Post-Translational Modification and Natural Mutation of TRPC Channels



Abstract
1. Introduction to TRPC
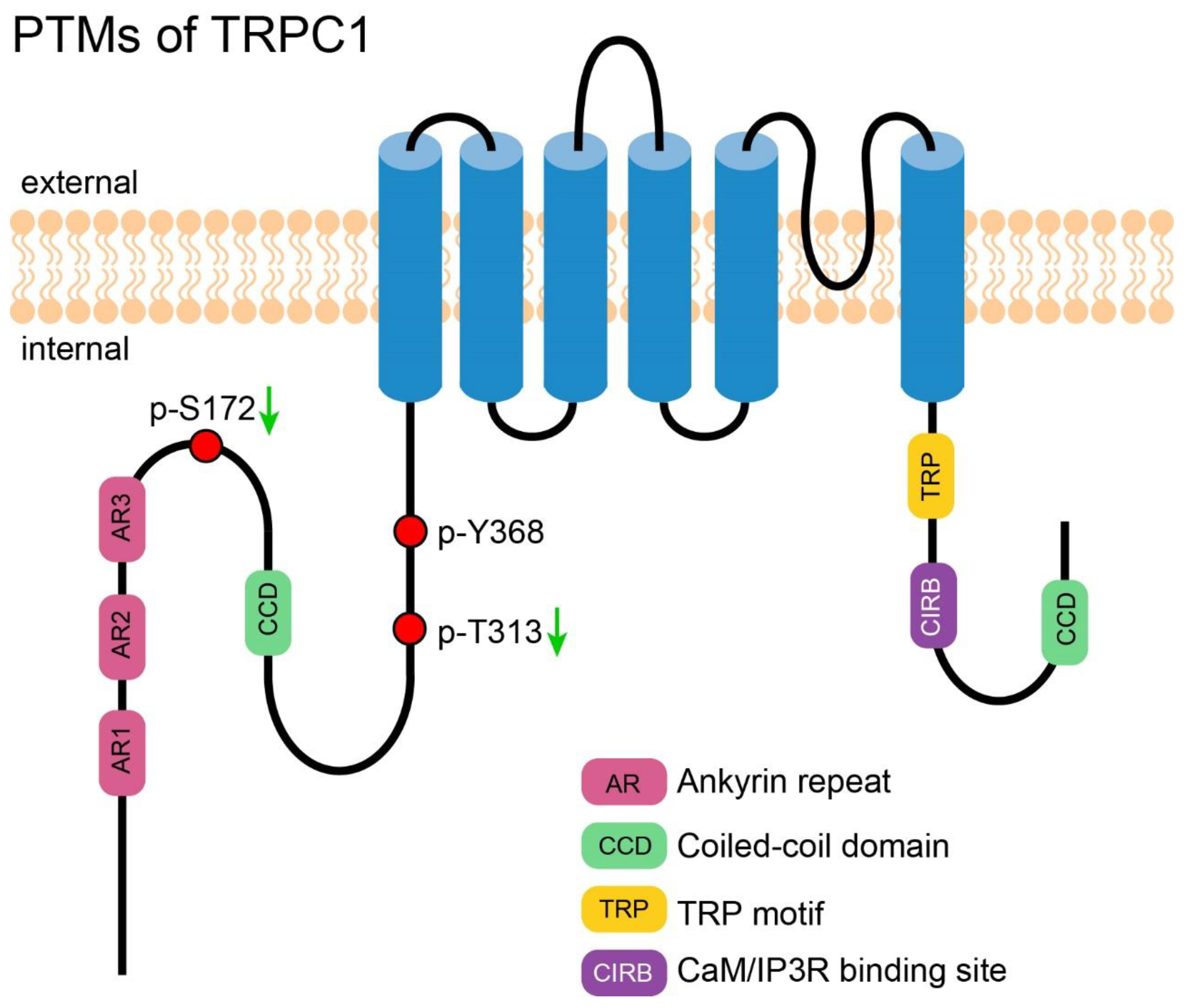
2. PTM of TRPC1
2.1. Phosphorylation-Induced Activation of TRPC1
2.2. Phosphorylation-Induced Inhibition of TRPC1
2.3. PTM of TRPC1 Discovered by High-Throughput Experiments
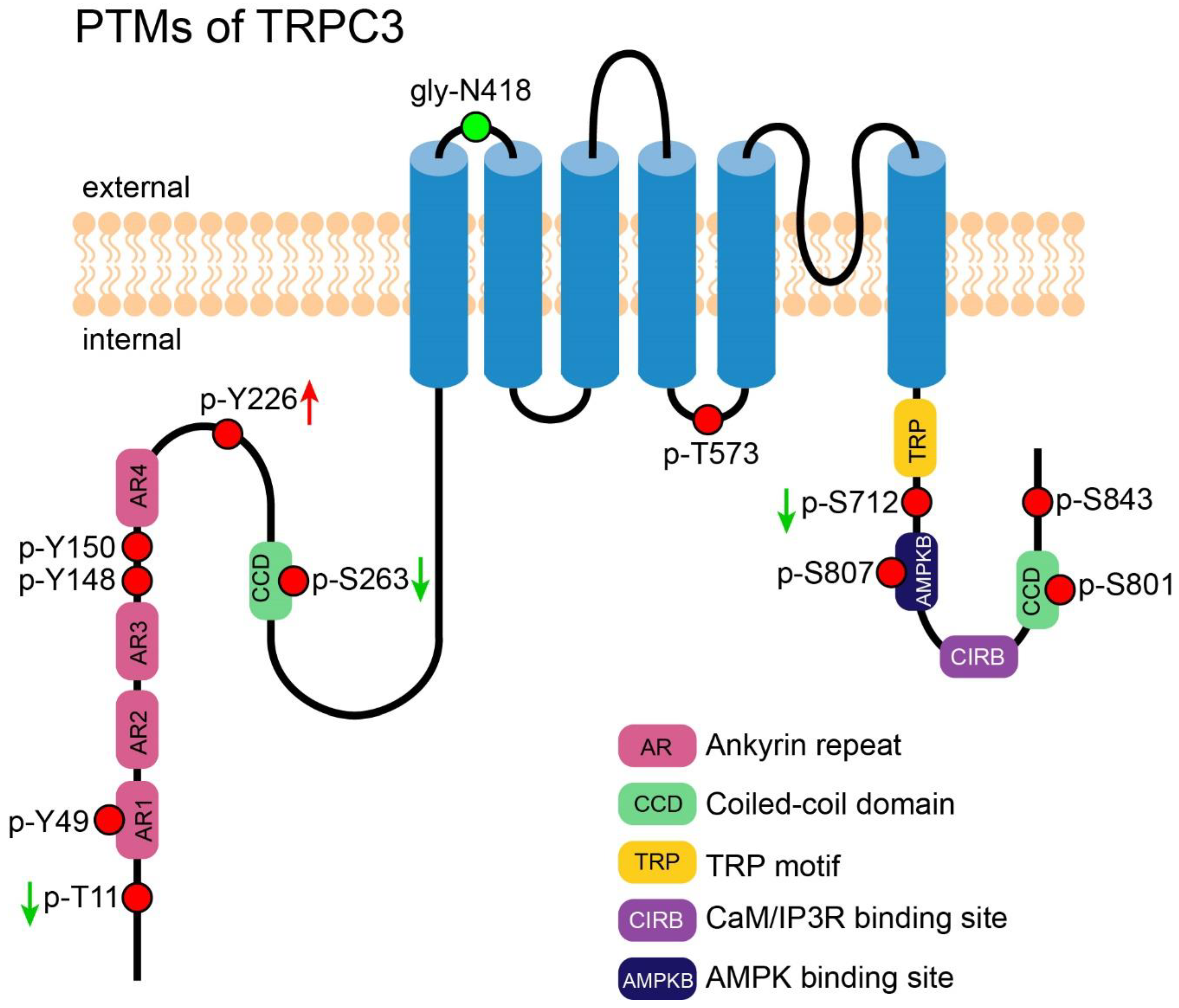
3. PTM of TRPC3
3.1. N-Glycosylation of TRPC3
3.2. Phosphorylation-Induced Activation of TRPC3
3.3. Phosphorylation-Induced Inhibition of TRPC3
3.4. Phosphorylation-Mediated Protein-Protein Interaction of TRPC3
3.5. PTM of TRPC3 Discovered by High-Throughput Experiments
4. PTM of TRPC4
4.1. Phosphorylation-Mediated Activation of TRPC4
4.2. PTM of TRPC4 Discovered by High-Throughput Experiments
4.3. Ubiquitination of TRPC4
4.4. Disulfide Bond Formation in TRPC4
5. PTM of TRPC5
5.1. Phosphorylation of TRPC5
5.2. Disulfide Bond Formation in TRPC5
5.3. S-Nitrosylation of TRPC5
5.4. S-Glutathionylation of TRPC5
6. PTM of TRPC6
6.1. Glycosylation of TRPC6
6.2. Acetylation of TRPC6
6.3. Phosphorylation-Induced Activation of TRPC6
6.4. Phosphorylation-Induced Inhibition of TRPC6
6.5. Phosphorylation-Induced Trafficking of TRPC6
6.6. Phosphorylation-Mediated Protein-Protein Interaction of TRPC6
6.7. PTM of TRPC6 Discovered by High-Throughput Experiments
7. PTM of TRPC7
7.1. Phosphorylation of TRPC7
7.2. PTM of TRPC7 Discovered by High-Throughput Experiments
8. TRPC6 Mutation and Focal Segmental Glomerulosclerosis (FSGS)
8.1. Introduction of FSGS
8.2. Overview of TRPC6 Mutations and SNP
8.3. Mechanism of TRPC6-Mutant Induced FSGS
9. TRPC6 Mutation/SNP and Other Diseases
9.1. TRPC6 and Infantile Hypertrophic Pyloric Stenosis (IHPS)
9.2. TRPC6 and Idiopathic Pulmonary Arterial Hypertension (IPAH)
9.3. TRPC6 and Neuropsychiatric Manifestations (NPSLE)
10. TRPC6 and Chronic Fatigue Syndrome (CFS)
11. Mutation/SNP of Other TRPC Channels and Corresponding Diseases
11.1. TRPC3 and Inherited Cerebellar Ataxia
11.2. TRPC4 and Myocardial Infarction (MI)
11.3. TRPC4/7 and Lung Cancer
12. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Okada, T.; Inoue, R.; Yamazaki, K.; Maeda, A.; Kurosaki, T.; Yamakuni, T.; Tanaka, I.; Shimizu, S.; Ikenaka, K.; Imoto, K.; et al. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7—Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J. Biol. Chem. 1999, 274, 27359–27370. [Google Scholar] [CrossRef]
- Okada, T.; Shimizu, S.; Wakamori, M.; Maeda, A.; Kurosaki, T.; Takada, N.; Imoto, K.; Mori, Y. Molecular cloning and functional characterization of a novel receptor-activated TRP Ca2+ channel from mouse brain. J. Biol. Chem. 1998, 273, 10279–10287. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. Trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 1996, 85, 661–671. [Google Scholar] [CrossRef]
- Philipp, S.; Cavalie, A.; Freichel, M.; Wissenbach, U.; Zimmer, S.; Trost, C.; Marquart, A.; Murakami, M.; Flockerzi, V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J. 1996, 15, 6166–6171. [Google Scholar] [CrossRef]
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656. [Google Scholar] [CrossRef]
- Boulay, G.; Zhu, X.; Peyton, M.; Jiang, M.; Hurst, R.; Stefani, E.; Birnbaumer, L. Cloning and expression of a novel mammalian homolog of Drosophila transient receptor potential (Trp) involved in calcium entry secondary to activation of receptors coupled by the Gq class of G protein. J. Biol. Chem. 1997, 272, 29672–29680. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W., Jr. The mammalian TRPC cation channels. Biochim. Biophys. Acta 2004, 1742, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.; Schaefer, M.; Schultz, G.; Gudermann, T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. USA 2002, 99, 7461–7466. [Google Scholar] [CrossRef] [PubMed]
- Liman, E.R.; Innan, H. Relaxed selective pressure on an essential component of pheromone transduction in primate evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 3328–3332. [Google Scholar] [CrossRef] [PubMed]
- Tsiokas, L.; Arnould, T.; Zhu, C.; Kim, E.; Walz, G.; Sukhatme, V.P. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc. Natl. Acad. Sci. USA 1999, 96, 3934–3939. [Google Scholar] [CrossRef]
- Schindl, R.; Fritsch, R.; Jardin, I.; Frischauf, I.; Kahr, H.; Muik, M.; Riedl, M.C.; Groschner, K.; Romanin, C. Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J. Biol. Chem. 2012, 287, 35612–35620. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cao, J.; Luo, J.; Nilius, B.; Huang, Y.; Ambudkar, I.S.; Yao, X. Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4-c1 heteromeric channels to the plasma membrane. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2249–2255. [Google Scholar] [CrossRef] [PubMed]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J. Biol. Chem. 2003, 278, 39014–39019. [Google Scholar] [CrossRef] [PubMed]
- Strubing, C.; Krapivinsky, G.; Krapivinsky, L.; Clapham, D.E. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 2001, 29, 645–655. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Lee, Y.; Kim, S.M.; Yang, Y.D.; Jung, J.; Oh, U. Quantitative analysis of TRP channel genes in mouse organs. Arch. Pharmacal Res. 2012, 35, 1823–1830. [Google Scholar] [CrossRef]
- Seth, M.; Zhang, Z.S.; Mao, L.; Graham, V.; Burch, J.; Stiber, J.; Tsiokas, L.; Winn, M.; Abramowitz, J.; Rockman, H.A.; et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 2009, 105, 1023–1030. [Google Scholar] [CrossRef]
- Bergdahl, A.; Gomez, M.F.; Dreja, K.; Xu, S.Z.; Adner, M.; Beech, D.J.; Broman, J.; Hellstrand, P.; Sward, K. Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ. Res. 2003, 93, 839–847. [Google Scholar] [CrossRef]
- Kumar, B.; Dreja, K.; Shah, S.S.; Cheong, A.; Xu, S.Z.; Sukumar, P.; Naylor, J.; Forte, A.; Cipollaro, M.; McHugh, D.; et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ. Res. 2006, 98, 557–563. [Google Scholar] [CrossRef]
- Yu, P.C.; Gu, S.Y.; Bu, J.W.; Du, J.L. TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ. Res. 2010, 106, 1221–1232. [Google Scholar] [CrossRef]
- Lopez, E.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. FKBP52 is involved in the regulation of SOCE channels in the human platelets and MEG 01 cells. Biochim. Biophys. Acta 2013, 1833, 652–662. [Google Scholar] [CrossRef]
- Louis, M.; Zanou, N.; Van Schoor, M.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121, 3951–3959. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Dragicevic, E.; Adelsberger, H.; Henning, H.A.; Sumser, M.; Abramowitz, J.; Blum, R.; Dietrich, A.; Freichel, M.; Flockerzi, V.; et al. TRPC3 channels are required for synaptic transmission and motor coordination. Neuron 2008, 59, 392–398. [Google Scholar] [CrossRef]
- Mottl, A.K.; Lu, M.; Fine, C.A.; Weck, K.E. A novel TRPC6 mutation in a family with podocytopathy and clinical variability. BMC Nephrol. 2013, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Ben-Mabrouk, F.; Tryba, A.K. Substance P modulation of TRPC3/7 channels improves respiratory rhythm regularity and ICAN-dependent pacemaker activity. Eur. J. Neurosci. 2010, 31, 1219–1232. [Google Scholar] [CrossRef]
