGCGTTTTTTTTTTTT-3′) was added to the reaction solutions to anneal at the end of poly(A) tails and then incubated for 2 h at 12 °C to complete ligation. The ligated primers were used to prime RT by M-MLV (Promega). Afterwards, PCR reaction was performed while using the anchor primer as well as a gene specific sense primer. The PCR products in the TBE buffer were resolved on an 8% polyacrylamide gel, stained with GelSafe (YPH), and then visualized by ChemiDoc Touch Imaging System (Bio-Rad). The sequences of the PAT primer of MDM2 is MDM2-primer 1, 5′-GGGTGGATGCTGAATTACATTTTG-3′; MDM2-primer 2, 5′-TTGTGATCATATTGTCTACCATGTAGCCAGCTTTC-3′.
2.20. Statistical Analysis
Most of the experiments were performed and analyzed with at least three independent biological replicates, which were separately done while using different sets of cells. The tissue distribution study was performed with two independent experiments and each contained three biological replicates. Statistical analysis of the data was performed by the Student t-test or two-way ANOVA test while using GraphPad Prism (GraphPad Software, San Diego, CA) 7.04 or OriginLab 9.0 (OriginLab Corporation, Northampton, MA, USA). A p-value less than 0.05 was considered to be significant.
4. Discussion
The concept that translation is spatiotemporally regulated has been well-documented since the early 1970s [
38]. The compartmentalization of mRNAs and ribosomes involves two predominant localizations, the ER surface, and cytosol. ER-associated mRNAs are believed to translate with a high efficiency and translation repression will release the ER-bound mRNAs back to the cytosol [
