RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs



Abstract
1. Introduction
1.1. Therapeutic Nucleic Acids
1.2. The Emergence of RNA-Based Drugs
2. RNA Therapeutics: An Expanding Repertoire
2.1. Common RNA Drugs
2.1.1. Antisense Oligonucleotide
2.1.2. Short Interfering RNA
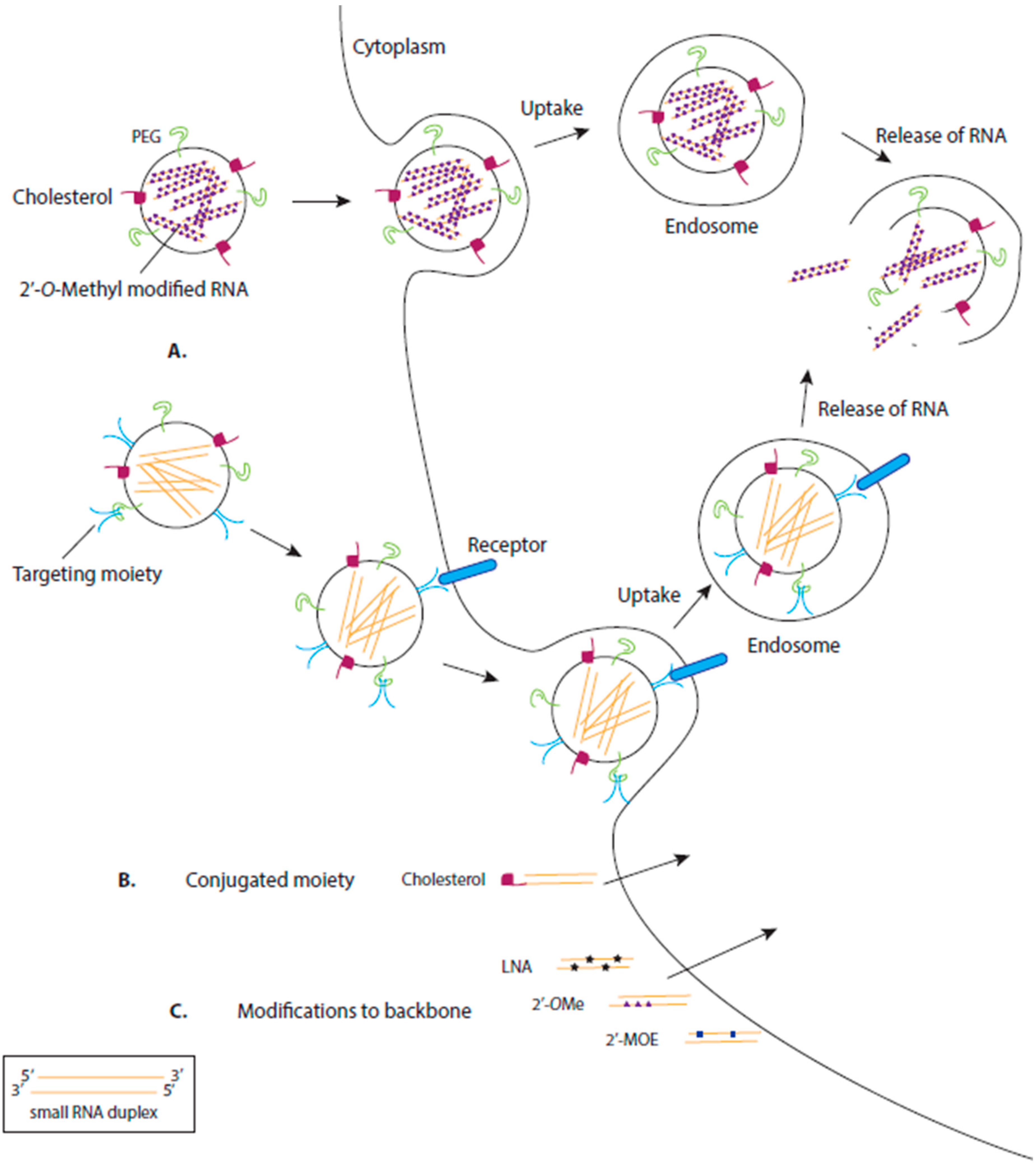
3. Delivery of RNA Drugs
3.1. Chemical Modification of RNA
3.2. Delivery by Ligand-Based Targeting Molecules
3.3. Delivery by Lipids
3.4. Delivery by Polymers
3.5. Delivery by Viral Vectors
3.6. Delivery by Bacterial Mini Cells
4. RNA-Based Therapeutic Success
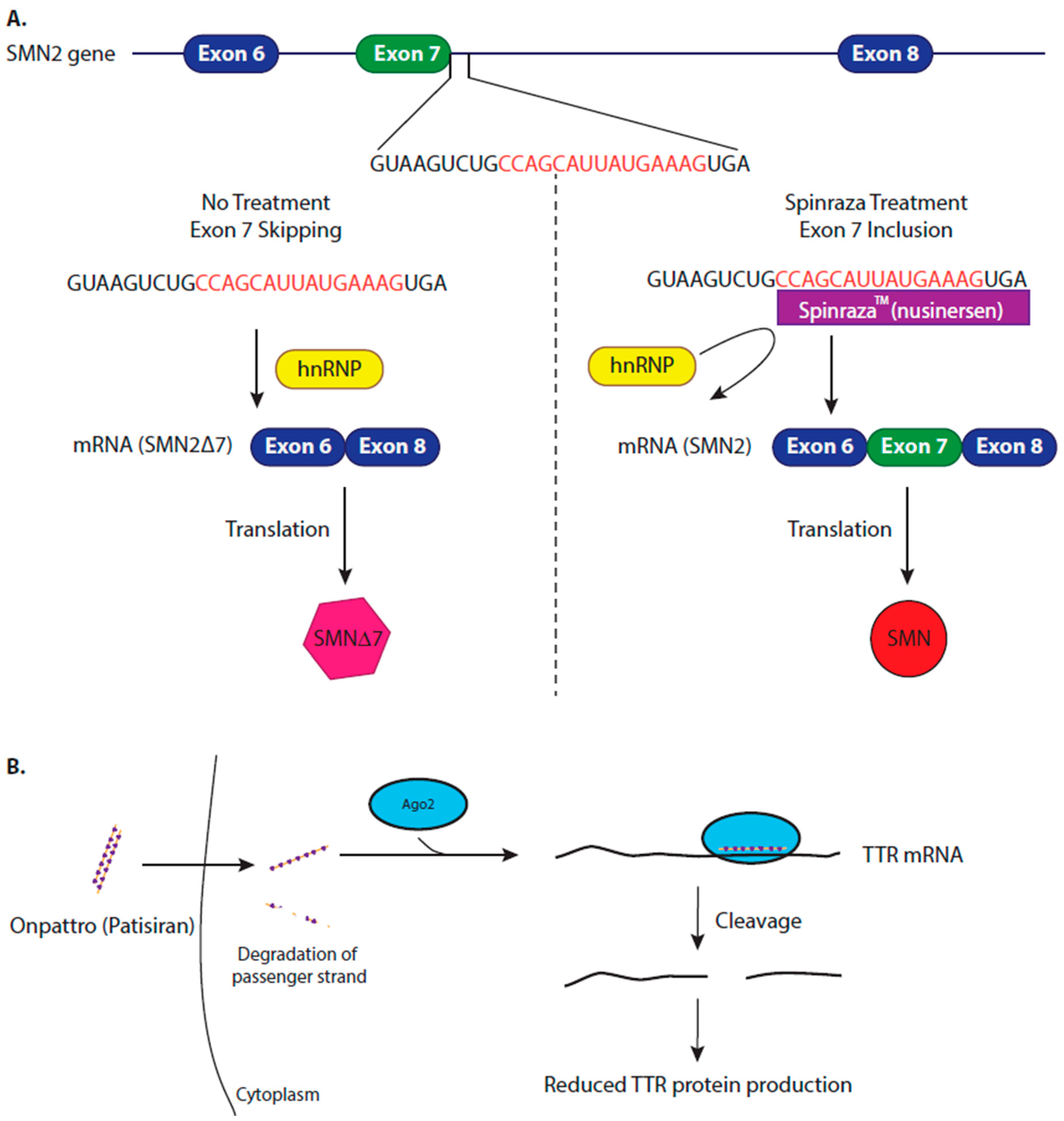
4.1. Modifying Alternative Splicing to Increase Protein Production
4.2. Inhibiting Protein Production to Reduce Amyloidosis
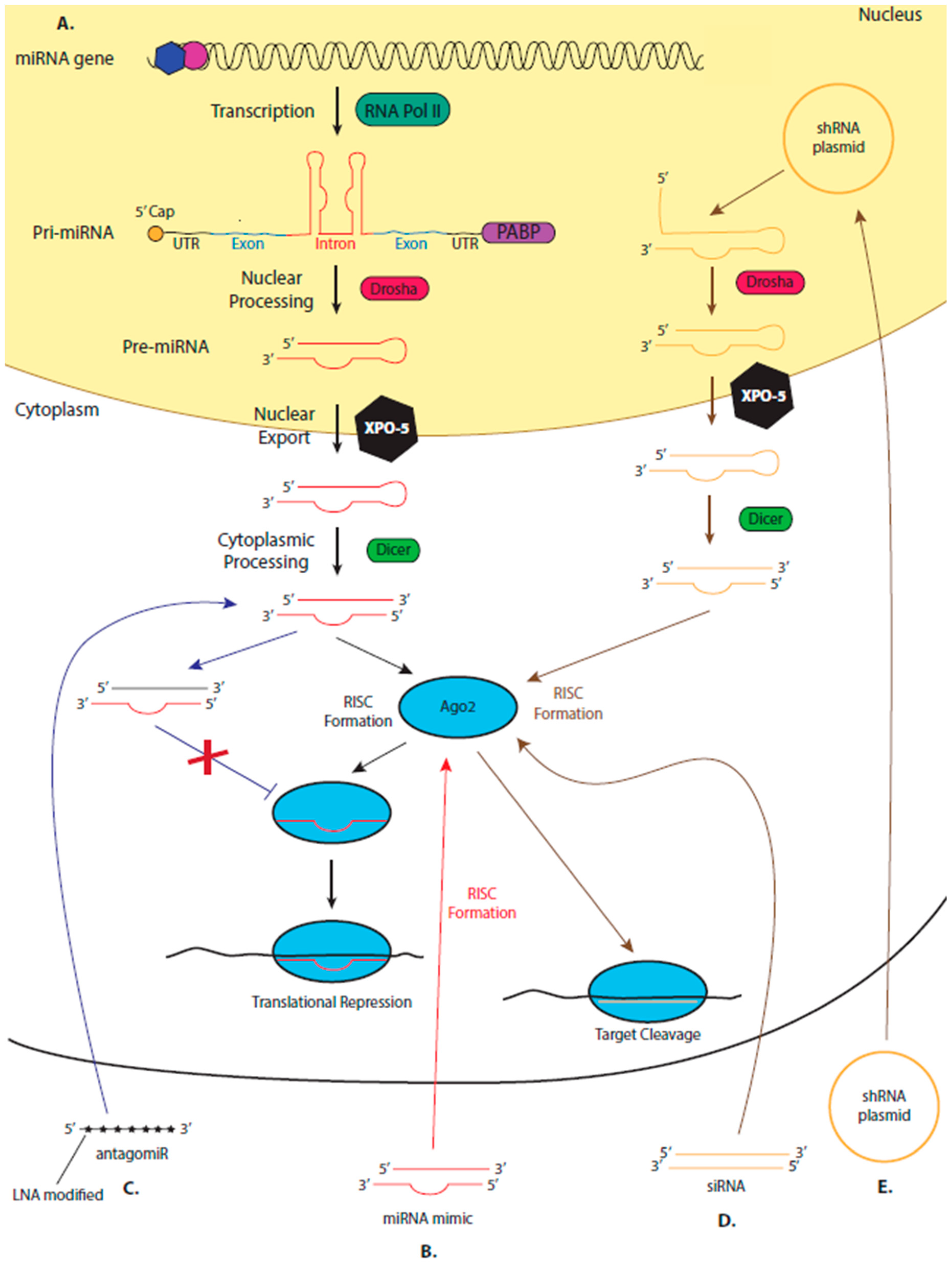
5. miRNA Therapeutics
5.1. The Suitability of miRNAs as Diagnostic Biomarkers
5.2. The Suitability of miRNAs as Therapeutics
5.3. miRNA Mimics and AntagomiRs
5.4. Leading the miRNA Therapeutic Field
5.5. miRNA Mimic Drugs in Development
6. Conclusions
Funding
Conflicts of Interest
References
- SoRelle, R. Who Owns you DNA? Who Will own it? Circulation 2000, 101, e67–e68. [Google Scholar]
- Anderson, W.F. Human gene therapy. Nature 1998, 392, 25–30. [Google Scholar] [CrossRef]
- Otsu, M.; Candotti, F. Gene therapy in infants with severe combined immunodeficiency. BioDrugs 2002, 16, 229–239. [Google Scholar] [CrossRef]
- Baekelandt, V.; De Strooper, B.; Nuttin, B.; Debyser, Z. Gene therapeutic strategies for neurodegenerative diseases. Curr. Opin. Mol. Ther. 2000, 2, 540–554. [Google Scholar]
- Galanis, E.; Russell, S. Cancer gene therapy clinical trials: Lessons for the future. Br. J. Cancer 2001, 85, 1432–1436. [Google Scholar] [CrossRef]
- Saraswat, P.; Soni, R.R.; Bhandari, A.; Nagori, B.P. DNA as therapeutics; an update. Indian J. Pharm. Sci. 2009, 71, 488–498. [Google Scholar] [CrossRef]
- Myhr, A.I. DNA Vaccines: Regulatory Considerations and Safety Aspects. Curr. Issues Mol. Biol. 2007, 79–88. [Google Scholar] [CrossRef]
- Perry, C.M.; Balfour, J.A. Fomivirsen. Drugs 1999, 57, 375–380. [Google Scholar] [CrossRef]
- Grillone, L.R.; Lanz, R. Fomivirsen. Drugs Today 2001, 37, 245–255. [Google Scholar] [CrossRef]
- Liu, B.; Montgomery, S.B. Identifying causal variants and genes using functional genomics in specialized cell types and contexts. Hum. Genet. 2019. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–221. [Google Scholar] [CrossRef]
- Harries, L.W. RNA biology provides new therapeutic targets for human disease. Front. Genet. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Lee, R.; Feinbaum, R.; Ambros, V. The C. elegans Heterochronic Gene lin-4 Encodes Small RNAs with Antisense Complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Olsen, P.H.; Ambros, V. The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev. Biol. 1999, 216, 671–680. [Google Scholar] [CrossRef]
- Wilson, R.; Doudna, J.A. Molecular mechanisms of RNA interference A BIOLOGICAL VIEW OF RNA INTERFERENCE • Small regulatory RNAs in cellular function and dysfunction HHS Public Access. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Hammond, S.M. An overview of microRNAs Scott. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. Regulation of miRNA Processing and miRNA Mediated Gene Repression in Cancer. MicroRNA 2014, 3, 10–17. [Google Scholar] [CrossRef]
- Gorabi, A.M.; Bianconi, V.; Pirro, M.; Banach, M. Biomedicine & Pharmacotherapy Regulation of cardiac stem cells by microRNAs: State-of-the-art. Biomed. Pharmacother. 2019, 120, 109447. [Google Scholar]
- Yang, Y.; Luo, C. MicroRNAs in acute pancreatitis: From pathogenesis to novel diagnosis and therapy. J. Cell. Physiol. 2019, 235, 1948–1961. [Google Scholar] [CrossRef]
- Hrach, H.C.; Mangone, M. miRNA Profiling for Early Detection and Treatment of Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4638. [Google Scholar] [CrossRef]
- Evangelatos, G.; Fragoulis, G.E.; Koulouri, V.; Lambrou, G.I. Micrornas in rheumatoid arthritis: From pathogenesis to clinical impact. Autoimmun. Rev. 2019, 2019, 102391. [Google Scholar] [CrossRef]
- Bonneau, E.; Neveu, B.; Kostantin, E.; Tsongalis, G.J.; De Guire, V. How close are miRNAs from clinical practice? A perspective on the diagnostic and therapeutic market. Ejifcc 2019, 30, 114–127. [Google Scholar]
- Rani, A.; O’Shea, A.; Ianov, L.; Cohen, R.A.; Woods, A.J.; Foster, T.C. miRNA in circulating microvesicles as biomarkers for age-related cognitive decline. Front. Aging Neurosci. 2017, 9, 1–10. [Google Scholar] [CrossRef]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Invest. 2017, 127, 761–771. [Google Scholar] [CrossRef]
- Blake, S.J.; Bokhari, F.F.; McMillan, N.A. RNA Interference for Viral Infections. Curr. Drug Targets 2012, 13, 1411–1420. [Google Scholar] [CrossRef]