- Salido, G.M.; Sage, S.O.; Rosado, J.A. TRPC channels and store-operated Ca2+ entry. Biochim. Biophys. Acta 2009, 1793, 223–230. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yeromin, A.V.; Zhang, X.H.; Yu, Y.; Safrina, O.; Penna, A.; Roos, J.; Stauderman, K.A.; Cahalan, M.D. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc. Natl. Acad. Sci. USA 2006, 103, 9357–9362. [Google Scholar] [CrossRef]
- Vig, M.; Peinelt, C.; Beck, A.; Koomoa, D.L.; Rabah, D.; Koblan-Huberson, M.; Kraft, S.; Turner, H.; Fleig, A.; Penner, R.; et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 2006, 312, 1220–1223. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E., Jr.; Meyer, T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Erxleben, C.; Yildirim, E.; Abramowitz, J.; Armstrong, D.L.; Birnbaumer, L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 4682–4687. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Zeng, W.; Huang, G.N.; Worley, P.F.; Muallem, S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007, 9, 636–645. [Google Scholar] [CrossRef]
- Ong, H.L.; Subedi, K.P.; Son, G.Y.; Liu, X.; Ambudkar, I.S. Tuning store-operated calcium entry to modulate Ca2+-dependent physiological processes. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1037–1045. [Google Scholar] [CrossRef]
- Storch, U.; Forst, A.L.; Pardatscher, F.; Erdogmus, S.; Philipp, M.; Gregoritza, M.; Mederos, Y.S.M.; Gudermann, T. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc. Natl. Acad. Sci. USA 2017, 114, E37–E46. [Google Scholar] [CrossRef]
- Lintschinger, B.; Balzer-Geldsetzer, M.; Baskaran, T.; Graier, W.F.; Romanin, C.; Zhu, M.X.; Groschner, K. Coassembly of Trp1 and Trp3 proteins generates diacylglycerol- and Ca2+-sensitive cation channels. J. Biol. Chem. 2000, 275, 27799–27805. [Google Scholar] [CrossRef]
- Storch, U.; Forst, A.L.; Philipp, M.; Gudermann, T.; Mederos y Schnitzler, M. Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J. Biol. Chem. 2012, 287, 3530–3540. [Google Scholar] [CrossRef]
- Kerstein, P.C.; Jacques-Fricke, B.T.; Rengifo, J.; Mogen, B.J.; Williams, J.C.; Gottlieb, P.A.; Sachs, F.; Gomez, T.M. Mechanosensitive TRPC1 channels promote calpain proteolysis of talin to regulate spinal axon outgrowth. J. Neurosci. 2013, 33, 273–285. [Google Scholar] [CrossRef]
- Gomis, A.; Soriano, S.; Belmonte, C.; Viana, F. Hypoosmotic- and pressure-induced membrane stretch activate TRPC5 channels. J. Physiol. 2008, 586, 5633–5649. [Google Scholar] [CrossRef]
- Sharif-Naeini, R.; Dedman, A.; Folgering, J.H.; Duprat, F.; Patel, A.; Nilius, B.; Honore, E. TRP channels and mechanosensory transduction: Insights into the arterial myogenic response. Pflug. Arch. 2008, 456, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Jemal, I.; Soriano, S.; Conte, A.L.; Morenilla, C.; Gomis, A. G protein-coupled receptor signalling potentiates the osmo-mechanical activation of TRPC5 channels. Pflug. Arch. Eur. J. Physiol. 2014, 466, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Mederos y Schnitzler, M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103. [Google Scholar] [CrossRef] [PubMed]
- Paria, B.C.; Vogel, S.M.; Ahmmed, G.U.; Alamgir, S.; Shroff, J.; Malik, A.B.; Tiruppathi, C. Tumor necrosis factor-alpha-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1303–L1313. [Google Scholar] [CrossRef] [PubMed]
- Paria, B.C.; Malik, A.B.; Kwiatek, A.M.; Rahman, A.; May, M.J.; Ghosh, S.; Tiruppathi, C. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J. Biol. Chem. 2003, 278, 37195–37203. [Google Scholar] [CrossRef] [PubMed]
- Paria, B.C.; Bair, A.M.; Xue, J.; Yu, Y.; Malik, A.B.; Tiruppathi, C. Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J. Biol. Chem. 2006, 281, 20715–20727. [Google Scholar] [CrossRef] [PubMed]
- Ahmmed, G.U.; Mehta, D.; Vogel, S.; Holinstat, M.; Paria, B.C.; Tiruppathi, C.; Malik, A.B. Protein kinase Calpha phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J. Biol. Chem. 2004, 279, 20941–20949. [Google Scholar] [CrossRef]
- Bodiga, V.L.; Kudle, M.R.; Bodiga, S. Silencing of PKC-alpha, TRPC1 or NF-kappaB expression attenuates cisplatin-induced ICAM-1 expression and endothelial dysfunction. Biochem. Pharmacol. 2015, 98, 78–91. [Google Scholar] [CrossRef]
- Bergdahl, A.; Gomez, M.F.; Wihlborg, A.K.; Erlinge, D.; Eyjolfson, A.; Xu, S.Z.; Beech, D.J.; Dreja, K.; Hellstrand, P. Plasticity of TRPC expression in arterial smooth muscle: Correlation with store-operated Ca2+ entry. Am. J. Physiol. Cell Physiol. 2005, 288, C872–C880. [Google Scholar] [CrossRef]
- Kunichika, N.; Yu, Y.; Remillard, C.V.; Platoshyn, O.; Zhang, S.; Yuan, J.X. Overexpression of TRPC1 enhances pulmonary vasoconstriction induced by capacitative Ca2+ entry. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L962–L969. [Google Scholar] [CrossRef]
- Saleh, S.N.; Albert, A.P.; Large, W.A. Obligatory role for phosphatidylinositol 4,5-bisphosphate in activation of native TRPC1 store-operated channels in vascular myocytes. J. Physiol. 2009, 587, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Miralles, F.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Store depletion induces Galphaq-mediated PLCbeta1 activity to stimulate TRPC1 channels in vascular smooth muscle cells. FASEB J. 2016, 30, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Miralles, F.; Birnbaumer, L.; Large, W.A.; Albert, A.P. Store-operated interactions between plasmalemmal STIM1 and TRPC1 proteins stimulate PLCbeta1 to induce TRPC1 channel activation in vascular smooth muscle cells. J. Physiol. 2017, 595, 1039–1058. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, W.; Cui, W.; Shi, B.; Wang, H. PKCalpha promotes insulin secretion via TRPC1 phosphorylation in INS-1E cells. Biosci. Biotechnol. Biochem. 2019, 83, 1676–1682. [Google Scholar] [CrossRef]
- Zhang, P.; Ma, Y.; Wang, Y.; Ma, X.; Huang, Y.; Li, R.A.; Wan, S.; Yao, X. Nitric oxide and protein kinase G act on TRPC1 to inhibit 11,12-EET-induced vascular relaxation. Cardiovasc. Res. 2014, 104, 138–146. [Google Scholar] [CrossRef]
- Zhang, P.; Mao, A.Q.; Sun, C.Y.; Zhang, X.D.; Pan, Q.X.; Yang, D.T.; Jin, J.; Tang, C.L.; Yang, Z.Y.; Yao, X.Q.; et al. Translocation of PKG1alpha acts on TRPV4-C1 heteromeric channels to inhibit endothelial Ca2+ entry. Acta Pharmacol. Sin. 2016, 37, 1199–1207. [Google Scholar] [CrossRef]
- Li, H.; Ren, Z.; Kang, X.; Zhang, L.; Li, X.; Wang, Y.; Xue, T.; Shen, Y.; Liu, Y. Identification of tyrosine-phosphorylated proteins associated with metastasis and functional analysis of FER in human hepatocellular carcinoma cells. BMC Cancer 2009, 9, 366. [Google Scholar] [CrossRef]
- Vannier, B.; Zhu, X.; Brown, D.; Birnbaumer, L. The membrane topology of human transient receptor potential 3 as inferred from glycosylation-scanning mutagenesis and epitope immunocytochemistry. J. Biol. Chem. 1998, 273, 8675–8679. [Google Scholar] [CrossRef]