43,
44,
69]. It is worth noting that, during ER stress, IRE1, an ER-localized endoribonuclease, can be activated to splice
XBP1 mRNA to a translatable form [
72], which implies that the ER surface might provide an interface not only for efficient translation, but also for prerequisite translation regulation. Herein, we further asked whether ribonucleases could target the ER-bound mRNAs and modulate their decay and translation efficiency in situ on the ER surface.
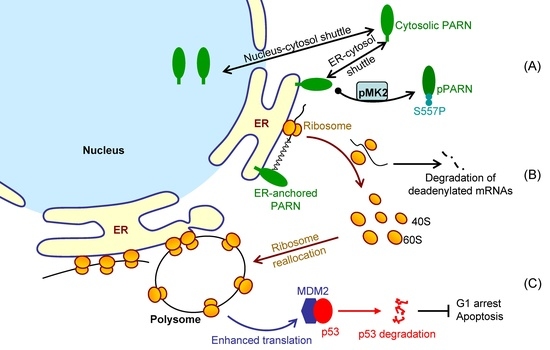
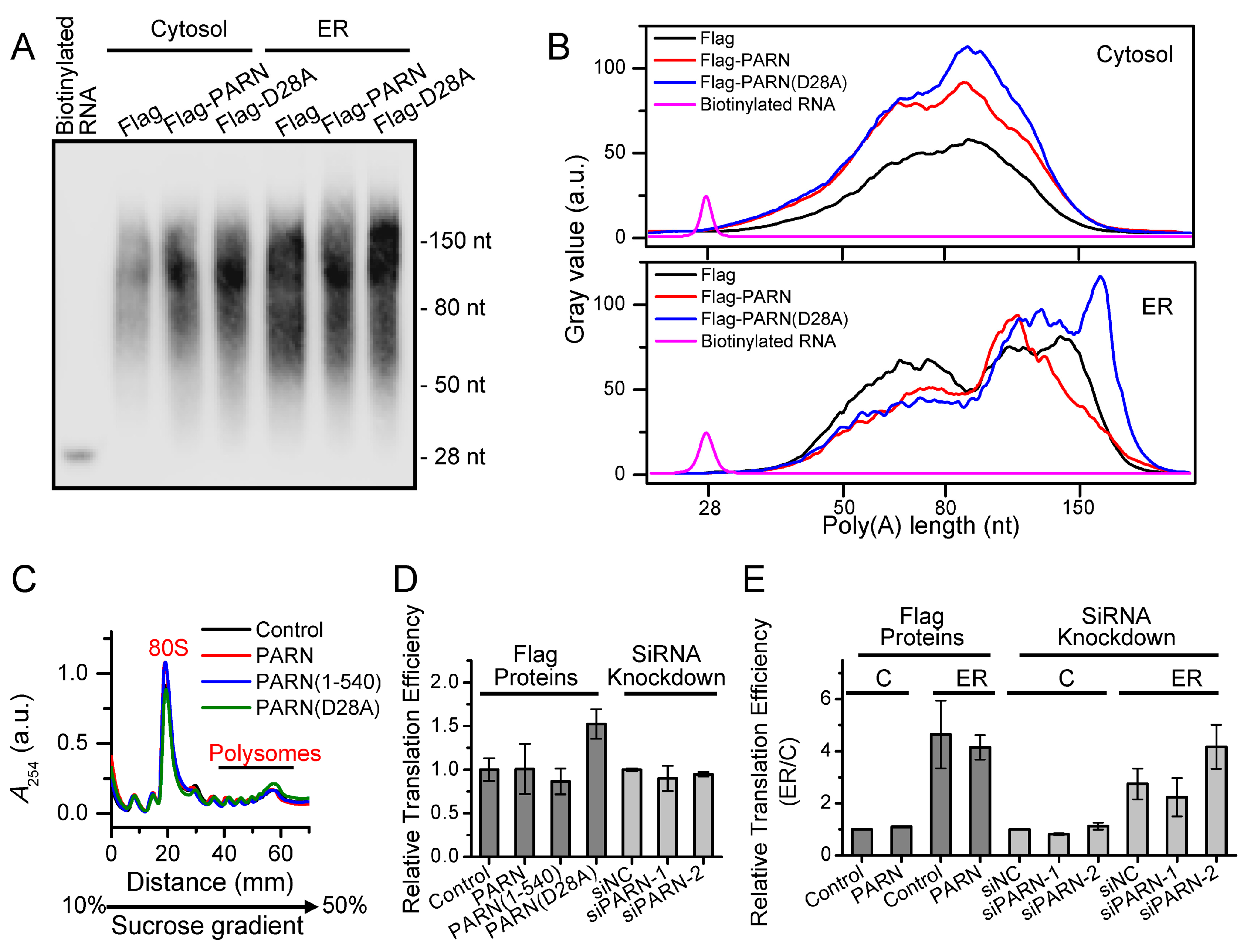
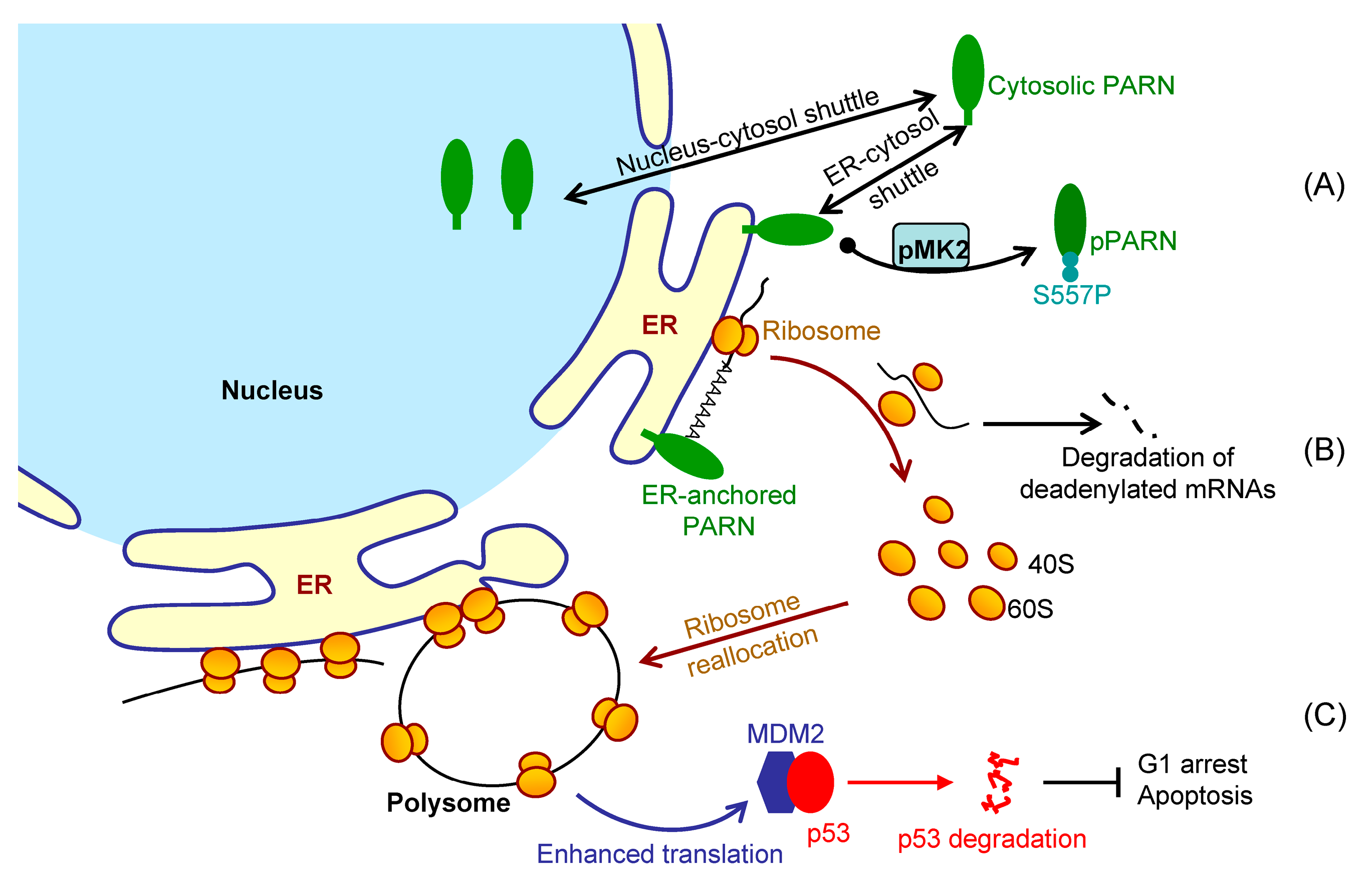
It is difficult to track the fates of ER-bound mRNAs since the bulk of mRNAs shuttled between the ER surface and cytosol (
