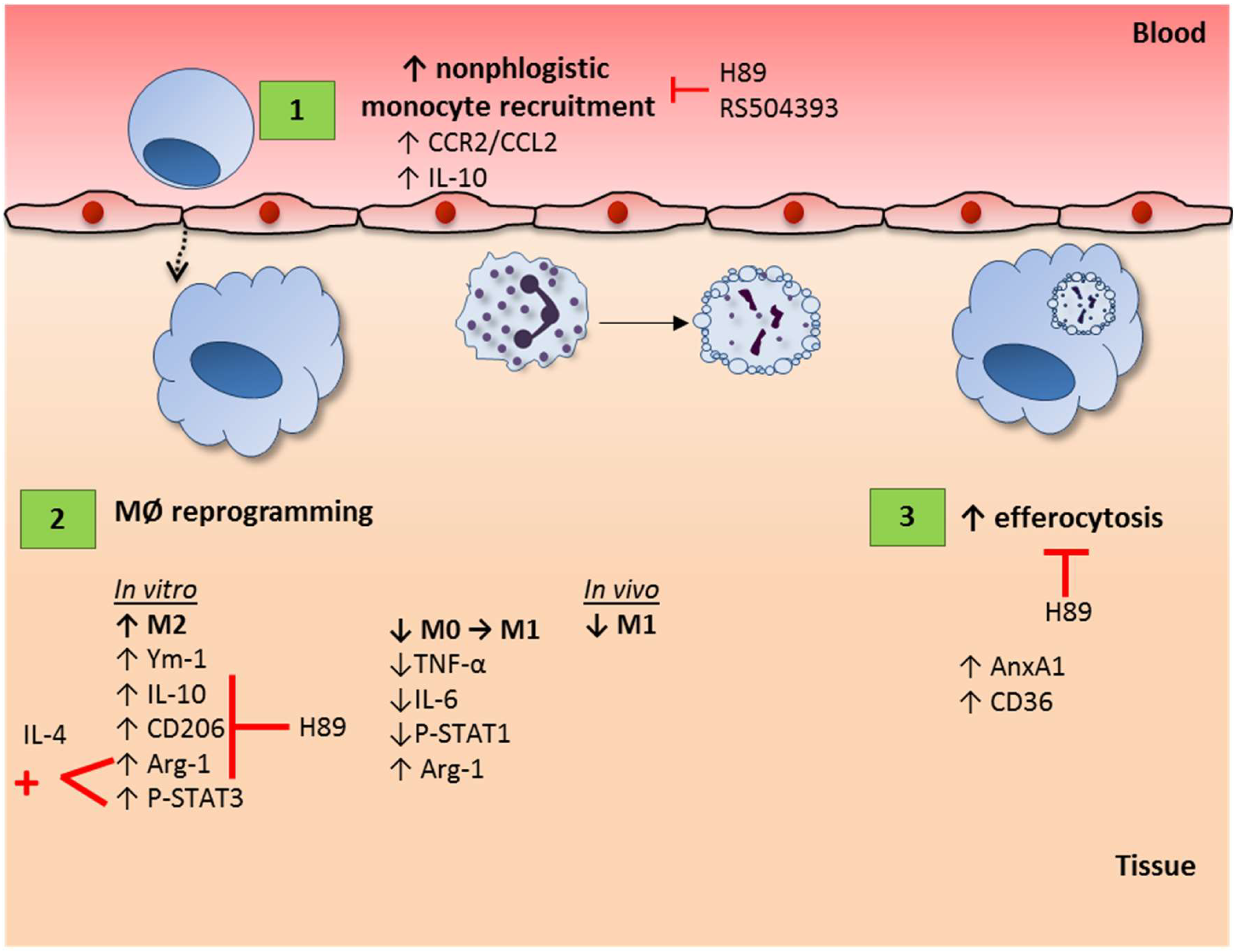
Cyclic AMP Regulates Key Features of Macrophages via PKA: Recruitment, Reprogramming and Efferocytosis

 , , , , , ,
, , , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drugs, Reagents, and Antibodies
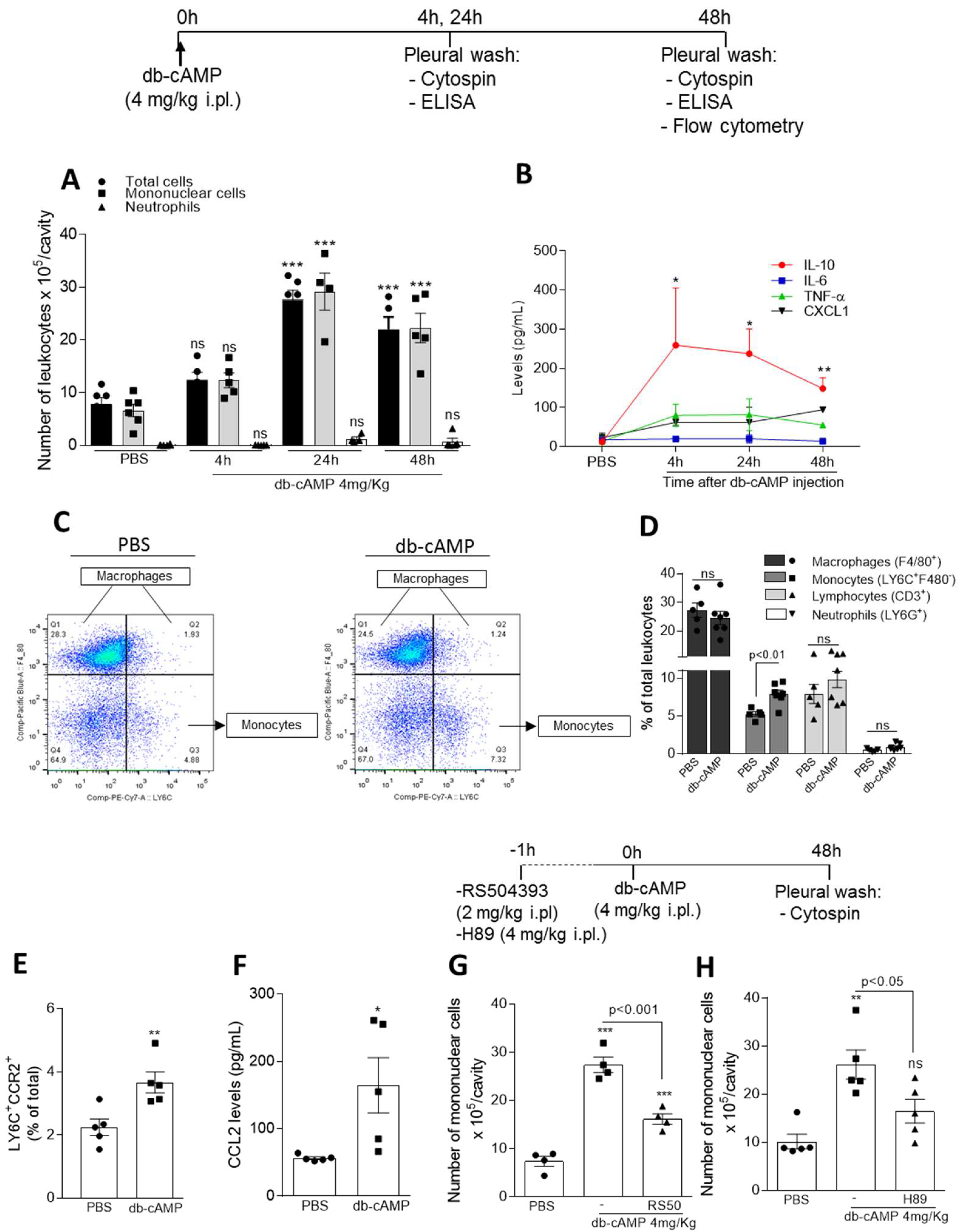
2.3. Leukocyte Migration to the Pleural Cavity Induced by db-cAMP
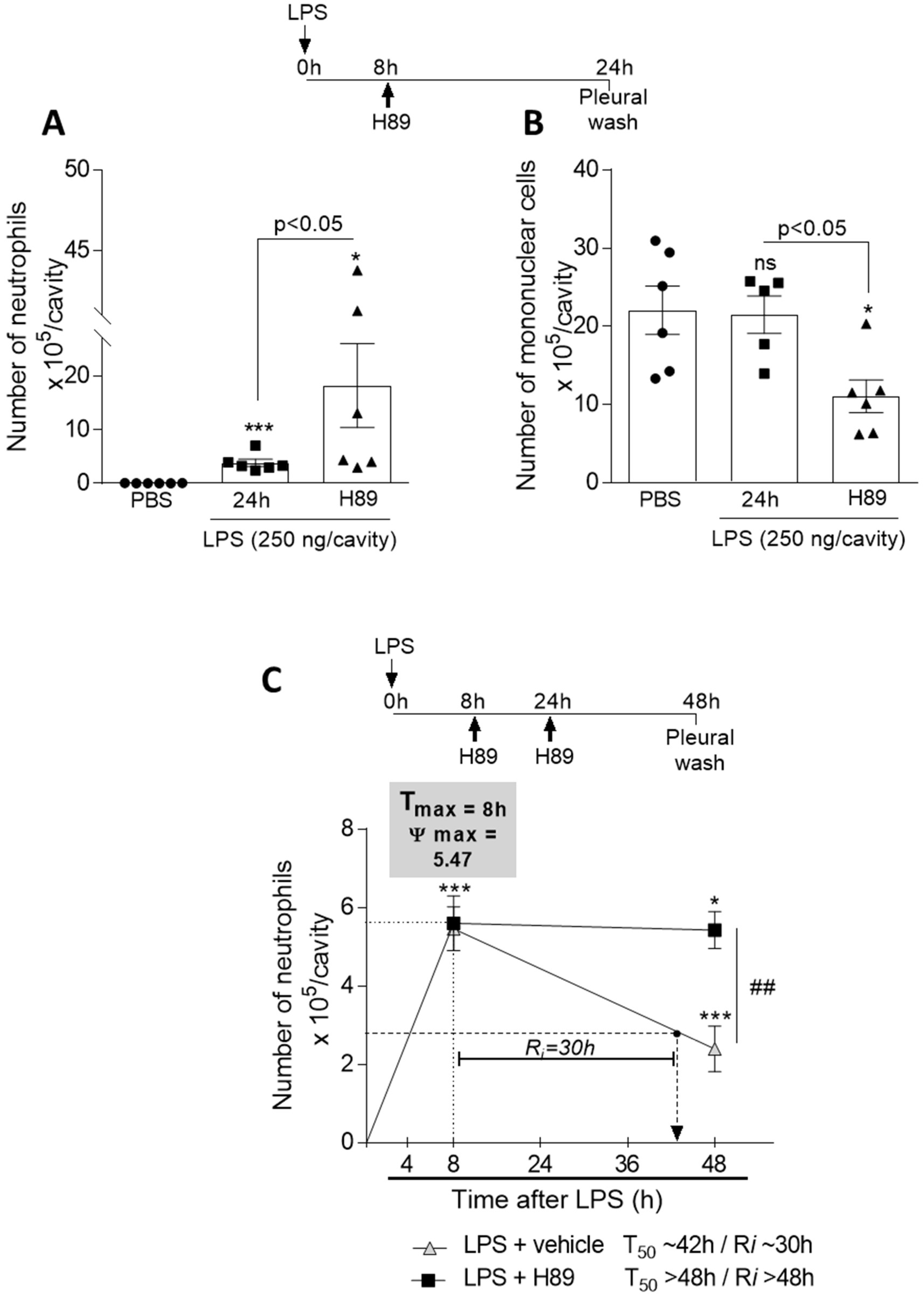
2.4. LPS-Induced Pleurisy Model and Treatment with db-cAMP or Inihibition of PKA Using H89
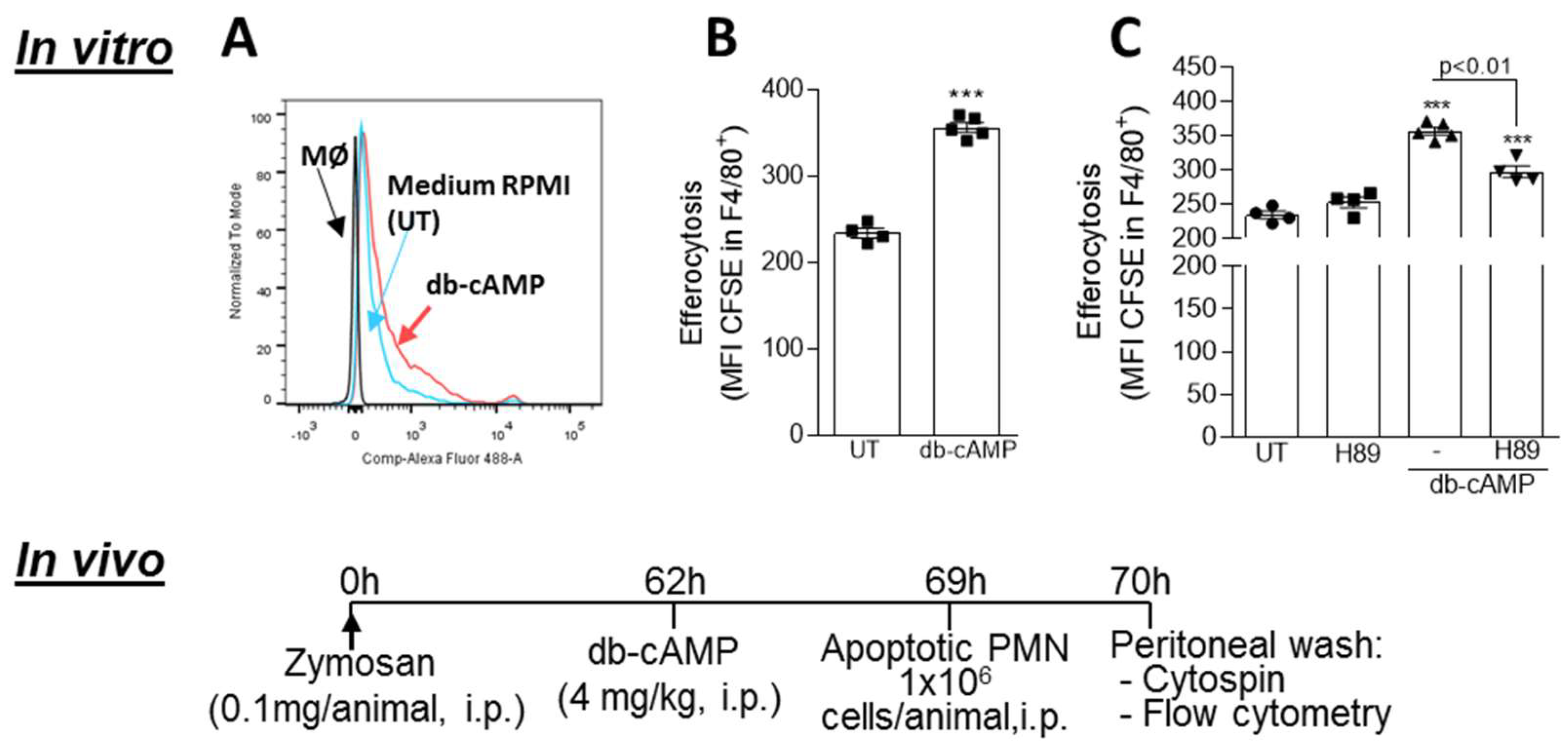
2.5. In Vivo Efferocytosis Assay
2.6. Isolation and Culture of Murine Bone Marrow-Derived Macrophages (BMDMs)
2.7. RAW264.7 Cell Culture
2.8. In Vitro Efferocytosis Assay
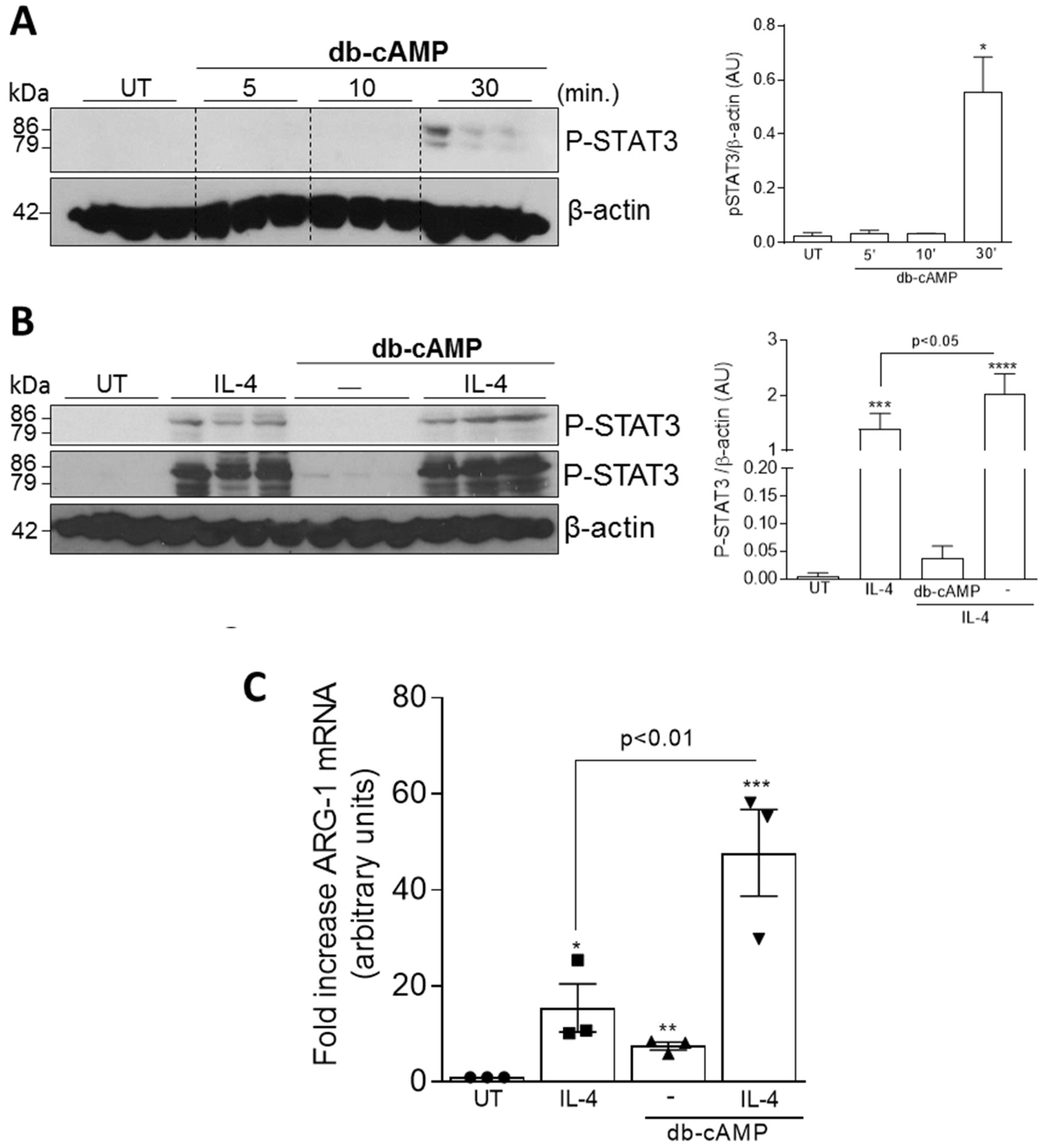
2.9. Western Blotting
2.10. qPCR Analysis of M1 and M2 Macrophage Markers
2.11. ELISA Assay
2.12. Statistical Analysis
3. Results
3.1. Db-cAMP Promotes Nonphlogistic Monocyte Recruitment to the Pleural Cavity of Mice Associated with Increased CCR2 Expression and CCL2 Levels and Depend on CCR2 and PKA