- Dietrich, A.; Mederos y Schnitzler, M.; Emmel, J.; Kalwa, H.; Hofmann, T.; Gudermann, T. N-linked protein glycosylation is a major determinant for basal TRPC3 and TRPC6 channel activity. J. Biol. Chem. 2003, 278, 47842–47852. [Google Scholar] [CrossRef]
- Vazquez, G.; Wedel, B.J.; Kawasaki, B.T.; Bird, G.S.; Putney, J.W., Jr. Obligatory role of Src kinase in the signaling mechanism for TRPC3 cation channels. J. Biol. Chem. 2004, 279, 40521–40528. [Google Scholar] [CrossRef]
- Kawasaki, B.T.; Liao, Y.; Birnbaumer, L. Role of Src in C3 transient receptor potential channel function and evidence for a heterogeneous makeup of receptor- and store-operated Ca2+ entry channels. Proc. Natl. Acad. Sci. USA 2006, 103, 335–340. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Huang, Y.; Yao, X. Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc. Natl. Acad. Sci. USA 2004, 101, 2625–2630. [Google Scholar] [CrossRef]
- Trebak, M.; Hempel, N.; Wedel, B.J.; Smyth, J.T.; Bird, G.S.; Putney, J.W., Jr. Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol. Pharmacol. 2005, 67, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.Y.; Huang, Y.; Yao, X. Protein kinase C can inhibit TRPC3 channels indirectly via stimulating protein kinase G. J. Cell. Physiol. 2006, 207, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Poteser, M.; Schleifer, H.; Lichtenegger, M.; Schernthaner, M.; Stockner, T.; Kappe, C.O.; Glasnov, T.N.; Romanin, C.; Groschner, K. PKC-dependent coupling of calcium permeation through transient receptor potential canonical 3 (TRPC3) to calcineurin signaling in HL-1 myocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10556–10561. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Nagaraj, N.; Zougman, A.; Gnad, F.; Mann, M. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 2010, 9, 3280–3289. [Google Scholar] [CrossRef]
- Trinidad, J.C.; Barkan, D.T.; Gulledge, B.F.; Thalhammer, A.; Sali, A.; Schoepfer, R.; Burlingame, A.L. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol. Cell Proteom. 2012, 11, 215–229. [Google Scholar] [CrossRef]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef]
- Chen, R.Q.; Yang, Q.K.; Lu, B.W.; Yi, W.; Cantin, G.; Chen, Y.L.; Fearns, C.; Yates, J.R., 3rd; Lee, J.D. CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res. 2009, 69, 2663–2668. [Google Scholar] [CrossRef]
- Odell, A.F.; Scott, J.L.; Van Helden, D.F. Epidermal growth factor induces tyrosine phosphorylation, membrane insertion, and activation of transient receptor potential channel 4. J. Biol. Chem. 2005, 280, 37974–37987. [Google Scholar] [CrossRef]
- Wie, J.; Jeong, S.; Kwak, M.; Myeong, J.; Chae, M.; Park, J.K.; Lee, S.W.; So, I. The regulation of transient receptor potential canonical 4 (TRPC4) channel by phosphodiesterase 5 inhibitor via the cyclic guanosine 3’5’-monophosphate. Pflug. Arch. Eur. J. Physiol. 2017, 469, 693–702. [Google Scholar] [CrossRef]
- Lee, J.E.; Song, M.Y.; Shin, S.K.; Bae, S.H.; Park, K.S. Mass spectrometric analysis of novel phosphorylation sites in the TRPC4beta channel. Rapid Commun. Mass Spectrom. 2012, 26, 1965–1970. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteom. 2014, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Wegierski, T.; Hill, K.; Schaefer, M.; Walz, G. The HECT ubiquitin ligase AIP4 regulates the cell surface expression of select TRP channels. EMBO J. 2006, 25, 5659–5669. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Li, J.; Zeng, B.; Chen, G.L.; Peng, X.; Zhang, Y.; Wang, J.; Clapham, D.E.; Li, Z.; Zhang, J. Structure of the mouse TRPC4 ion channel. Nat. Commun. 2018, 9, 3102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.H.; Chae, M.; Kim, H.J.; Lee, Y.M.; Kim, M.J.; Jin, N.G.; Yang, D.K.; So, I.; Kim, K.W. Desensitization of canonical transient receptor potential channel 5 by protein kinase C. Am. J. Physiol. Cell Physiol. 2005, 289, C591–C600. [Google Scholar] [CrossRef]
- Sung, T.S.; Jeon, J.P.; Kim, B.J.; Hong, C.; Kim, S.Y.; Kim, J.; Jeon, J.H.; Kim, H.J.; Suh, C.K.; Kim, S.J.; et al. Molecular determinants of PKA-dependent inhibition of TRPC5 channel. Am. J. Physiol. Cell Physiol. 2011, 301, C823–C832. [Google Scholar] [CrossRef]
- Xu, S.Z.; Sukumar, P.; Zeng, F.; Li, J.; Jairaman, A.; English, A.; Naylor, J.; Ciurtin, C.; Majeed, Y.; Milligan, C.J.; et al. TRPC channel activation by extracellular thioredoxin. Nature 2008, 451, 69–72. [Google Scholar] [CrossRef]
- Hong, C.; Kwak, M.; Myeong, J.; Ha, K.; Wie, J.; Jeon, J.H.; So, I. Extracellular disulfide bridges stabilize TRPC5 dimerization, trafficking, and activity. Pflug. Arch. Eur. J. Physiol. 2015, 467, 703–712. [Google Scholar] [CrossRef]
- Duan, J.; Li, J.; Chen, G.L.; Ge, Y.; Liu, J.; Xie, K.; Peng, X.; Zhou, W.; Zhong, J.; Zhang, Y.; et al. Cryo-EM structure of TRPC5 at 2.8-A resolution reveals unique and conserved structural elements essential for channel function. Sci. Adv. 2019, 5, eaaw7935. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Inoue, R.; Morii, T.; Takahashi, N.; Yamamoto, S.; Hara, Y.; Tominaga, M.; Shimizu, S.; Sato, Y.; Mori, Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2006, 2, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.O.; Sukumar, P.; Beech, D.J.; Yao, X. Nitric oxide lacks direct effect on TRPC5 channels but suppresses endogenous TRPC5-containing channels in endothelial cells. Pflug. Arch. Eur. J. Physiol. 2010, 460, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. J. Physiol. 2002, 539, 77–91. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Huang, Y.; Yao, X. Store-operated calcium entry in vascular endothelial cells is inhibited by cGMP via a protein kinase G-dependent mechanism. J. Biol. Chem. 2000, 275, 6758–6763. [Google Scholar] [CrossRef]
- Takahashi, S.; Lin, H.; Geshi, N.; Mori, Y.; Kawarabayashi, Y.; Takami, N.; Mori, M.X.; Honda, A.; Inoue, R. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J. Physiol. 2008, 586, 4209–4223. [Google Scholar] [CrossRef]
- Hong, C.; Seo, H.; Kwak, M.; Jeon, J.; Jang, J.; Jeong, E.M.; Myeong, J.; Hwang, Y.J.; Ha, K.; Kang, M.J.; et al. Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 2015, 138, 3030–3047. [Google Scholar] [CrossRef]
- Zhang, L.; Saffen, D. Muscarinic acetylcholine receptor regulation of TRP6 Ca2+ channel isoforms. Molecular structures and functional characterization. J. Biol. Chem. 2001, 276, 13331–13339. [Google Scholar] [CrossRef]
- Talbot, B.E.; Vandorpe, D.H.; Stotter, B.R.; Alper, S.L.; Schlondorff, J.S. Transmembrane insertases and N-glycosylation critically determine synthesis, trafficking, and activity of the nonselective cation channel TRPC6. J. Biol. Chem. 2019, 294, 12655–12669. [Google Scholar] [CrossRef]
- Yu, Y.; Sweeney, M.; Zhang, S.; Platoshyn, O.; Landsberg, J.; Rothman, A.; Yuan, J.X. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am. J. Physiol. Cell Physiol. 2003, 284, C316–C330. [Google Scholar] [CrossRef]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Poole, K.; Gimple, M.; Benz, R. Modification of the conductance, selectivity and concentration-dependent saturation of Pseudomonas aeruginosa protein P channels by chemical acetylation. Biochim. Biophys. Acta 1983, 735, 137–144. [Google Scholar] [CrossRef]