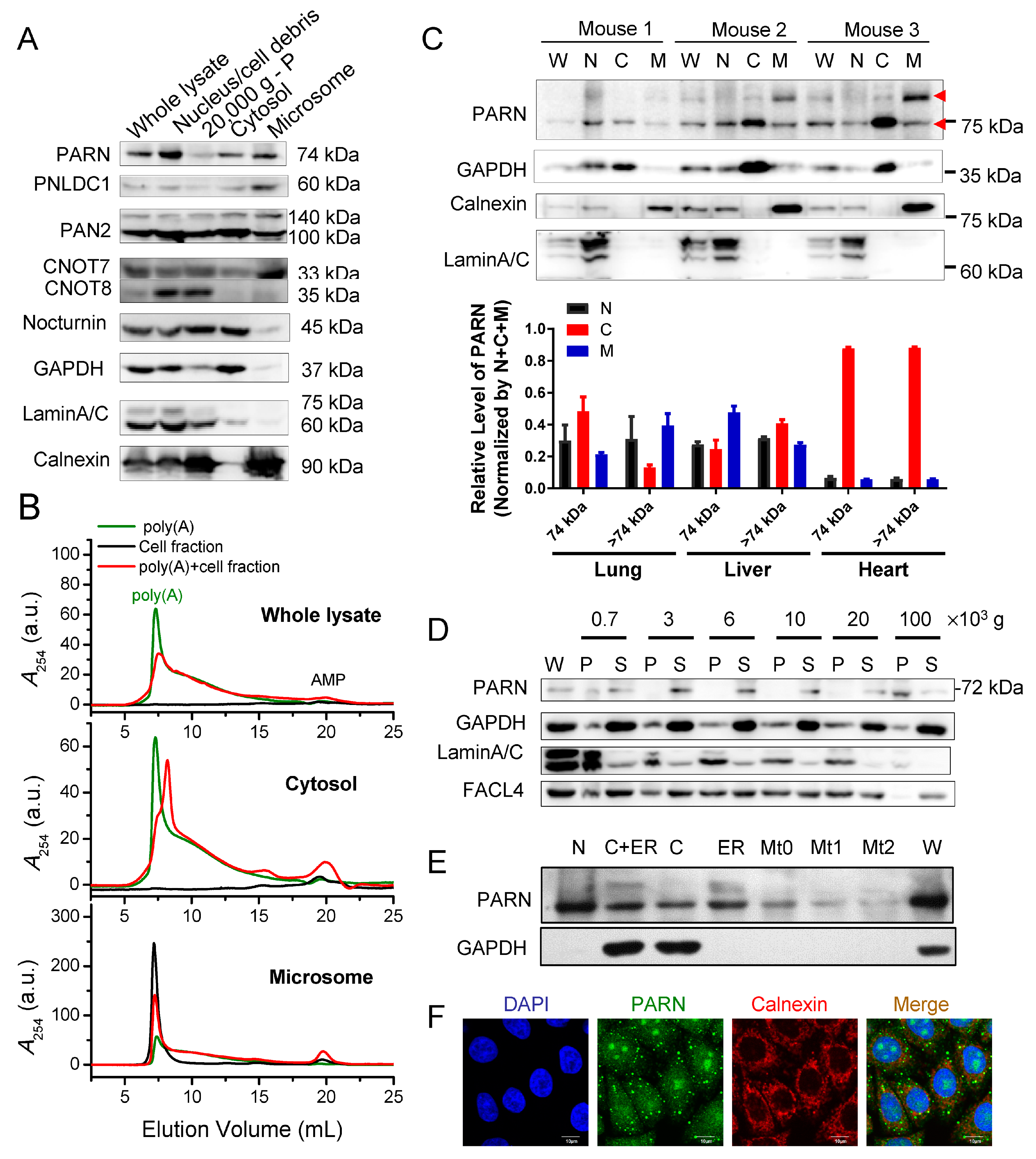
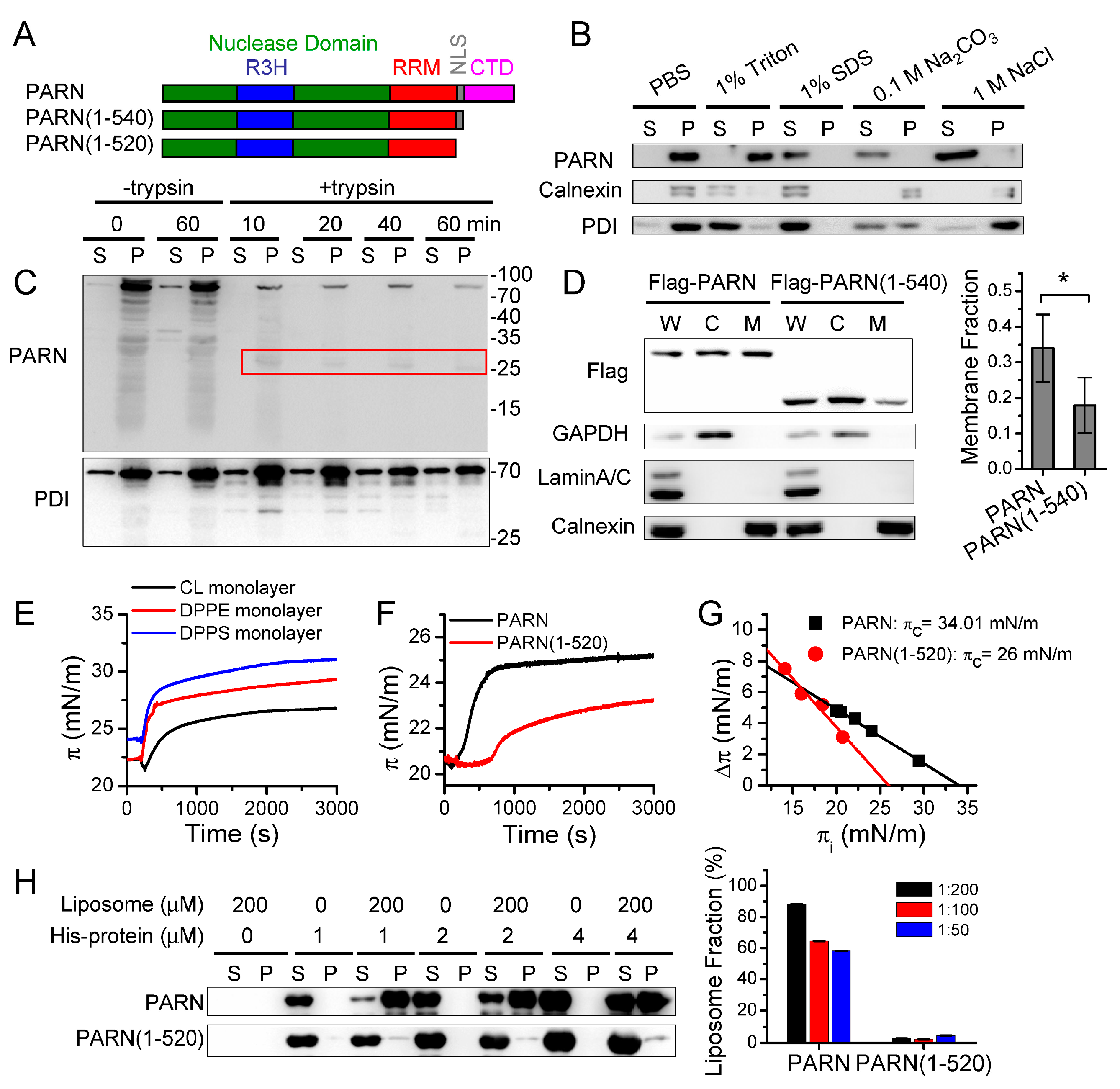
Figure 5). We then addressed this problem by identifying ER-anchored deadenylases exhibiting deadenylation activities in somatic cells and by verifying the membrane-binding ability of PARN in vitro (
Figure 1 and
Figure 2). We showed that the ER-anchored PARN requisitely orchestrated translation by modulating the poly(A) length distribution and mRNA abundance on the ER. Our findings shed new lights on the mechanistic understanding regarding the emerging concept of crosstalk between mRNA degradation and translation by showing that this crosstalk can occur not only in cytosol, but also on the ER surface, the workplace of highly efficient translation.