- Rossor, A.M.; Reilly, M.M.; Sleigh, J.N. Antisense oligonucleotides and other genetic therapies made simple. Pract. Neurol. 2018, 18, 126–131. [Google Scholar] [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Schuster, S.; Miesen, P.; van Rij, R.P. Antiviral RNAi in insects and mammals: Parallels and differences. Viruses 2019, 11, 448. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.A. RNAi and double-strand RNA. Genes. Dev. 1999, 139–141. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Ding, H.; Kennington, L.; Moore, J.T.; Schelter, J.; Burchard, J.; Linsley, P.S.; Aronin, N.; Xu, Z.; Zamore, P.D. Designing siRNA that distinguish between genes that differ by a single nucleotide. PLoS Genet. 2006, 2, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Grimm, D. The dose can make the poison: Lessons learned from adverse in vivo toxicities caused by RNAi overexpression. Silence 2011, 2, 8. [Google Scholar] [CrossRef]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther. Nucl. Acid. 2017, 8, 132–143. [Google Scholar] [CrossRef]
- Liu, Y.P.; Haasnoot, J.; Berkhout, B. Design of extended short hairpin RNAs for HIV-1 inhibition. Nucl. Acid. Res. 2007, 35, 5683–5693. [Google Scholar] [CrossRef]
- Liu, Y.P.; von Eije, K.J.; Schopman, N.C.T.; Westerink, J.T.; ter Brake, O.; Haasnoot, J.; Berkhout, B. Combinatorial RNAi against HIV-1 using extended short hairpin RNAs. Mol. Ther. 2009, 17, 1712–1723. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef]
- Haussecker, D. Current issues of RNAi therapeutics delivery and development. J. Control. Release 2014, 195, 49–54. [Google Scholar] [CrossRef]
- Shukla, S.; Sumaria, C.S.; Pradeepkumar, P.I. Exploring chemical modifications for siRNA therapeutics: A structural and functional outlook. ChemMedChem 2010, 5, 328–349. [Google Scholar] [CrossRef]
- Egli, M.; Manoharan, M. Re-Engineering RNA Molecules into Therapeutic Agents. Acc. Chem. Res. 2019, 52, 1036–1047. [Google Scholar] [CrossRef]
- Judge, A.D.; Bola, G.; Lee, A.C.H.; MacLachlan, I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther. 2006, 13, 494–505. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef]
- Davis, S.; Lollo, B.; Freier, S.; Esau, C. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006, 34, 2294–2304. [Google Scholar] [CrossRef] [PubMed]
- Esau, C.; Davis, S.; Murray, S.F.; Yu, X.X.; Pandey, S.K.; Pear, M.; Watts, L.; Booten, S.L.; Graham, M.; McKay, R.; et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006, 3, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ørom, U.A.; Kauppinen, S.; Lund, A.H. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene 2006, 372, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Lennox, K.A.; Behlke, M.A. A direct comparison of anti-microRNA oligonucleotide potency. Pharm. Res. 2010, 27, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Elmén, J.; Lindow, M.; Schütz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-mediated microRNA silencing in non-human primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Ørum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010, 327, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Gelck, A.; Hadwiger, P.; Harborth, J.; et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.S.; Chernikov, I.V.; Meschaninova, M.I.; Dovydenko, I.S.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Carrier-free cellular uptake and the gene-silencing activity of the lipophilic siRNAs is strongly affected by the length of the linker between siRNA and lipophilic group. Nucleic Acids Res. 2012, 40, 2330–2344. [Google Scholar] [CrossRef]
- Letsinger, R.L.; Zhang, G.; Sun, D.K.; Ikeuchi, T.; Sarin, P.S. Cholesteryl-conjugated oligonucleotides: Synthesis, properties, and activity as inhibitors of replication of human immunodeficiency virus in cell culture. Proc. Natl. Acad. Sci. USA 1989, 86, 6553–6556. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef]
- Peer, D.; Lieberman, J. Special delivery: Targeted therapy with small RNAs. Gene Ther. 2011, 18, 1127–1133. [Google Scholar] [CrossRef]
- Chiriboga, C.A. Expert Review of Neurotherapeutics Nusinersen for the treatment of spinal muscular atrophy Nusinersen for the treatment of spinal muscular atrophy. Expert Rev. Neurother. 2017, 17, 955–962. [Google Scholar] [CrossRef]