- Kim, S.J.; Widenmaier, S.B.; Choi, W.S.; Nian, C.; Ao, Z.; Warnock, G.; McIntosh, C.H. Pancreatic beta-cell prosurvival effects of the incretin hormones involve post-translational modification of Kv2.1 delayed rectifier channels. Cell Death Differ. 2012, 19, 333–344. [Google Scholar] [CrossRef]
- Butler, P.L.; Staruschenko, A.; Snyder, P.M. Acetylation stimulates the epithelial sodium channel by reducing its ubiquitination and degradation. J. Biol. Chem. 2015, 290, 12497–12503. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Diao, H.; Wang, C.; Lin, Y.; Yu, F.; Lu, H.; Xu, W.; Li, Z.; Shi, H.; Zhao, S.; et al. Acetylproteomic analysis reveals functional implications of lysine acetylation in human spermatozoa (sperm). Mol. Cell. Proteom. 2015, 14, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, C.; Kuroda, Y.; Nakamura, K.; Inoue, T.; Nakamura, T.; Michikawa, T.; Mizutani, A.; Mikoshiba, K. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J. Biol. Chem. 2004, 279, 18887–18894. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Geshi, N.; Takahashi, S.; Kiyonaka, S.; Ichikawa, J.; Hu, Y.; Mori, Y.; Ito, Y.; Inoue, R. Molecular determinants for cardiovascular TRPC6 channel regulation by Ca2+/calmodulin-dependent kinase II. J. Physiol. 2013, 591, 2851–2866. [Google Scholar] [CrossRef]
- Shen, B.; Kwan, H.Y.; Ma, X.; Wong, C.O.; Du, J.; Huang, Y.; Yao, X. cAMP activates TRPC6 channels via the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB)-mitogen-activated protein kinase kinase (MEK)-ERK1/2 signaling pathway. J. Biol. Chem. 2011, 286, 19439–19445. [Google Scholar] [CrossRef]
- Onohara, N.; Nishida, M.; Inoue, R.; Kobayashi, H.; Sumimoto, H.; Sato, Y.; Mori, Y.; Nagao, T.; Kurose, H. TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy. EMBO J. 2006, 25, 5305–5316. [Google Scholar] [CrossRef]
- Nishida, M.; Watanabe, K.; Sato, Y.; Nakaya, M.; Kitajima, N.; Ide, T.; Inoue, R.; Kurose, H. Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J. Biol. Chem. 2010, 285, 13244–13253. [Google Scholar] [CrossRef]
- Koitabashi, N.; Aiba, T.; Hesketh, G.G.; Rowell, J.; Zhang, M.; Takimoto, E.; Tomaselli, G.F.; Kass, D.A. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J. Mol. Cell. Cardiol. 2010, 48, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Kuwahara, K.; Nishida, M.; Jian, Z.; Rong, X.; Kiyonaka, S.; Kuwabara, Y.; Kurose, H.; Inoue, R.; Mori, Y.; et al. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ. Res. 2010, 106, 1849–1860. [Google Scholar] [CrossRef] [PubMed]
- Hammers, D.W.; Sleeper, M.M.; Forbes, S.C.; Shima, A.; Walter, G.A.; Sweeney, H.L. Tadalafil Treatment Delays the Onset of Cardiomyopathy in Dystrophin-Deficient Hearts. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Nishida, M.; Ariyoshi, M.; Jian, Z.; Saiki, S.; Hirano, M.; Nakaya, M.; Sato, Y.; Kita, S.; Iwamoto, T.; et al. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2278–2286. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, T.; Higa, T.; Aoyagi, H.; Nishiya, T.; Terada, K.; Miwa, S. Adenylate cyclase/cAMP/protein kinase A signaling pathway inhibits endothelin type A receptor-operated Ca2+ entry mediated via transient receptor potential canonical 6 channels. J. Pharmacol. Exp. Ther. 2012, 340, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Oberwinkler, H.; Werner, F.; Gassner, B.; Nakagawa, H.; Feil, R.; Hofmann, F.; Schlossmann, J.; Dietrich, A.; Gudermann, T.; et al. Atrial natriuretic peptide-mediated inhibition of microcirculatory endothelial Ca2+ and permeability response to histamine involves cGMP-dependent protein kinase I and TRPC6 channels. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2121–2129. [Google Scholar] [CrossRef]
- Bousquet, S.M.; Monet, M.; Boulay, G. Protein kinase C-dependent phosphorylation of transient receptor potential canonical 6 (TRPC6) on serine 448 causes channel inhibition. J. Biol. Chem. 2010, 285, 40534–40543. [Google Scholar] [CrossRef]
- Hall, G.; Rowell, J.; Farinelli, F.; Gbadegesin, R.A.; Lavin, P.; Wu, G.; Homstad, A.; Malone, A.; Lindsey, T.; Jiang, R.; et al. Phosphodiesterase 5 inhibition ameliorates angiontensin II-induced podocyte dysmotility via the protein kinase G-mediated downregulation of TRPC6 activity. Am. J. Physiol. 2014, 306, F1442–F1450. [Google Scholar] [CrossRef]
- Kanda, S.; Harita, Y.; Shibagaki, Y.; Sekine, T.; Igarashi, T.; Inoue, T.; Hattori, S. Tyrosine phosphorylation-dependent activation of TRPC6 regulated by PLC-gamma1 and nephrin: Effect of mutations associated with focal segmental glomerulosclerosis. Mol. Biol. Cell 2011, 22, 1824–1835. [Google Scholar] [CrossRef]
- Hagmann, H.; Mangold, N.; Rinschen, M.M.; Koenig, T.; Kunzelmann, K.; Schermer, B.; Benzing, T.; Brinkkoetter, P.T. Proline-dependent and basophilic kinases phosphorylate human TRPC6 at serine 14 to control channel activity through increased membrane expression. FASEB J. 2018, 32, 208–219. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, S.M.; Monet, M.; Boulay, G. The serine 814 of TRPC6 is phosphorylated under unstimulated conditions. PLoS ONE 2011, 6, e18121. [Google Scholar] [CrossRef]
- Kim, J.Y.; Saffen, D. Activation of M1 muscarinic acetylcholine receptors stimulates the formation of a multiprotein complex centered on TRPC6 channels. J. Biol. Chem. 2005, 280, 32035–32047. [Google Scholar] [CrossRef] [PubMed]
- Moritz, A.; Li, Y.; Guo, A.; Villen, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 2010, 3, ra64. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, K.; Matsuda, T.; Tsuji, A. Functional regulation of transient receptor potential canonical 7 by cGMP-dependent protein kinase Ialpha. Cell Signal. 2011, 23, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Sogaard, P.; Multhaupt, H.A.; Pataki, C.; Okina, E.; Xian, X.; Pedersen, M.E.; Stevens, T.; Griesbeck, O.; Park, P.W.; et al. Transmembrane proteoglycans control stretch-activated channels to set cytosolic calcium levels. J. Cell Biol. 2015, 210, 1199–1211. [Google Scholar] [CrossRef]
- Ruse, C.I.; McClatchy, D.B.; Lu, B.; Cociorva, D.; Motoyama, A.; Park, S.K.; Yates, J.R., 3rd. Motif-specific sampling of phosphoproteomes. J. Proteome Res. 2008, 7, 2140–2150. [Google Scholar] [CrossRef]
- Zielinska, D.F.; Gnad, F.; Wisniewski, J.R.; Mann, M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 2010, 141, 897–907. [Google Scholar] [CrossRef]
- Manes, N.P.; Dong, L.; Zhou, W.; Du, X.; Reghu, N.; Kool, A.C.; Choi, D.; Bailey, C.L.; Petricoin, E.F., 3rd; Liotta, L.A.; et al. Discovery of mouse spleen signaling responses to anthrax using label-free quantitative phosphoproteomics via mass spectrometry. Mol. Cell. Proteom. 2011, 10, M110 000927. [Google Scholar] [CrossRef]