Besides PARN, our results also indicated that the other types of deadenylases might also have ER distributions. Further research is needed to verify this hypothesis. An unresolved question is the fate of the deadenylated mRNAs that are generated by the action of ER-anchored deadenylases. Are these deadenylated ER-associated mRNAs released to the cytosol or degraded by the decay machinery on ER in situ? Furthermore, will the poly(A) length affect the ER-localization of mRNAs? Further study using single-molecule RNA imaging in living cells might provide clues to these unresolved problems.
Among various deadenylases, PARN is evolved later than the other deadenylases and it only exists in vertebrates and higher plants [
1,
4,
60]. The transcriptome-level study has revealed that basal deadenylation in human cells is unaffected by the depletion of PARN [
25]. Our results showed that PARN overexpression did not affect the poly(A) length profile of cytosolic mRNAs, while the length distribution of the ER-bound mRNAs was reshaped (
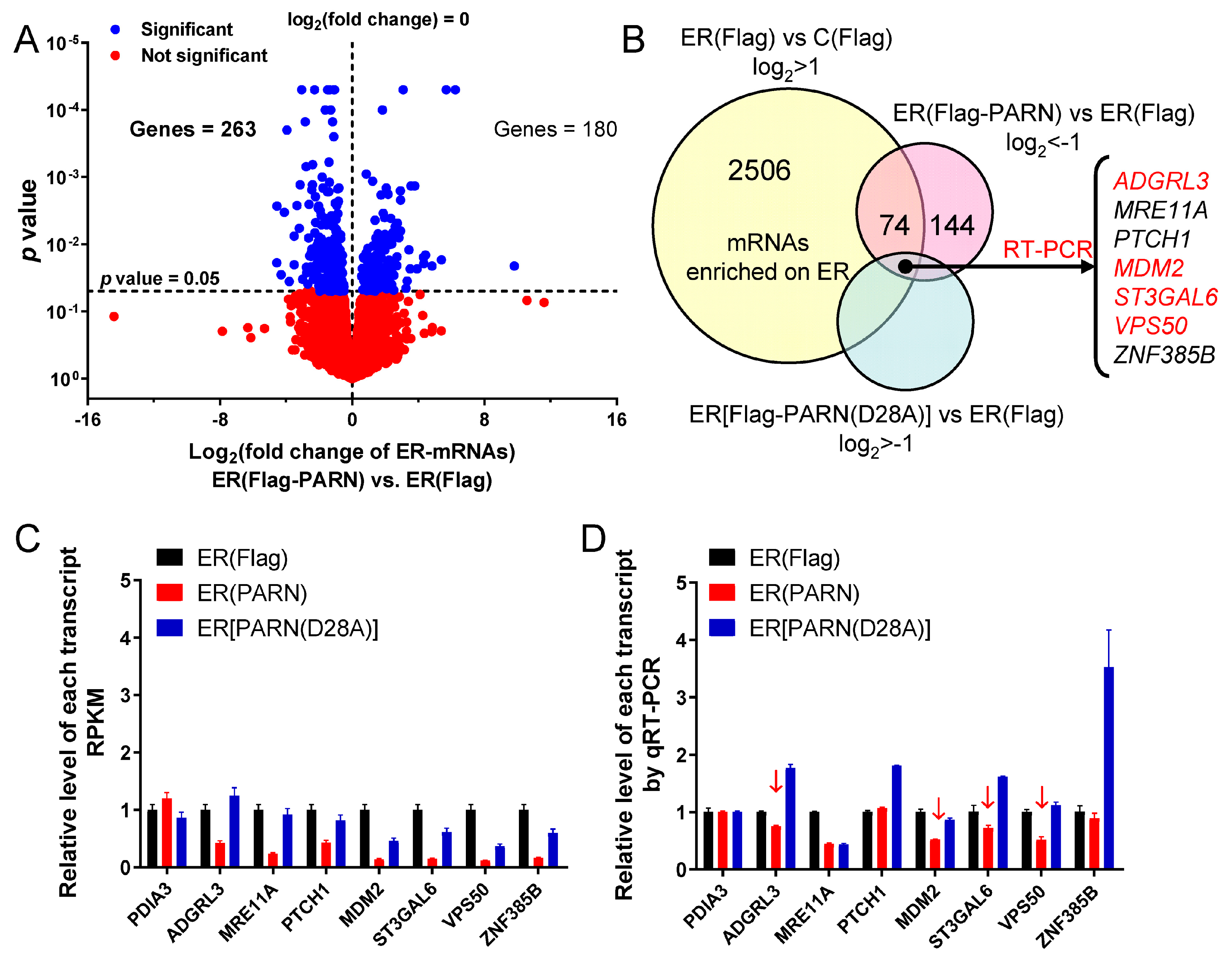
Figure 4). Transcriptome analysis indicated that only a small portion of the ER-bound mRNAs had significant changes in their abundance (
Figure 5). A combined analysis of the results that are shown in
Figure 4 and
Figure 5 suggested that the ER-associated PARN triggered the decay of only a small subset of ER-bound transcripts, while it might play a non-specific global role in the shortening of long poly(A) tails without the destabilization of most ER-bound mRNAs. PARN had little impact on the poly(A) length distribution of cytosolic mRNAs, which implied that the basal deadenylation in the cytosol is probably achieved by the PAN2-PAN3 and CCR4-NOT complexes but not PARN. Consistently, it has been reported that PARN knockdown in mouse myoblasts affects the stability of a discrete subset of cell-mobility related mRNAs [
73].