3.2. Db-cAMP Induces Macrophage Polarization towards M2 Phenotype in a PKA Dependent Manner
3.3. Db-cAMP has Synergic Effect with IL-4 to Induce Phosphorylation of STAT3 and Arg-1 Expression
3.4. Db-cAMP Re-educates M1 Macrophages toward an M2-like Phenotype Associated with Inhibition of STAT1 Phosphorylation
3.5. Treatment of LPS-Inflamed Mice with db-cAMP Decreased the Numbers of M1 Macrophages without Modifying M2 and Mres, and Increases the Engulfment-Related Molecules AnxA1 and CD36
3.6. Db-cAMP Enhances Efferocytosis of Apoptotic Neutrophils in a PKA Dependent Manner
3.7. Inhibition of the cAMP Pathway with a PKA Antagonist H89 Prevents Natural Resolution of Pleurisy and Decreases the Numbers of Mononuclear Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.A.; Vago, J.P.; Perretti, M.; Teixeira, M.M. Mediators of the Resolution of the Inflammatory Response. Trends Immunol. 2019, 40, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Feehan, K.T.; Gilroy, D.W. Is Resolution the End of Inflammation? Trends Mol. Med. 2019, 25, 198–214. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Hallett, J.M.; Leitch, A.E.; Riley, N.A.; Duffin, R.; Haslett, C.; Rossi, A.G. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol. Sci. 2008, 29, 250–257. [Google Scholar] [CrossRef]
- Poon, I.K.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef]
- Labonte, A.C.; Tosello-Trampont, A.C.; Hahn, Y.S. The role of macrophage polarization in infectious and inflammatory diseases. Mol. Cells 2014, 37, 275–285. [Google Scholar] [CrossRef]
- Ariel, A.; Serhan, C.N. New Lives Given by Cell Death: Macrophage Differentiation Following Their Encounter with Apoptotic Leukocytes during the Resolution of Inflammation. Front. Immunol. 2012, 3, 4. [Google Scholar] [CrossRef]
- Lichtnekert, J.; Kawakami, T.; Parks, W.C.; Duffield, J.S. Changes in macrophage phenotype as the immune response evolves. Curr. Opin. Pharmacol. 2013, 13, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Schif-Zuck, S.; Gross, N.; Assi, S.; Rostoker, R.; Serhan, C.N.; Ariel, A. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: Modulation by resolvins and glucocorticoids. Eur. J. Immunol. 2011, 41, 366–379. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Harris, R.A.; Ley, K. Macrophage Polarization: Decisions That Affect Health. J. Clin. Cell Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Lee, C.H.; Kim, Y.J.; Jang, J.H.; Park, J.W. Modulating macrophage polarization with divalent cations in nanostructured titanium implant surfaces. Nanotechnology 2016, 27, 085101. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Tran, T.H.; Amiji, M. Macrophage repolarization with targeted alginate nanoparticles containing IL-10 plasmid DNA for the treatment of experimental arthritis. Biomaterials 2015, 61, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O. Regulators of macrophage activation. Eur. J. Immunol. 2011, 41, 1531–1534. [Google Scholar] [CrossRef]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef]
- Korns, D.; Frasch, S.C.; Fernandez-Boyanapalli, R.; Henson, P.M.; Bratton, D.L. Modulation of macrophage efferocytosis in inflammation. Front. Immunol. 2011, 2, 57. [Google Scholar] [CrossRef]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef]
- Freire-de-Lima, C.G.; Xiao, Y.Q.; Gardai, S.J.; Bratton, D.L.; Schiemann, W.P.; Henson, P.M. Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J. Biol. Chem. 2006, 281, 38376–38384. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, Y.X.; Qiao, S.L.; An, H.W.; Ma, Y.; Qiao, Z.Y.; Rajapaksha, R.P.; Wang, H. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials 2017, 112, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Vidyarthi, A.; Khan, N.; Agnihotri, T.; Negi, S.; Das, D.K.; Aqdas, M.; Chatterjee, D.; Colegio, O.R.; Tewari, M.K.; Agrewala, J.N. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-αβ Signaling and Restricts Tumor Progression. Front. Immunol. 2018, 9, 1650. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Sunil, V.R.; Gardner, C.R.; Laskin, J.D. Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharmacol. Toxicol. 