- Shiromizu, T.; Adachi, J.; Watanabe, S.; Murakami, T.; Kuga, T.; Muraoka, S.; Tomonaga, T. Identification of missing proteins in the neXtProt database and unregistered phosphopeptides in the PhosphoSitePlus database as part of the Chromosome-centric Human Proteome Project. J. Proteome Res. 2013, 12, 2414–2421. [Google Scholar] [CrossRef]
- Schweppe, D.K.; Rigas, J.R.; Gerber, S.A. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J. Proteom. 2013, 91, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Sacco, F.; Humphrey, S.J.; Cox, J.; Mischnik, M.; Schulte, A.; Klabunde, T.; Schafer, M.; Mann, M. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat. Commun. 2016, 7, 13250. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.A.; Symons, J.M. Nephrotic syndrome in childhood. Lancet 2003, 362, 629–639. [Google Scholar] [CrossRef]
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef]
- Rood, I.M.; Deegens, J.K.; Wetzels, J.F. Genetic causes of focal segmental glomerulosclerosis: Implications for clinical practice. Nephrol Dial. Transpl. 2012, 27, 882–890. [Google Scholar] [CrossRef]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef]
- Reiser, J.; Polu, K.R.; Moller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef]
- Gaudet, R. A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 2008, 4, 372–379. [Google Scholar] [CrossRef]
- Schindl, R.; Romanin, C. Assembly domains in TRP channels. Biochem. Soc. Trans. 2007, 35, 84–85. [Google Scholar] [CrossRef]
- Lepage, P.K.; Lussier, M.P.; Barajas-Martinez, H.; Bousquet, S.M.; Blanchard, A.P.; Francoeur, N.; Dumaine, R.; Boulay, G. Identification of two domains involved in the assembly of transient receptor potential canonical channels. J. Biol. Chem. 2006, 281, 30356–30364. [Google Scholar] [CrossRef]
- Dionisio, N.; Albarran, L.; Berna-Erro, A.; Hernandez-Cruz, J.M.; Salido, G.M.; Rosado, J.A. Functional role of the calmodulin- and inositol 1,4,5-trisphosphate receptor-binding (CIRB) site of TRPC6 in human platelet activation. Cell. Signal. 2011, 23, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Friedlova, E.; Grycova, L.; Holakovska, B.; Silhan, J.; Janouskova, H.; Sulc, M.; Obsilova, V.; Obsil, T.; Teisinger, J. The interactions of the C-terminal region of the TRPC6 channel with calmodulin. Neurochem. Int. 2010, 56, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Hofmann, T.; Montell, C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol. Cell 2007, 25, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Hirschler-Laszkiewicz, I.; Tong, Q.; Waybill, K.; Conrad, K.; Keefer, K.; Zhang, W.; Chen, S.J.; Cheung, J.Y.; Miller, B.A. The transient receptor potential (TRP) channel TRPC3 TRP domain and AMP-activated protein kinase binding site are required for TRPC3 activation by erythropoietin. J. Biol. Chem. 2011, 286, 30636–30646. [Google Scholar] [CrossRef] [PubMed]
- Bandell, M.; Dubin, A.E.; Petrus, M.J.; Orth, A.; Mathur, J.; Hwang, S.W.; Patapoutian, A. High-throughput random mutagenesis screen reveals TRPM8 residues specifically required for activation by menthol. Nat. Neurosci. 2006, 9, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Riehle, M.; Buscher, A.K.; Gohlke, B.O.; Kassmann, M.; Kolatsi-Joannou, M.; Brasen, J.H.; Nagel, M.; Becker, J.U.; Winyard, P.; Hoyer, P.F.; et al. TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J. Am. Soc. Nephrol. 2016, 27, 2771–2783. [Google Scholar] [CrossRef]
- Heeringa, S.F.; Moller, C.C.; Du, J.; Yue, L.; Hinkes, B.; Chernin, G.; Vlangos, C.N.; Hoyer, P.F.; Reiser, J.; Hildebrandt, F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE 2009, 4, e7771. [Google Scholar] [CrossRef]
- Sun, Z.J.; Ng, K.H.; Liao, P.; Zhang, Y.; Ng, J.L.; Liu, I.D.; Tan, P.H.; Chong, S.S.; Chan, Y.H.; Liu, J.; et al. Genetic Interactions Between TRPC6 and NPHS1 Variants Affect Posttransplant Risk of Recurrent Focal Segmental Glomerulosclerosis. Am. J. Transpl. 2015, 15, 3229–3238. [Google Scholar] [CrossRef]
- Azumaya, C.M.; Sierra-Valdez, F.; Cordero-Morales, J.F.; Nakagawa, T. Cryo-EM structure of the cytoplasmic domain of murine transient receptor potential cation channel subfamily C member 6 (TRPC6). J. Biol. Chem. 2018, 293, 10381–10391. [Google Scholar] [CrossRef]
- Gigante, M.; Caridi, G.; Montemurno, E.; Soccio, M.; d’Apolito, M.; Cerullo, G.; Aucella, F.; Schirinzi, A.; Emma, F.; Massella, L.; et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin. J. Am. Soc. Nephrol. 2011, 6, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Kuang, X.Y.; Huang, W.Y.; Xu, H.; Shi, Y.; Zhang, X.L.; Niu, X.L.; Wu, Y.; Mei, C.Z.; Zha, X.L.; Zhao, Z.H.; et al. 254C>G: A TRPC6 promoter variation associated with enhanced transcription and steroid-resistant nephrotic syndrome in Chinese children. Pediatric Res. 2013, 74, 511–516. [Google Scholar] [CrossRef][Green Version]
- Krall, P.; Canales, C.P.; Kairath, P.; Carmona-Mora, P.; Molina, J.; Carpio, J.D.; Ruiz, P.; Mezzano, S.A.; Li, J.; Wei, C.; et al. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS ONE 2010, 5, e12859. [Google Scholar] [CrossRef]
- Schlondorff, J.; Del Camino, D.; Carrasquillo, R.; Lacey, V.; Pollak, M.R. TRPC6 mutations associated with focal segmental glomerulosclerosis cause constitutive activation of NFAT-dependent transcription. Am. J. Physiol. Cell Physiol. 2009, 296, C558–C569. [Google Scholar] [CrossRef]
- Chiluiza, D.; Krishna, S.; Schumacher, V.A.; Schlondorff, J. Gain-of-function mutations in transient receptor potential C6 (TRPC6) activate extracellular signal-regulated kinases 1/2 (ERK1/2). J. Biol. Chem. 2013, 288, 18407–18420. [Google Scholar] [CrossRef]
- Zhang, H.; Ding, J.; Fan, Q.; Liu, S. TRPC6 up-regulation in Ang II-induced podocyte apoptosis might result from ERK activation and NF-kappaB translocation. Exp. Biol. Med. (MaywoodN. J.) 2009, 234, 1029–1036. [Google Scholar] [CrossRef]
- Verheijden, K.A.T.; Sonneveld, R.; Bakker-van Bebber, M.; Wetzels, J.F.M.; van der Vlag, J.; Nijenhuis, T. The Calcium-Dependent Protease Calpain-1 Links TRPC6 Activity to Podocyte Injury. J. Am. Soc. Nephrol. 2018, 29, 2099–2109. [Google Scholar] [CrossRef]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019. [Google Scholar] [CrossRef]
- Polat, O.K.; Uno, M.; Maruyama, T.; Tran, H.N.; Imamura, K.; Wong, C.F.; Sakaguchi, R.; Ariyoshi, M.; Itsuki, K.; Ichikawa, J.; et al. Contribution of Coiled-Coil Assembly to Ca2+/Calmodulin-Dependent Inactivation of TRPC6 Channel and its Impacts on FSGS-Associated Phenotypes. J. Am. Soc. Nephrol. 2019, 30, 1587–1603. [Google Scholar] [CrossRef]