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Pico de Coaña, Y.; Shmushkovich, T.; Yoshimoto, Y.; Truxova, I.; Yang, Y.; Betancur-Boissel, M.; Eliseev, A.V.; Wolfson, A.D.; Kiessling, R. Self-Delivering RNAi Targeting PD-1 Improves Tumor-Specific T Cell Functionality for Adoptive Cell Therapy of Malignant Melanoma. Mol. Ther. 2018, 26, 1482–1493. [Google Scholar] [CrossRef]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O.; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Swiderski, P.; Li, H.; Zhang, J.; Neff, C.P.; Akkina, R.; Rossi, J.J. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009, 37, 3094–3109. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T Cell-Specific siRNA Delivery Suppresses HIV-1 Infection in Humanized Mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Peer, D.; Park, E.J.; Morishita, Y.; Carman, C.V.; Shimaoka, M. Systemic Leukocyte-Directed siRNA Delivery Revealing Cyclin D1 as an Anti-Inflammatory Target. Science 2008, 319, 627–630. [Google Scholar] [CrossRef]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Frank-Kamenetsky, M.; Grefhorst, A.; Anderson, N.N.; Racie, T.S.; Bramlage, B.; Akinc, A.; Butler, D.; Charisse, K.; Dorkin, R.; Fan, Y.; et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. USA 2008, 105, 11915–11920. [Google Scholar] [CrossRef]
- Sonoke, S.; Ueda, T.; Fujiwara, K.; Sato, Y.; Takagaki, K.; Hirabayashi, K.; Ohgi, T.; Yano, J. Tumor regression in mice by delivery of Bcl-2 small interfering RNA with pegylated cationic liposomes. Cancer Res. 2008, 68, 8843–8851. [Google Scholar] [CrossRef]
- Azuma, K.; Nakashiro, K.I.; Sasaki, T.; Goda, H.; Onodera, J.; Tanji, N.; Yokoyama, M.; Hamakawa, H. Anti-tumor effect of small interfering RNA targeting the androgen receptor in human androgen-independent prostate cancer cells. Biochem. Biophys. Res. Commun. 2010, 391, 1075–1079. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, W.; Chen, Y.; Huang, K.; Shuai, X.; Chen, Q.; Li, X.; Lian, G. The investigation of polymer-siRNA nanoparticle for gene therapy of gastric cancer in vitro. Int. J. Nanomed. 2010, 5, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.K.; Ji, H.J.; Kim, T.I.; Sung, W.K.; Bull, D.A. VEGF siRNA delivery system using arginine-grafted bioreducible poly(disulfide amine). Mol. Pharm. 2009, 6, 718–726. [Google Scholar]
- Yam, P.Y.; Li, S.; Wu, J.; Hu, J.; Zaia, J.A.; Yee, J.K. Design of HIV vectors for efficient gene delivery into human hematopoietic cells. Mol. Ther. 2002, 5, 479–484. [Google Scholar] [CrossRef]
- Miyoshi, H.; Blömer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a self-inactivating lentivirus vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar] [CrossRef] [PubMed]
- VandenDriessche, T.; Thorrez, L.; Naldini, L.; Follenzi, A.; Moons, L.; Berneman, Z.; Collen, D.; Chuah, M.K.L. Lentiviral vectors containing the human immunodeficiency virus type-1 central polypurine tract can efficiently transduce nondividing hepatocytes and antigen-presenting cells in vivo. Blood 2002, 100, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2- Associated Clocal T cell Prolferation in Two Patients after Gene Therapy for SCID-X1. Science 2003, 302, 1181–1185. [Google Scholar]
- Gijsbers, R.; Ronen, K.; Vets, S.; Malani, N.; De Rijck, J.; McNeely, M.; Bushman, F.D.; Debyser, Z. LEDGF hybrids efficiently retarget lentiviral integration into heterochromatin. Mol. Ther. 2010, 18, 552–560. [Google Scholar] [CrossRef]
- Michelfelder, S.; Trepel, M. Chapter 2-Adeno-Associated Viral Vectors and Their Redirection to Cell-Type Specific Receptors. In Tissue-Specific Vascular Endothelial Signals and Vector Targeting, Part A; Advances in Genetics; Academic Press: Cambridge, MA, USA, 2009; Volume 67, pp. 29–60. [Google Scholar]
- MacDiarmid, J.A.; Brahmbhatt, H. Minicells: Versatile vectors for targeted drug or si/shRNA cancer therapy. Curr. Opin. Biotechnol. 2011, 22, 909–916. [Google Scholar] [CrossRef]
- MacDiarmid, J.A.; Mugridge, N.B.; Weiss, J.C.; Phillips, L.; Burn, A.L.; Paulin, R.P.P.; Haasdyk, J.E.; Dickson, K.A.; Brahmbhatt, V.N.; Pattison, S.T.; et al. Bacterially Derived 400 nm Particles for Encapsulation and Cancer Cell Targeting of Chemotherapeutics. Cancer Cell 2007, 11, 431–445. [Google Scholar] [CrossRef] [PubMed]
- MacDiarmid, J.A.; Amaro-Mugridge, N.B.; Madrid-Weiss, J.; Sedliarou, I.; Wetzel, S.; Kochar, K.; Brahmbhatt, V.N.; Phillips, L.; Pattison, S.T.; Petti, C.; et al. Sequential treatment of drug-resistant tumors with targeted minicells containing siRNA or a cytotoxic drug. Nat. Biotechnol. 2009, 27, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Marques, J.T.; Williams, B.R.G. Activation of the mammalian immune system by siRNAs. Nat. Biotechnol. 2005, 23, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Poeck, H.; Besch, R.; Maihoefer, C.; Renn, M.; Tormo, D.; Morskaya, S.S.; Kirschnek, S.; Gaffal, E.; Landsberg, J.; Hellmuth, J.; et al. 5′-triphosphate-siRNA: Turning gene silencing and Rig-I activation against melanoma. Nat. Med. 2008, 14, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Gantier, M.P.; Tong, S.; Behlke, M.A.; Irving, A.T.; Lappas, M.; Nilsson, U.W.; Latz, E.; McMillan, N.A.J.; Williams, B.R.G. Rational design of immunostimulatory siRNAs. Mol. Ther. 2010, 18, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Sugarman, E.A.; Nagan, N.; Zhu, H.; Akmaev, V.R.; Zhou, Z.; Rohlfs, E.M.; Flynn, K.; Hendrickson, B.C.; Scholl, T.; Sirko-Osadsa, D.A.; et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72 400 specimens. Eur. J. Hum. Genet. 2012, 20, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef]