Intriguingly, PARN can modulate the decay of mRNAs encoding proteins that are crucial to cell cycle regulation in both the ER and cytosol fractions. The importance of PARN in cell cycle progression has been revealed by its essential role during early development in both vertebrates and higher plants [
1,
60] and the proliferation of several types of cancer cells [
12,
16,
17]. PARN can modulate cell cycle progression by targeting a couple of RNAs, including
Gadd45α [
66],
p21 [
16],
p53 [
16,
74], genes that are involved in telomere maintenance [
75], miR-21 [
76], miR-125b [
77], and multiple miRNAs repressing p53 [
78]. Although it is clear that PARN activity is highly regulated to precisely control cell cycle progression, it remains elusive as to how PARN defines its targets.
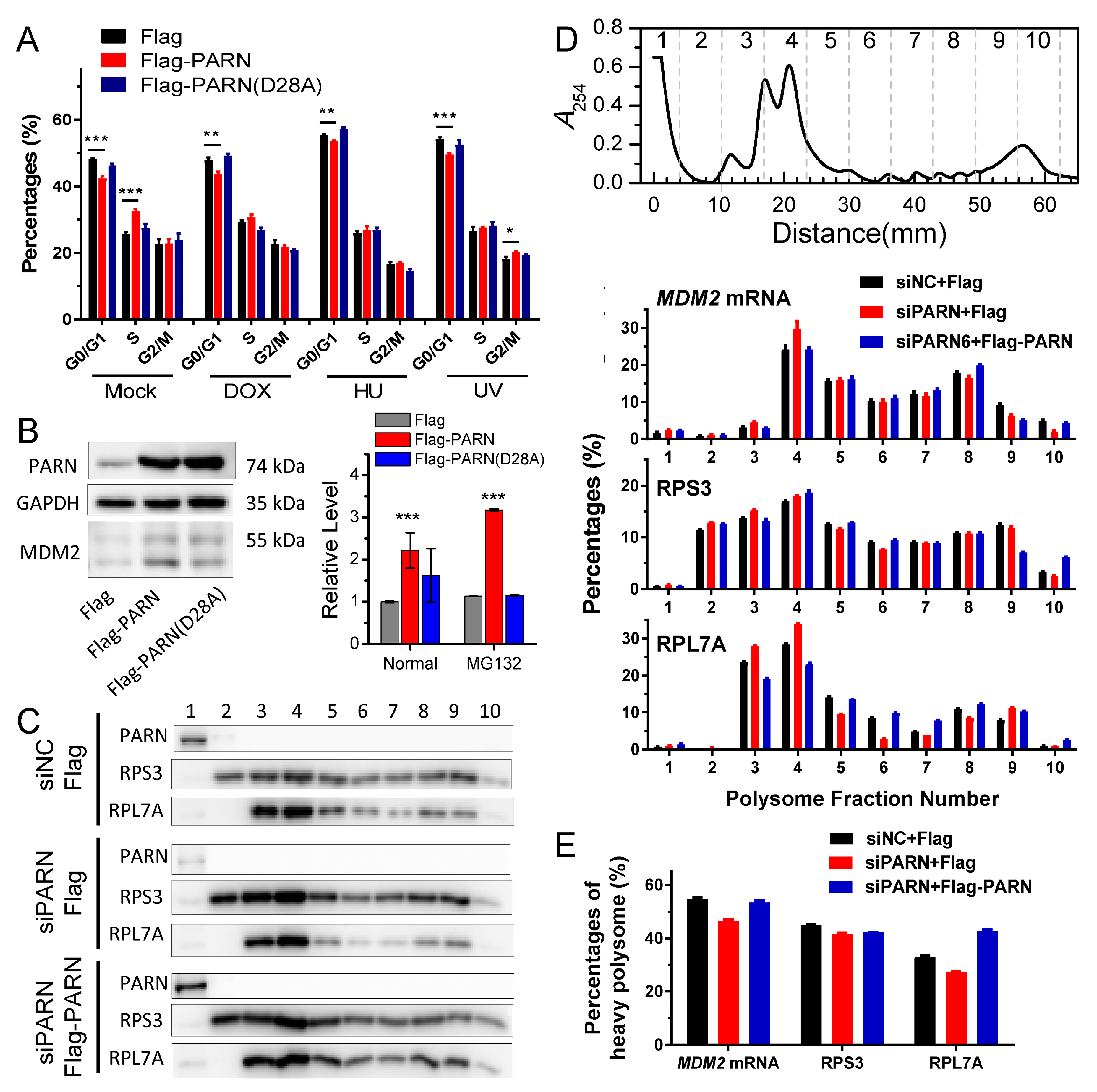
Inconsistency between the changes in mRNA and protein levels is frequently observed in cellular studies of gene expression. We also found that PARN overexpression resulted in opposing effects on the
MDM2 mRNA and protein levels. The poly(A) tail length distribution of the remained
MDM2 mRNAs in the PARN-overexpression group was indistinguishable from the control group. The decreased transcript abundance and increased decay rate implied that the deadenylated
MDM2 transcripts were quickly removed by mRNA degradation machinery. Meanwhile, the remained
MDM2 transcripts were concentrated in heavy polysomes with high ribosome occupancies (
Figure 6). These observations led to a proposal that PARN selectively promoted the degradation of the
MDM2 transcripts with relatively low translation efficiency. Ribosomes were released from these deadenylated transcripts and then reallocated to transcripts with high ribosome occupancies.