- Everett, K.V.; Chioza, B.A.; Georgoula, C.; Reece, A.; Gardiner, R.M.; Chung, E.M. Infantile hypertrophic pyloric stenosis: Evaluation of three positional candidate genes, TRPC1, TRPC5 and TRPC6, by association analysis and re-sequencing. Hum. Genet. 2009, 126, 819–831. [Google Scholar] [CrossRef]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, G.A.; Coletto, L.A.; Bozzolo, E.P.; Citterio, L.; Delli Carpini, S.; Zagato, L.; Rovere-Querini, P.; Lanzani, C.; Manunta, P.; Manfredi, A.A.; et al. The TRPC6 intronic polymorphism, associated with the risk of neurological disorders in systemic lupus erythematous, influences immune cell function. J. Neuroimmunol. 2018, 325, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Marshall-Gradisnik, S.; Johnston, S.; Chacko, A.; Nguyen, T.; Smith, P.; Staines, D. Single nucleotide polymorphisms and genotypes of transient receptor potential ion channel and acetylcholine receptor genes from isolated B lymphocytes in myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Int. Med. Res. 2016, 44, 1381–1394. [Google Scholar] [CrossRef]
- Vercoulen, J.H.; Swanink, C.M.; Fennis, J.F.; Galama, J.M.; van der Meer, J.W.; Bleijenberg, G. Dimensional assessment of chronic fatigue syndrome. J. Psychosom. Res. 1994, 38, 383–392. [Google Scholar] [CrossRef]
- Becker, E.B.; Oliver, P.L.; Glitsch, M.D.; Banks, G.T.; Achilli, F.; Hardy, A.; Nolan, P.M.; Fisher, E.M.; Davies, K.E. A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in moonwalker mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6706–6711. [Google Scholar] [CrossRef]
- Jung, C.; Gene, G.G.; Tomas, M.; Plata, C.; Selent, J.; Pastor, M.; Fandos, C.; Senti, M.; Lucas, G.; Elosua, R.; et al. A gain-of-function SNP in TRPC4 cation channel protects against myocardial infarction. Cardiovasc. Res. 2011, 91, 465–471. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; He, J.; Zeng, X.; Chen, X.; Xiong, M.; Zhou, Q.; Guo, M.; Li, D.; Lu, W. Identification of TRPCs genetic variants that modify risk for lung cancer based on the pathway and two-stage study. Meta Gene 2016, 9, 191–196. [Google Scholar] [CrossRef][Green Version]
- Buscher, A.K.; Kranz, B.; Buscher, R.; Hildebrandt, F.; Dworniczak, B.; Pennekamp, P.; Kuwertz-Broking, E.; Wingen, A.M.; John, U.; Kemper, M.; et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2010, 5, 2075–2084. [Google Scholar] [CrossRef]
- Santin, S.; Ars, E.; Rossetti, S.; Salido, E.; Silva, I.; Garcia-Maset, R.; Gimenez, I.; Ruiz, P.; Mendizabal, S.; Luciano Nieto, J.; et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transpl. 2009, 24, 3089–3096. [Google Scholar] [CrossRef]
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013, 83, 316–322. [Google Scholar] [CrossRef]
- Gheissari, A.; Meamar, R.; Kheirollahi, M.; Rouigari, M.; Dehbashi, M.; Dehghani, L.; Abedini, A. TRPC6 Mutational Analysis in Iranian Children With Focal Segmental Glomerulosclerosis. Iran. J. Kidney Dis. 2018, 12, 341–349. [Google Scholar]
- Hofstra, J.M.; Lainez, S.; van Kuijk, W.H.; Schoots, J.; Baltissen, M.P.; Hoefsloot, L.H.; Knoers, N.V.; Berden, J.H.; Bindels, R.J.; van der Vlag, J.; et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol. Dial. Transpl. 2013, 28, 1830–1838. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, Y.; Mao, J.; Yu, Z.; Yi, Z.; Yu, L.; Sun, J.; Wei, X.; Ding, F.; Zhang, H.; et al. Spectrum of mutations in Chinese children with steroid-resistant nephrotic syndrome. Pediatr. Nephrol. 2017, 32, 1181–1192. [Google Scholar] [CrossRef]
- Yan, K.; Wang, K.; Li, P. The role of post-translational modifications in cardiac hypertrophy. J. Cell. Mol. Med. 2019, 23, 3795–3807. [Google Scholar] [CrossRef]
- Buscher, A.K.; Konrad, M.; Nagel, M.; Witzke, O.; Kribben, A.; Hoyer, P.F.; Weber, S. Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin. Nephrol. 2012, 78, 47–53. [Google Scholar] [CrossRef]
- Mir, S.; Yavascan, O.; Berdeli, A.; Sozeri, B. TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol. Dial. Transpl. 2012, 27, 205–209. [Google Scholar] [CrossRef]
- Bullich, G.; Trujillano, D.; Santin, S.; Ossowski, S.; Mendizabal, S.; Fraga, G.; Madrid, A.; Ariceta, G.; Ballarin, J.; Torra, R.; et al. Targeted next-generation sequencing in steroid-resistant nephrotic syndrome: Mutations in multiple glomerular genes may influence disease severity. Eur. J. Hum. Genet. 2015, 23, 1192–1199. [Google Scholar] [CrossRef]
- Zhu, B.; Chen, N.; Wang, Z.H.; Pan, X.X.; Ren, H.; Zhang, W.; Wang, W.M. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat. Res. 2009, 664, 84–90. [Google Scholar] [CrossRef]
- Vinayagam, D.; Mager, T.; Apelbaum, A.; Bothe, A.; Merino, F.; Hofnagel, O.; Gatsogiannis, C.; Raunser, S. Electron cryo-microscopy structure of the canonical TRPC4 ion channel. Elife 2018, 7. [Google Scholar] [CrossRef]
- Tang, Q.; Guo, W.; Zheng, L.; Wu, J.X.; Liu, M.; Zhou, X.; Zhang, X.; Chen, L. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 2018, 28, 746–755. [Google Scholar] [CrossRef]
- Sierra-Valdez, F.; Azumaya, C.M.; Romero, L.O.; Nakagawa, T.; Cordero-Morales, J.F. Structure-function analyses of the ion channel TRPC3 reveal that its cytoplasmic domain allosterically modulates channel gating. J. Biol. Chem. 2018, 293, 16102–16114. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Channel | Modification | Species and Accession Number | Site | Sequence | Enzyme | Cell | Effect | Reference |
|---|---|---|---|---|---|---|---|---|
| TRPC1 | Phosphorylation | Human n.a. | S/T/Y | n.a. | PKCα | HUVEC and HMEC | Increased TRPC1 activity, enhanced permeability of endothelial monolayers | [47] |
| TRPC1 | Phosphorylation | Rabbit n.a. | S/T | n.a. | PKC | Rabbit portal vein myocytes | Increased TRPC1 activity, promoted PIP2-mediated SOCE | [51] |
| TRPC1 | Phosphorylation | Human P48995.2 | S172 T313 | SAKNKKDSLRHSRFR SGYRRKPTCKKIMTV | PKG | Porcine coronary arterial smooth muscle cell | Decreased TRPC1 activity, inhibited NO-, PKG-induced smooth muscle hyperpolarization and relaxation | [55] |
| TRPC1 | Phosphorylation | Human | S/T/Y | n.a. | PKCα | HUVEC | Increased TRPC1 activity, promoted monocyte binding to endothelial cells and increased endothelial hyperpermeability | [48] |
| TRPC1 | Phosphorylation | Rabbit n.a. | S/T | n.a. | PKC | Rabbit vascular smooth muscle cell | Increased TRPC1 activity | [52] |
| TRPC1 | Phosphorylation | Human P48995.2 | S172 T313 | SAKNKKDSLRHSRFR SGYRRKPTCKKIMTV | PKG | Primary mesenteric artery endothelial cells | Decreased TRPC1 activity | [56] |
| TRPC1 | Phosphorylation | Rat | S/T | n.a. | PKCα | INS-1E | Increased TRPC1 activity, promoted insulin secretion | [54] |
| TRPC3 | N-glycosylation | Human Q13507.3 | N416 (N418 *) | EGITTLPNITVTDYP | n.a. | COS-M6 and HEK-293 | n.a. | [58,59] |
| TRPC3 | Phosphorylation | Human Q13507.3 | S712 | PPFSLVPSPKSFVYF | PKC | HEK-293 | Decreased TRPC3 activity | [63] |
| TRPC3 | Phosphorylation | Human Q13507.3 | T11 S263 | SPSLRRMTVMREKGR KNDYRKLSMQCKDFV | PKG | HEK-293 | Decreased TRPC3 activity, abolished the SOCE mediated by TRPC3 | [62,64] |
| TRPC3 | Phosphorylation | Human Q13507.3 | Y49 Y148 Y150 Y226 | RFLDAAEYGNIPVVR ELQDDDFYAYDEDGT QDDDFYAYDEDGTRF KGLASPAYLSLSSED | Src | HEK | Phosphorylation at Y226 but not others increased TRPC3 activity | [61] |
| TRPC3 | Phosphorylation | Human Q13507.3 | T573 | LQISLGRTVKDIFKF | PKC | HL-1 | Coupled TRPC3 signaling to the activation of the NFAT pathway | [65] |