- Monani, U.R.; Lorson, C.L.; Parsons, D.W.; Prior, T.W.; Androphy, E.J.; Burghes, A.H.M.; McPherson, J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999, 8, 1177–1183. [Google Scholar] [CrossRef]
- Mailman, M.D.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Wirth, B.; Burghes, A.H.M.; Prior, T.W. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar] [CrossRef]
- Wirth, B.; Herz, M.; Wetter, A.; Moskau, S.; Hahnen, E.; Wienker, T.; Zerres, K. Quantitative Analysis of Survival Motor Neuron Copies: Identification of Subtle SMN1 Mutations in Patients with Spinal Muscular Atrophy, Genotype-Phenotype Correlation and Implications for Genetic Counseling. Am. J. Hum. Genet. 1999, 64, 1340–1356. [Google Scholar] [CrossRef]
- FDA, U.S. Department of Health and Human Services, Center for Drug Evaluation and Research. Application Number: 209531Orig1s000. Med. Rev. 2017, 2017, 4026626. [Google Scholar]
- Kole, R.; Krieg, A.M. Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Deliv. Rev. 2015, 87, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Singh, N.K.; Singh, R.N.; Singh, R.N. Splicing of a Critical Exon of Human Survival Motor Neuron is Regulated by a Unique Silencer Element Located in the Last Intron. Mol. Cell. Biol. 2006, 26, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Karpati, G. Duchenne Muscular Dystophy: Plasma Membrane Loss Initiates Muscle Cell Necrosis Unless it is Repaired. Brain 1979, 102, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E.H. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; Den Dunnen, J.T.; Koop, K.; Van Der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef]
- Goemans, N.M.; Tulinius, M.; Van Den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef]
- FDA. ONPATTRO (patisiran) lipid complex injection, for intravenous use. Highlights Prescrib. Inf. 2018, 2018, 4305330. [Google Scholar]
- European Medicines Agency. Onpattro (Patisiran): An Overview of Onpattro and Why It Is Authorised in the EU; Assessment Report: EMA/554262/2018; Committee for Medicinal Products for Human Use (CHMP): London, UK, 2018. [Google Scholar]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef]
- Yang, J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev. Clin. Pharmacol. 2019, 12, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Plante-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Waddington-Cruz, M.; Ackermann, E.J.; Polydefkis, M.; Heitner, S.B.; Dyck, P.J.; Barroso, F.A.; Wang, A.K.; Berk, J.L.; Dyck, P.J.B.; Monia, B.P.; et al. Hereditary transthyretin amyloidosis: Baseline characteristics of patients in the NEURO-TTR trial. Amyloid 2018, 25, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Rodrigues, C.; Fonseca, I.; Sousa, A.; Branco, M.; Coelho, T.; Sequeiros, J.; Freitas, P. Family dynamics in transthyretin-related familial amyloid polyneuropathy Val30Met: Does genetic risk affect family functioning? Clin. Genet. 2018, 94, 401–408. [Google Scholar] [CrossRef]
- Hawkins, P.N.; Ando, Y.; Dispenzeri, A.; Gonzalez-Duarte, A.; Adams, D.; Suhr, O.B. Evolving landscape in the management of transthyretin amyloidosis. Ann. Med. 2015, 47, 625–638. [Google Scholar] [CrossRef]
- Buxbaum, J.N. Oligonucleotide Drugs for Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 82–85. [Google Scholar] [CrossRef]
- Maia, L.F.; Magalhães, R.; Freitas, J.; Taipa, R.; Pires, M.M.; Osório, H.; Dias, D.; Pessegueiro, H.; Correia, M.; Coelho, T. CNS involvement in V30M transthyretin amyloidosis: Clinical, neuropathological and biochemical findings. J. Neurol. Neurosurg. Psychiatry 2015, 86, 159–167. [Google Scholar] [CrossRef]
- Lavigne-Moreira, C.; Marques, V.D.; Gonçalves, M.V.M.; de Oliveira, M.F.; Tomaselli, P.J.; Nunez, J.C.; do Nascimento, O.J.M.; Barreira, A.A.; Marques, W. The genetic heterogeneity of hereditary transthyretin amyloidosis in a sample of the Brazilian population. J. Peripher. Nerv. Syst. 2018, 23, 134–137. [Google Scholar] [CrossRef]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef]
- Walker, S. New Medications in the Treatment of Hereditary Transthyretin Amyloidosis. Hosp. Pharm. 2018, 53, 236–238. [Google Scholar] [CrossRef]