Figure 7 shows a proposed model. By this means, the cells could remove the redundant transcripts to optimize the costs for efficient protein production. This ribosome reallocation mechanism could also explain why the protein levels either increased or unchanged, even though the levels of the five tested cytosolic mRNAs were significantly decreased by overexpressed PARN.
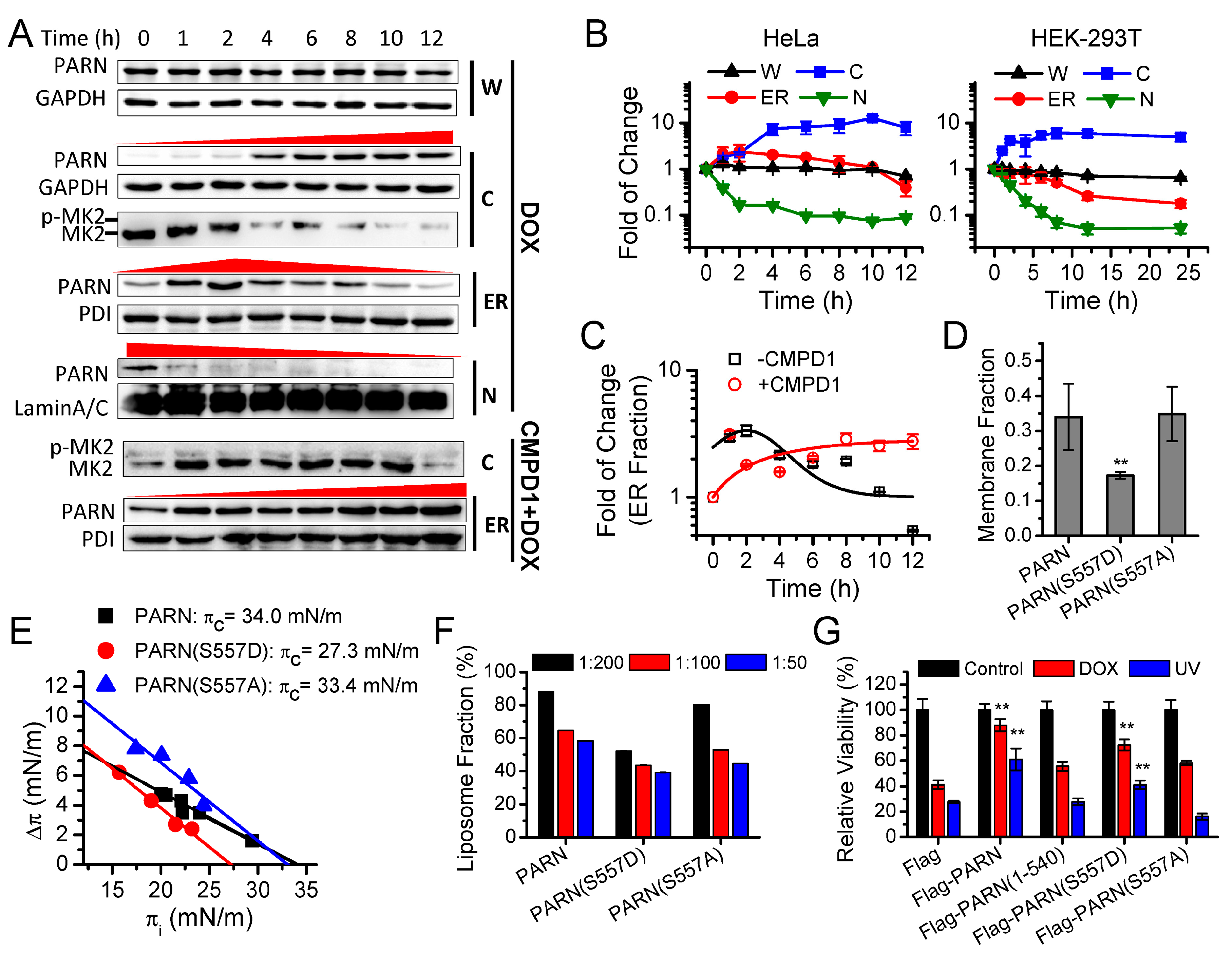
Although PARN predominantly locates in the nucleus, it is a nuclear-cytoplasmic shuttling protein [
79]. Herein, we identified that the cytoplasmic PARN can shuttle between the cytosol and ER. Under DNA damaging conditions induced by DOX, the anterior nucleus-localized PARN was quickly translocated to cytoplasm, while the DNA damage-mimic reagent hydroxyl urea did not induce such a translocation [
8]. This suggested that the action of PARN during DDR might involve step-wise processes. The step-wise process can also be observed for the action of PARN in the cytoplasm. The ER-anchored PARN had an apex value after 4 h DOX treatment. A possible method is posttranslational modification to achieve such an exquisitely spatiotemporal regulation of PARN activity. The phosphorylation of PARN has been observed in cells with serum starvation [
80], DNA damage [
66], and pathological conditions, such as acute leukemia [
12]. Moreover, Ser557 phosphorylation by MK2 prevents
Gadd45α mRNA degradation [
66]. However, it remains unclear for the underlying mechanism and exact function of PARN phosphorylation. We found that the intrinsically disordered CTD was responsible for the membrane binding of PARN, probably via ionic interactions by the abundant positively charged residues in the CTD. Ser557 is located at the CTD and phosphorylation introduces additional negative charges in the CTD, which probably weaken the ionic interactions between the CTD and ER membrane. The translocation of PARN from the ER to cytosol was strongly dependent on the level of phosphorylated MK2 (