| TRPC4 | Phosphorylation | Human Q9UBN4.1 | Y959 Y972 | EEDSSIDYDLNLPDT DTVTHEDYVTTRL | Src | COS-7 | Induced insertion of TRPC4 into plasma membrane and its association with NHERF | [70] |
| TRPC4 | ubiquitination | Mouse Q9QUQ5.2 | N terminus | n.a. | AIP4 | HEK-293T | Promoted endocytosis of TRPC4 and reduced its basal activity | [75] |
| TRPC4 | Phosphorylation | Mouse Q9QUQ5.2 | S688 | KMRRKPESFGTIGRR | PKG | HEK-293 | Increased TRPC4 activity | [71] |
| TRPC4 | Disulfide bond | Mouse Q9QUQ5.2 | C549 C554 | TKGLSCKGIRCEKQNN | n.a. | n.a. | Constrained TRPC4 activity | [76] |
| TRPC5 | Phosphorylation | Mouse Q9QX29.1 | T972 | DGQEEQVTTRL | PKC | HEK-293 | Accelerated desensitization of TRPC5, necessary for the association with NHERF | [36,77] |
| TRPC5 | S-nitrosylation | Mouse Q9QX29.1 | C553 C558 | DEPNNCKGIRCEKQNN | n.a. | HEK, vascular endothelial cells | Increased TRPC5 activity | [82] |
| TRPC5 | Disulfide bond | Human Q9UL62.1 | C553 C558 | DEPNNCKGIRCEKQNN | n.a. | HEK-293 | Constrained TRPC5 activity | [79] |
| TRPC5 | Phosphorylation | Human Q9UL62.1 | S794 S796 | SGGARAKSKSVSFNL | PKA | HEK-293 | Decreased TRPC5 activity | [78] |
| TRPC5 | S-glutathionylation | Human Q9UL62.1 | C176 C178 | RPHQIRCNCVECVSS | n.a. | HEK-293, Clonal striatal cells | Increased TRPC5 activity, activated the calmodulin- dependent protein kinase and calpain-caspase pathway | [87] |
| TRPC5 | Disulfide bond | Human Q9UL62.1 | C553 C558 | DEPNNCKGIRCEKQNN | n.a. | HEK-293 | Stabilized TRPC5 multimerization, promoted its plasma membrane insertion | [80] |
| TRPC5 | Disulfide bond | Mouse Q9QX29.1 | C553 C558 | DEPNNCKGIRCEKQNN | n.a. | n.a. | Essential for TRPC5 activation | [81] |
| TRPC6 | N-glycosylation | Mouse Q61143.1 | N472 N560 | EGTKLLPNETSTDNA AQSIIDANDTLKDLT | n.a. | COS | n.a. | [6] |
| TRPC6 | N-glycosylation | Rat Q99N78.1 | N711 | YVLYGVYNVTMVIVL | n.a. | COS | n.a. | [88] |
| TRPC6 | N-glycosylation | Human Q9Y210.1 | N473 N561 | EGTKLLPNETSTDNA AQSIIDANDTLKDLT | n.a. | HEK-293 | Decreased TRPC6 constitutive activity | [59] |
| TRPC6 | Phosphorylation | n.a. | Y | n.a. | src | COS-7 | Increased TRPC6 activity | [96] |
| TRPC6 | Phosphorylation | Rat Q99N78.1 | S768 | VPFNLVPSPKSLLYL | PKC | PC12D | Decreased TRPC6 activity, required for protein complex formation upon activation of the M1 muscarinic acetylcholine receptor | [113] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | T69 | RLTHRRQTILREKGR | PKG | HEK-293 and A7r5 | Decreased TRPC6 activity | [86] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | S448 | KFVAHAASFTIFLGL | PKC | HEK-293T and A7r5 | Decreased TRPC6 activity, reduced vasopressin-induced Ca2+ entry | [107] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | T69 | RLTHRRQTILREKGR | PKG | NRVM | Decreased TRPC6 activity, suppressed NFAT activation, prevented cardiac hypertrophy | [100,102] |
| TRPC6 | Phosphorylation | Human Q9Y210.1 | T70 S322 | RLAHRRQTVLREKGR KNDYKKLSMQCKDFV | PKG | NRVM and AMVM | Decrease TRPC6 activity, suppressed NFAT activation, prevented cardiac hypertrophy | [101] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | T69 | RLTHRRQTILREKGR | PKA | RAoSMC | Decreased TRPC6 activity, attenuated angiotensin II-induced vasoconstriction | [104] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | S814 | KKFGISGSHEDLSKF | n.a., but not casein kinase II | HEK-293 | No effect on TRPC6 activity | [112] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | S281 | NAYKGLASPAYLSLS | ERK1/2 | HEK-293 and Rat Glomerular mesangial cell | Increased TRPC6 activity, necessary for cAMP-induced Ca2+ influx | [98] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | Y284 | KGLASPAYLSLSSED | Src | HEK-293T, podocyte | Enhanced plasma membrane trafficking of TRPC6 | [109] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | S28 T69 | AGTRRNESQDYLLMD RLTHRRQTILREKGR | PKA | HEK-293 | S28 but not T69 decreased TRPC6 activity | [105] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | T69 | RLTHRRQTILREKGR | cGKI | MLEC and HDMEC | Decreased TRPC6 activity, prevented endothelial cell hyperpermeability responses to hyperforin | [106] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | T487 | RQLFRMKTSCFSWME | CaMKII | HEK293 and A7r5 | Increased TRPC6 activity, enhanced arginine vasopressin-induced cation current in smooth muscle cells | [97] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | T69 S321 | RLTHRRQTILREKGR KNDYKKLSMQCKDFV | PKG | Murine podocyte | Decreased TRPC6 activity, reduced podocyte motility | [108] |
| TRPC6 | Phosphorylation | Dog | T | n.a. | n.a. | Canine heart left ventricle | Decreased TRPC6 activity, underpinned the tadalafil-caused delay of dystrophic cardiomyopathy onset | [103] |
| TRPC6 | Phosphorylation | Human Q9Y210.1 | S4 S13, S14 S814 | MSQSPAFGPRR AFGPRRGSSPRGAAG KKLGILGSHEDLSKL | MAPK Cdk-5 | HEK-293T | S14 enhanced TRPC6 plasma membrane trafficking and constitutive activity | [110] |
| TRPC6 | N-glycosylation | Human Q9Y210.1 | N473 N561 | EGTKLLPNETSTDNA AQSIIDANDTLKDLT | n.a. | TRex293, podocyte | Required for channel activity, ERK activation and cytotoxicity of the GOF TRPC6 mutant (R895C) | [89] |
| TRPC7 | Phosphorylation | Mouse Q9WVC5.1 | T15 | KNMQRRHTTLREKGR | cGKI | COS-7 and HEK-293T | Decreased TRPC7 activity | [115] |
| TRPC7 | Phosphorylation | Mouse Q9WVC5.1 | S714 | APFNLVPSPKSFYYL | PKC | MEF | Decreased TRPC7 activity, regulated cytoskeleton organization and myofibroblast phenotype | [116] |
| Channel | Modification | Species and Accession Number | Site | Sequence | Cell/Tissue | Remark | Reference |
|---|---|---|---|---|---|---|---|
| TRPC1 | Phosphorylation | Human P48995.1 | Y368 | WPVLSLCYLIAPKS | Hep3B and MHCC97H | Found in nonmetastatic instead of metastatic human hepatocellular carcinoma cells | [57] |
| TRPC3 | N-glycosylation | Mouse Q9QZC1.1 | N404 | EGITTLPNITVIDYP | Mouse brain | Detected only in brain | [118] |
| TRPC3 | Phosphorylation | Human Q13507.3 | S843 | LSEKLNPSMLRCE | HeLa | Phosphorylation of TRPC3 decreased after mammalian target of rapamycin (mTOR) inhibition | [69] |
| TRPC3 | Phosphorylation | Mouse B1ATV3.1 | S807 | LELGMGNSKSRLNLF | Mouse brain | n.a. | [66] |
| TRPC3 | Phosphorylation | Mouse B1ATV3.1 | S56 | PCPRAPPSPGPDASS | Mouse brain synaptic membrane | n.a. | [67] |
| TRPC3 | Phosphorylation | Rat Q9JMI9.1 | S801 | EIKQDISSLRYELLE | Rat multiple organs | Detected in intestine | [68] |
| TRPC4 | Phosphorylation | Human Q9UBN4.1 | S193 | DVDSLRHSRSRLNIY | Hela | n.a. | [73] |
| TRPC4 | Phosphorylation | Mouse Q9QUQ5.2 | S688 T691, S875 T879 | KMRRKPESFGTIGRR RKPESFGTIGRRAAD SSIDYDLSPTDTAAH YDLSPTDTAAHEDYV | HEK-293 | n.a. | [72] |
| TRPC4 | Phosphorylation | Human Q9UBN4.1 | S955 | KHAKEEDSSIDYDLN | Human liver | n.a. | [74] |
| TRPC6 | Phosphorylation | Human Q9Y210.1 | T70 | RLAHRRQTVLREKGR | Hela | n.a. | [73] |
| TRPC6 | Phosphorylation | Human Q9Y210.1 | S13 S14 Y31 T70 | AFGPRRGSSPRGAAG RRNESQDYLLMDSEL RLAHRRQTVLREKGR | NCI-H3255 | Phosphorylation changed after Gefitinib treatment | [114] |