- Suhr, O.B.; Coelho, T.; Buades, J.; Pouget, J.; Conceicao, I.; Berk, J.; Schmidt, H.; Waddington-Cruz, M.; Campistol, J.M.; Bettencourt, B.R.; et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 2015, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Coelho, T.; Conceicao, E.; Waddington-Cruz, M.; Schmidt, H.; Buades, J.; Campistol, J.; Pouget, J.; Berk, J.; Polydefkis, M.; et al. Phase 2 Open-Label Extension (Ole) Study of Patisiran, an Investigational Rna Interference (Rnai) Therapeutic for the Treatment of Hereditary Attr Amyloidosis with Polyneuropathy. Value Heal. 2017. [Google Scholar] [CrossRef]
- European Medicines Agency. Summary of Product Characteristics: Tegsedi 284mg Solution for Injection in Pre-Filled Syringe; Akcea Therapeutics UK Ltd.: Surrey, UK, 2018. [Google Scholar]
- Akcea Therapeutics Inc. US Prescribing Information: TEGSEDITM (Inotersen) Injection, for Subcutaneous Use; Akcea Therapeutics UK Ltd.: Surrey, UK, 2018. [Google Scholar]
- Aboul-Fadl, T. Antisense Oligonucleotides: The State of the Art. Curr. Med. Chem. 2005, 12, 2193–2214. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Garba, A.O.; Mousa, S.A. Bevasiranib for the Treatment of Wet, Age-Related Macular Degeneration. Ophthalmol. Eye Dis. 2010, 2, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Singerman, L. Combination therapy using the small interfering RNA bevasiranib. Retina 2009, 29, S49–S50. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, L.; Weinreb, R.N. Ophthalmic drug discovery: Novel targets and mechanisms for retinal diseases and glaucoma. Nat. Rev. Drug Discov. 2012, 11, 541–559. [Google Scholar] [CrossRef]
- Worringer, K.A.; Rand, T.A.; Hayashi, Y.; Sami, S.; Takahashi, K.; Tanabe, K.; Narita, M.; Srivastava, D.; Yamanaka, S. The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 2014, 14, 40–52. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nat. 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Calin, G.A.; Lopez-Berestein, G.; Sood, A.K. MiRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 2016, 6, 235–246. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Gregory, R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 2015, 15, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L.S. Delivery of siRNA therapeutics: Barriers and carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yue, W.; Xie, Y.; Liu, L.; Li, S.; Dang, W.; Xin, S.; Yang, L.; Zhai, X.; Cao, P.; et al. The four-microRNA signature identified by bioinformatics analysis predicts the prognosis of nasopharyngeal carcinoma patients. Oncol. Rep. 2019, 1767–1780. [Google Scholar] [CrossRef]
- Andersen, G.B.; Tost, J. Circulating miRNAs as Biomarker in Cancer. In Tumor Liquid Biopsies; Schaffner, F., Merlin, J.L., von Bubnoff, N., Eds.; Recent Results in Cancer Research; Springer: Cham, Switzerland, 2020; Volume 215. [Google Scholar]
- Cavalcante, P.; Mizrachi, T.; Barzago, C.; Scandiffio, L.; Bortone, F.; Bonanno, S.; Frangiamore, R.; Mantegazza, R.; Bernasconi, P.; Brenner, T.; et al. MicroRNA signature associated with treatment response in myasthenia gravis: A further step towards precision medicine. Pharmacol. Res. 2019, 148, 104388. [Google Scholar] [CrossRef]
- Wang, Y.; Ru, J.; Jin, T.; Sun, M.; Jia, L.; Sun, G. An Approach to Identify Individual Functional Single Nucleotide Polymorphisms and Isoform MicroRNAs. Biomed. Res. Int. 2019, 2019, 6193673. [Google Scholar] [CrossRef]
- Rooij, E.; Kauppinen, S. Development of micro RNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef]
- Jazbutyte, V. Specific, Robust, Reproducible: The Hunt for the Ideal Biomarker. J. Clin. Exp. Cardiol. 2012, 01, 9880. [Google Scholar] [CrossRef]
- De Guire, V.; Robitaille, R.; Tétreault, N.; Guérin, R.; Ménard, C.; Bambace, N.; Sapieha, P. Circulating miRNAs as sensitive and specific biomarkers for the diagnosis and monitoring of human diseases: Promises and challenges. Clin. Biochem. 2013, 46, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Schaefer, A.; Steiner, I.; Kempkensteffen, C.; Stephan, C.; Erbersdobler, A.; Jung, K. Robust MicroRNA stability in degraded RNA preparations from human tissue and cell samples. Clin. Chem. 2010, 56, 998–1006. [Google Scholar] [CrossRef]
- Kumar, M.S.; Erkeland, S.J.; Pester, R.E.; Chen, C.Y.; Ebert, M.S.; Sharp, P.A.; Jacks, T. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc. Natl. Acad. Sci. USA 2008, 105, 3903–3908. [Google Scholar] [CrossRef]
- Gustafson, D.; Tyryshkin, K.; Renwick, N. microRNA-guided diagnostics in clinical samples. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 563–575. [Google Scholar] [CrossRef]
- Kim, Y.K.; Yeo, J.; Kim, B.; Ha, M.; Kim, V.N. Short Structured RNAs with Low GC Content Are Selectively Lost during Extraction from a Small Number of Cells. Mol. Cell 2012, 46, 893–895. [Google Scholar] [CrossRef]