Figure 3). Kinase-triggered translocation endows PARN with the potency to respond to diverse signaling pathways. It is worthy noting that the complicated behaviors of the ER-bound PARN imply that the phosphorylation of Ser557 by MK2 is probably only one of the regulators. Further research is needed for identifying the additional regulators modulating the membrane-binding ability of PARN.
Previously, PARN has been shown to participate into DNA damage response (DDR) via multiple pathways, including switching off PARN activity by MAPKAP kinase 2 (MK2)-induced Ser557 phosphorylation to stabilize the
Gadd45α mRNA in the p38MAPK/MK2 pathway [
66], degrading a subset of mRNAs by recruiting to the CstF/BARD1 complex in the nucleus [
81], modulating protein-protein interaction network by phosphorylation at S557 [
8], and regulating
p53 [
74] or
p21 mRNA stability [
16]. Herein, we also found that PARN is involved in the p38MAPK/MK2 signaling pathway and identified an additional target of PARN during DDR. ER-anchored PARN decreased
MDM2 mRNA stability, but enhanced its translation efficiency. The enhanced MDM2 protein level sequestered more p53 molecules and inhibited the p53 transcriptional activities, which facilitated the HeLa cells to bypass the checkpoints and thereby promoted cell cycle progression (
Figure 6). These findings provide a mechanistic understanding of the delicate control of cell cycle progression by PARN-mediated translation regulation and expand the complicated actions of PARN in DDR and carcinogenesis.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