| TRPC6 | N-glycosylation | Mouse Q61143.1 | N472 | EGTKLLPNETSTDNA | Mouse brain | Class I N-glycosylated sites, detected only in the brain | [118] |
| TRPC6 | N-glycosylation | Mouse Q61143.1 | N560 | AQSIIDANDTLKDLT | Mouse brain, liver, plasma | Class II N-glycosylated sites, detected in multiple organs | [118] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | S812 S814 | KKFGISGSHEDLSKF | Mouse multiple organs | S812 was detected in the spleen. S814 was detected in the spleen and lung | [111] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | S814 | KKFGISGSHEDLSKF | Mouse spleen | n.a. | [119] |
| TRPC6 | Acetylation | Rat Q99N78.1 | K170 K370 | ALLLAISKGYVRIVE SRLKLAIKYEVKKFV | Rat multiple organs | K170 was detected in muscles. K370 was detected in the brain, heart, and stomach | [91] |
| TRPC6 | Phosphorylation | Rat Q99N78.1 | S4 S814 | MSQSPGFVTRR KKFGILGSHEDLSKF | Rat multiple organs | S4 was detected in the lung and spleen. S814 was detected in the spleen, thymus, lung, and blood | [68] |
| TRPC6 | Phosphorylation | Human Q9Y210.1 | S815 | KKLGILGSHEDLSKL | Colorectal cancer samples | n.a. | [120] |
| TRPC6 | Phosphorylation | Human Q9Y210.1 | S815 | KKLGILGSHEDLSKL | Non-small cell lung cancer tumors | n.a. | [121] |
| TRPC6 | Phosphorylation | Mouse Q61143.1 | S814 | KKFGISGSHEDLSKF | Min6 | n.a. | [122] |
| TRPC7 | Phosphorylation | Human Q9HCX4.1 | S775 T778 | LTANNTLSKPTRYQK | HEK | n.a. | [117] |
| TRPC7 | N-glycosylation | Mouse Q9WVC5.1 | N418 | EGVKTLPNETFTDYP | Mouse brain | Class II N-glycosylated sites, detected only in the brain | [118] |
| Channel | cDNA and Protein Accession Numbers | Position | Nucleotide Change | Amino Acid Change | Effect | Disease | Reference |
|---|---|---|---|---|---|---|---|
| TRPC3 | NM_019510 NP_062383 | Exon | 1903A>G | T635A | Increased TRPC3 activity | Hereditary cerebellar ataxias | [155] |
| TRPC4 | NM_016179 NP_057263 | Exon | 2869A>G (3104A>G *) | I957V | Increased TRPC4 activity | Reduced risk of MI | [156] |
| TRPC4 | NM_016179 NP_057263 | Intron | rs9547991: NG_029849.2:g.232148A>G | No change | n.a. | Lung cancer | [157] |
| TRPC4 | NM_016179 NP_057263 | Intron | rs978156: NG_029849.2:g.172052G>A | No change | n.a. | Lung cancer | [157] |
| TRPC6 | NM_004621 NP_004612 | Exon | 202C>T | R68W | Increased plasma membrane trafficking and activity of TRPC6 | Proteinuria and FSGS | [139] |
| TRPC6 | NM_004621 NP_004612 | Exon | 265delA | S89NfsX8 | n.a. | FSGS | [158] |
| TRPC6 | NM_004621 NP_004612 | Exon | 325G>A | G109S | Increased basal and maximum activity of TRPC6 | FSGS | [137,159] |
| TRPC6 | NM_004621 NP_004612 | Exon | 328T>G | N110H | Increased basal and maximum activity of TRPC6 | FSGS | [137,160] |
| TRPC6 | NM_004621 NP_004612 | Exon | 333C>T | I111I | n.a. | Proteinuria and FSGS | [161] |
| TRPC6 | NM_004621 NP_004612 | Exon | C335>A | P112Q | Increased plasma membrane trafficking, basal, and maximum activity of TRPC6 | FSGS | [127] |
| TRPC6 | NM_004621 NP_004612 | Exon | 362G>C | C121S | n.a. | Proteinuria and FSGS | [161] |
| TRPC6 | NM_004621 NP_004612 | Exon | 374A>G | N125S | Increased/decreased TRPC6 activity | FSGS | [137,141,159] |
| TRPC6 | NM_004621 NP_004612 | Exon | 389A>T | D130V | n.a. | Proteinuria and FSGS | [161] |
| TRPC6 | NM_004621 NP_004612 | Exon | 395T>C (495T>C *) | M132T | Delayed inactivation, increased basal and maximum activity of TRPC6 | FSGS | [137,138,158] |
| TRPC6 | NM_004621 NP_004612 | Exon | n.a. | N143S | Increased maximum activity of TRPC6 | FSGS | [128,137] |
| TRPC6 | NM_004621 NP_004612 | Exon | 484G>C | G162R | n.a. | Proteinuria and FSGS | [161] |
| TRPC6 | NM_004621 NP_004612 | Exon | 524G>A | R175Q | Increased TRPC6 activity | FSGS | [162] |
| TRPC6 | NM_004621 NP_004612 | Exon | 523C>T | R175W | Increased basal TRPC6 activity | SRNS | [137,163] |
| TRPC6 | NM_004621 NP_004612 | Exon | 653A>T | H218L | Increased maximum activity and expression of TRPC6 | FSGS | [137,141] |
| TRPC6 | NM_004621 NP_004612 | Exon | n.a. | S270T | Delayed TRPC6 inactivation | FSGS | [128,164] |
| TRPC6 | NM_004621 NP_004612 | Exon | 1079G>A | R360H | n.a. | FSGS | [165] |
| TRPC6 | NM_004621 NP_004612 | Exon | n.a. | L395A | Decreased TRPC6 activity | SRNS/FSGS | [137,166] |
| TRPC6 | NM_004621 NP_004612 | Exon | 1211C>T | A404V | Increased maximum activity of TRPC6 | IHPS | [137,150] |
| TRPC6 | NM_004621 NP_004612 | Exon | 2270G>A | G757D | Decreased TRPC6 activity | FSGS | [137,158] |
| TRPC6 | NM_004621 NP_004612 | Exon | 2339T>C | L780P | Decreased TRPC6 activity | FSGS | [137,159] |
| TRPC6 | NM_004621 NP_004612 | Exon | 2617_2620 delGATA | D873RfsX5 | n.a. | Heterogenous phenotype ranging from asymptomatic minimal change disease to end-stage kidney disease | [25] |
| TRPC6 | NM_004621 NP_004612 | Exon | n.a. | K874X | Delayed TRPC6 inactivation | FSGS | [128,138] |
| TRPC6 | NM_004621 NP_004612 | Exon | 2656G>A | E886K | n.a. | SRNS/FSGS | [167] |
| TRPC6 | NM_004621 NP_004612 | Exon | 2665C>A | Q889K | Increased basal and maximum activity of TRPC6 | FSGS | [137,168] |
| TRPC6 | NM_004621 NP_004612 | Exon | n.a. | R895C | Increased TRPC6 expression, increased/decreased TRPC6 activity | FSGS | [128,137] |
| TRPC6 | NM_004621 NP_004612 | Exon | 2684G>T | R895L | Increased TRPC6 activity | FSGS | [141] |
| TRPC6 | NM_004621 NP_004612 | Exon | n.a. | E897K | Increased basal and maximum activity of TRPC6 | FSGS | [128,137] |
| TRPC6 | NM_004621 NP_004612 | Promoter | rs3922961: NG_011476.2:g.4512A>C | No change | n.a. | IHPS | [150] |
| TRPC6 | NM_004621 NP_004612 | Promoter | rs3824934: NG_011476.2:g.5172C>G | No change | Increased TRPC6 expression | IPAH | [151] |
| TRPC6 | NM_004621 NP_004612 | Promoter | rs3824934: NG_011476.2:g.5172C>G | No change | Increased TRPC6 expression | SRNS | [142] |
| TRPC6 | NM_004621 NP_004612 | Intron | rs7118839: NG_011476.2:g.50182G>A | No change | n.a. | IHPS | [150] |
| TRPC6 | NM_004621 NP_004612 | Intron | rs7925662: NG_011476.2:g.51354A>G | No change | n.a. | NPSLE | [152] |
| TRPC6 | NM_004621 NP_004612 | Intron | rs11224816: NG_011476.1:g.63374A>G | No change | n.a. | ME/CFS | [153] |
| TRPC7 | NM_020389 NP_065122 | Intron | rs11748198: NC_000005.9:g.135575829C>A | No change | n.a. | Lung cancer | [157] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Yao, X.; Tsang, S.Y. Post-Translational Modification and Natural Mutation of TRPC Channels. Cells 2020, 9, 135. https://doi.org/10.3390/cells9010135
Liu X, Yao X, Tsang SY. Post-Translational Modification and Natural Mutation of TRPC Channels. Cells. 2020; 9(1):135. https://doi.org/10.3390/cells9010135
Chicago/Turabian StyleLiu, Xianji, Xiaoqiang Yao, and Suk Ying Tsang. 2020. "Post-Translational Modification and Natural Mutation of TRPC Channels" Cells 9, no. 1: 135. https://doi.org/10.3390/cells9010135
APA StyleLiu, X., Yao, X., & Tsang, S. Y. (2020). Post-Translational Modification and Natural Mutation of TRPC Channels. Cells, 9(1), 135. https://doi.org/10.3390/cells9010135