- Leichter, A.L.; Purcell, R.V.; Sullivan, M.J.; Eccles, M.R.; Chatterjee, A. Multi-platform microRNA profiling of hepatoblastoma patients using formalin fixed paraffin embedded archival samples. Gigascience 2015. [Google Scholar] [CrossRef]
- Git, A.; Dvinge, H.; Salmon-Divon, M.; Osborne, M.; Kutter, C.; Hadfield, J.; Bertone, P.; Caldas, C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 2001, 16, 991–1006. [Google Scholar] [CrossRef]
- Sato, F.; Tsuchiya, S.; Terasawa, K.; Tsujimoto, G. Intra-platform repeatability and inter-platform comparability of microRNA microarray technology. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.X.; Tuschl, T.; Singer, S. Empirical insights into the stochasticity of small RNA sequencing. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Guan, Z.; Cuk, K.; Brenner, H.; Zhang, Y. Circulating microRNA biomarkers for lung cancer detection in Western populations. Cancer Med. 2018, 7, 4849–4862. [Google Scholar] [CrossRef] [PubMed]
- Rawlings-Goss, R.A.; Campbell, M.C.; Tishkoff, S.A. Global population-specific variation in miRNA associated with cancer risk and clinical biomarkers. BMC Med. Genomics 2014, 7, 1–14. [Google Scholar] [CrossRef]
- Rissin, D.M.; López-Longarela, B.; Pernagallo, S.; Ilyine, H.; Vliegenthart, A.D.B.; Dear, J.W.; Díaz-Mochón, J.J.; Duffy, D.C. Polymerase-free measurement of microRNA-122 with single base specificity using single molecule arrays: Detection of drug-induced liver injury. PLoS ONE 2017, 12, e0179669. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Boran, A.; Iyengar, R. Systems approaches to polypharmacology and drug discovery. Curr. Opin Drug Discov. Dev. 2010, 13, 297–309. [Google Scholar]
- Lim, L.P.; Lau, N.C.; Garrett-engele, P.; Grimson, A. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 292, 288–292. [Google Scholar] [CrossRef]
- Singh, S.; Narang, A.S.; Mahato, R.I. Subcellular fate and off-target effects of siRNA, shRNA, and miRNA. Pharm. Res. 2011, 28, 2996–3015. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, H.; Tan, Z.; Zhang, C.; Fu, X. Bottleneck limitations for microRNA-based therapeutics from bench to the bedside. Pharmazie 2015, 70, 147–154. [Google Scholar] [PubMed]
- Lal, A.; Navarro, F.; Maher, C.A.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofmann, O.; et al. miR-24 Inhibits Cell Proliferation by Targeting E2F2, MYC, and Other Cell-Cycle Genes via Binding to “Seedless” 3′UTR MicroRNA Recognition Elements. Mol. Cell 2009, 35, 610–625. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H. Redefining MicroRNA Targets. Curr. Biol. 2009, 19, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, V.; Muth, D.C.; Witwer, K.W. Validated MicroRNA Target Databases: An Evaluation. Drug Dev. Res. 2015, 76, 389–396. [Google Scholar]
- Leclercq, M.; Diallo, A.B.; Blanchette, M. Prediction of human miRNA target genes using computationally reconstructed ancestral mammalian sequences. Nucleic Acids Res. 2017, 45, 556–566. [Google Scholar] [CrossRef]
- Metias, S.M.; Lianidou, E.; Yousef, G.M. MicroRNAs in clinical oncology: At the crossroads between promises and problems. J. Clin. Pathol. 2009, 62, 771–776. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Fayyaz, S.; Shatynska-Mytsyk, I.; Javed, Z.; Jabeen, S.; Yaylim, I.; Gasparri, M.L.; Panici, P.B. Is miR-34a a Well-equipped Swordsman to Conquer Temple of Molecular Oncology? Chem. Biol. Drug Des. 2016, 87, 321–334. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Trang, P.; Wiggins, J.F.; Patrawala, L.; Cheng, A.; Ford, L.; Weidhaas, J.B.; Brown, D.; Bader, A.G.; Slack, F.J. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle 2008, 7, 759–764. [Google Scholar] [CrossRef]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef]
- Boudreau, R.L.; Monteys, A.M.; Davidson, B.L. Minimizing variables among hairpin-based RNAi vectors reveals the potency of shRNAs. RNA 2008, 14, 1834–1844. [Google Scholar] [CrossRef]
- Bauer, M.; Kinkl, N.; Meixner, A.; Kremmer, E.; Riemenschneider, M.; Förstl, H.; Gasser, T.; Ueffing, M. Prevention of interferon-stimulated gene expression using microRNA-designed hairpins. Gene Ther. 2009, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, L.A.; Zhang, J.; von Eije, K.J.; Li, H.; Sætrom, P.; Amarzguioui, M.; Rossi, J.J. Engineering and optimization of the miR-106b cluster for ectopic expression of multiplexed anti-HIV RNAs. Gene Ther. 2008, 15, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Haasnoot, J.; ter Brake, O.; Berkhout, B.; Konstantinova, P. Inhibition of HIV-1 by multiple siRNAs expressed from a single microRNA polycistron. Nucleic Acids Res. 2008, 36, 2811–2824. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef]
- Grimm, D.; Wang, L.; Lee, J.S.; Schürmann, N.; Gu, S.; Börner, K.; Storm, T.A.; Kay, M.A. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J. Clin. Investig. 2010, 120, 3106–3119. [Google Scholar] [CrossRef]
- Diederichs, S.; Jung, S.; Rothenberg, S.M.; Smolen, G.A.; Mlody, B.G.; Haber, D.A. Coexpression of Argonaute-2 enhances RNA interference toward perfect match binding sites. Proc. Natl. Acad. Sci. USA 2008, 105, 9284–9289. [Google Scholar] [CrossRef]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef]
- Boudreau, R.L.; Martins, I.; Davidson, B.L. Artificial MicroRNAs as siRNA shuttles: Improved safety as compared to shRNAs in vitro and In vivo. Mol. Ther. 2009, 17, 169–175. [Google Scholar] [CrossRef]
- Beer, S.; Bellovin, D.I.; Lee, J.S.; Komatsubara, K.; Wang, L.S.; Koh, H.; Börner, K.; Storm, T.A.; Davis, C.R.; Kay, M.A.; et al. Low-level shRNA cytotoxicity can contribute to MYC-induced hepatocellular carcinoma in adult mice. Mol. Ther. 2010, 18, 161–170. [Google Scholar] [CrossRef]
- Singh, A.; Bhattacharyya, N.; Srivastava, A.; Pruett, N.; Ripley, R.T.; Schrump, D.S.; Hoang, C.D. MicroRNA-215-5p Treatment Suppresses Mesothelioma Progression via the MDM2-p53-Signaling Axis. Mol. Ther. 2019, 27, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Hede, K. MicroRNAs As Onco-miRs, Drivers of Cancer. J. Am. Med. Assoc. 2010, 102, 1306–1308. [Google Scholar]
- Ebert, M.S.; Neilson, J.R.; Sharp, P. A MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2013, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618. [Google Scholar] [CrossRef]
- Lindow, M.; Kauppinen, S. Discovering the first microrna-targeted drug. J. Cell Biol. 2012, 199, 407–412. [Google Scholar] [CrossRef]
- Umbach, J.L.; Cullen, B.R. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 2009, 23, 1151–1164. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; Rebhan, M.A.E.; Crivelli, S.E.M.; Denzler, R.; Stoffel, M.; Hall, J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014, 42, 609–621. [Google Scholar] [CrossRef]
- Jopling, C. Liver-specific microRNA-122. RNA Biol. 2012, 9, 137–142. [Google Scholar] [CrossRef]
- Jopling, C.L.; Schütz, S.; Sarnow, P. Position-Dependent Function for a Tandem MicroRNA miR-122-Binding Site Located in the Hepatitis C Virus RNA Genome. Cell Host Microbe 2008, 4, 77–85. [Google Scholar] [CrossRef]
- Baek, J.; Kang, S.; Min, H. MicroRNA-targeting therapeutics for hepatitis C. Arch. Pharm. Res. 2014, 37, 299–305. [Google Scholar] [CrossRef]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.; Pel, M.E.; Kirschner, M.B.; Cheng, Y.Y.; Mugridge, N.; Weiss, J.; Williams, M.; Wright, C.; Edelman, J.J.B.; Vallely, M.P.; et al. Restoring expression of miR-16: A novel approach to therapy for malignant pleural mesothelioma. Ann. Oncol. 2013, 24, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013, 31, 577. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Parsons, C.; Slack, F.J. The Tumor-Suppressive and Potential Therapeutic Functions of miR-34a in Epithelial Carcinomas. Expert Opin Ther Targets 2016, 20, 737–753. [Google Scholar] [CrossRef]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gullà, A.; Tagliaferri, P.; Tassone, P.; et al. Mir-34: A new weapon against cancer? Mol. Ther. Nucleic Acids 2014, 3, e195. [Google Scholar] [CrossRef]
- Ling, H.; Girnita, L.; Buda, O.; Calin, G.A. Non-coding RNAs: The cancer genome dark matter that matters! Clin. Chem. Lab. Med. 2017, 55, 705–714. [Google Scholar] [CrossRef]
- Sergeeva, O.V.; Koteliansky, V.E.; Zatsepin, T.S. mRNA Based Therapeutics—Advances and Perspectives. Biochemistry 2016. [Google Scholar] [CrossRef]
- Heiser, A.; Coleman, D.; Dannull, J.; Yancey, D.; Maurice, M.A.; Lallas, C.D.; Dahm, P.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J. Clin. Invest. 2002, 109, 311–312. [Google Scholar] [CrossRef]
- le Sage, C.; Lawo, S.; Cross, B.C.S. CRISPR: A Screener’s Guide. SLAS Discov. Adv. life Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Kurata, M.; Yamamoto, K.; Moriarity, B.S.; Kitagawa, M.; Largaespada, D.A. CRISPR/Cas9 library screening for drug target discovery. J. Hum. Genet. 2018, 63, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Gao, Z.; Berkhout, B. CRISPR therapy towards an HIV cure. Brief. Funct. Genomics 2019. [Google Scholar] [CrossRef] [PubMed]
- Beierlein, J.M.; McNamee, L.M.; Ledley, F.D. As Technologies for Nucleotide Therapeutics Mature, Products Emerge. Mol. Ther. Nucleic Acids 2017, 9, 379–386. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iwama, H.; Murao, K.; Imachi, H.; Ishida, T. MicroRNA networks alter to conform to transcription factor networks adding redundancy and reducing the repertoire of target genes for coordinated regulation. Mol. Biol. Evol. 2011, 28, 639–646. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neilsen, C.T.; Goodall, G.J.; Bracken, C.P. IsomiRs—The overlooked repertoire in the dynamic microRNAome. Trends Genet. 2012, 28, 544–549. [Google Scholar] [CrossRef]
- Fehlmann, T.; Backes, C.; Kahraman, M.; Haas, J.; Ludwig, N.; Posch, A.E.; Würstle, M.L.; Hübenthal, M.; Franke, A.; Meder, B.; et al. Web-based NGS data analysis using miRMaster: a large-scale meta-analysis of human miRNAs. Nucleic Acids Res. 2017, 45, 8731–8744. [Google Scholar] [CrossRef]
- Lan, C.; Peng, H.; Mcgowan, E.M.; Hutvagner, G.; Li, J. An isomiR expression panel based novel breast cancer classification approach using improved mutual information. BMC Med. Genomics 2018, 11, 73–85. [Google Scholar] [CrossRef]
- Telonis, A.G.; Magee, R.; Loher, P.; Chervoneva, I.; Londin, E.; Rigoutsos, I. Knowledge about the presence or absence of miRNA isoforms (isomiRs ) can successfully discriminate amongst 32 TCGA cancer types. Nuc. Acids. Res. 2017, 45, 2973–2985. [Google Scholar] [CrossRef]
- Hu, W.Z.; Tan, C.L.; He, Y.J.; Zhang, G.Q.; Xu, Y.; Tang, J.H. Functional miRNAs in breast cancer drug resistance. Onco. Targets. Ther. 2018, 11, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Van Roosbroeck, K. miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018, 37, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Li, M.J.; Kim, J.; Li, S.; Zaia, J.; Yee, J.K.; Anderson, J.; Akkina, R.; Rossi, J.J. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol. Ther. 2005, 12, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Digiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Yam, P.; Stinson, S.; Kalos, M.; Alvarnas, J.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS- related lymphoma. Sci. Transl Med. 2011. [Google Scholar] [CrossRef] [PubMed]
- Unwalla, H.J.; Li, H.-T.; Bahner, I.; Li, M.-J.; Kohn, D.; Rossi, J.J. Novel Pol II Fusion Promoter Directs Human Immunodeficiency Virus Type 1-Inducible Coexpression of a Short Hairpin RNA and Protein. J. Virol. 2006, 80, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Canadian Agency for Drugs and Technologies in Health. Pharmacoeconomic Review Report for Nusinsersin; Appendix 1, Cost Comparison. Nusinersen (Spinraza): (Biogen Canada Inc.): Indication: Treatment of patients with 5q SMA. CADTH Common Drug Review. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30480926 (accessed on 2 January 2020).
- King, N.M.P.; Bishop, C.E. New treatments for serious conditions: Ethical implications. Gene Ther. 2017, 24, 534–538. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNA-Based Therapeutics | |||||
|---|---|---|---|---|---|
| Company | Name | Therapeutic Agent | Delivery System | Target Disease | Stage in Drug Development Pipeline |
| Santaris Pharma/Roche | Miravirsen | AntimiR-122 | LNA antagomiR | Hepatitis C; Chronic hepatitis C | Phase II clinical trials (NCT02452814; NCT2508090) |
| Regulus Therapeutics | RG-101 | AntimiR-122 | GaLNAc-conjugated antagomiR | Chronic hepatitis C | Phase II clinical trials (discontinued) |
| RG-125 | AntimiR-103/107 | GaLNAc-conjugated antagomiR | Diabetic non-alcoholic steatohepatitis | Phase II (discontinued) | |
| RG-012 | AntimiR-21 | NA | Hereditary nephritis | Phase II (NCT02855268) | |
| RGLS4326 | AntimiR-17 | NA | Autosomal dominant polycystic kidney disease | Phase I (on hold) | |
| miRagen Therapeutics | MRG-106 | AntimiR-155 | LNA-modified antisense inhibitor | CTCL mycosis fungoides subtype; CLL; DLBCL; ATLL | Phase II (NCT03713320; NCT03837457); Phase I (NCT02580552) |
| MRG-107 | AntimiR-155 | NA | ALS; cardiac disorders; retinal disorders | Pre-Clinical | |
| MRG-110 | AntimiR-92 | LNA antagomiR | Wounds | Phase I (NCT03603431) | |
| MRG-201 | miR-29 mimic | Cholesterol-conjugated miRNA duplex | Keloid; fibrosis | Phase II (NCT03601052); Phase I (NCT02603224) | |
| EnGeneIC | MesomiR-1 | miR-16 mimic | EnGeneIC Dream Vector | Malignant pleural mesothelioma; non-small-cell lung cancer | Phase I (NCT02369198) |
| Mirna Therapeutics Inc. | MRX-34 | miR-34 mimic | dsRNA liposomal nanoparticle | Solid tumours; haematological malignancies | Phase 1 (terminated) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. https://doi.org/10.3390/cells9010137
Bajan S, Hutvagner G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells. 2020; 9(1):137. https://doi.org/10.3390/cells9010137
Chicago/Turabian StyleBajan, Sarah, and Gyorgy Hutvagner. 2020. "RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs" Cells 9, no. 1: 137. https://doi.org/10.3390/cells9010137
APA StyleBajan, S., & Hutvagner, G. (2020). RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells, 9(1), 137. https://doi.org/10.3390/cells9010137