2011, 51, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Leng, X.; Zhang, Q. The Current State of Nanoparticle-Induced Macrophage Polarization and Reprogramming Research. Int. J. Mol. Sci. 2017, 18, 336. [Google Scholar] [CrossRef]
- Gerlo, S.; Kooijman, R.; Beck, I.M.; Kolmus, K.; Spooren, A.; Haegeman, G. Cyclic AMP: A selective modulator of NF-κB action. Cell. Mol. Life Sci. 2011, 68, 3823–3841. [Google Scholar] [CrossRef]
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef]
- Stables, M.J.; Newson, J.; Ayoub, S.S.; Brown, J.; Hyams, C.J.; Gilroy, D.W. Priming innate immune responses to infection by cyclooxygenase inhibition kills antibiotic-susceptible and -resistant bacteria. Blood 2010, 116, 2950–2959. [Google Scholar] [CrossRef]
- Bystrom, J.; Evans, I.; Newson, J.; Stables, M.; Toor, I.; van Rooijen, N.; Crawford, M.; Colville-Nash, P.; Farrow, S.; Gilroy, D.W. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood 2008, 112, 4117–4127. [Google Scholar] [CrossRef]
- Veremeyko, T.; Yung, A.W.Y.; Dukhinova, M.; Kuznetsova, I.S.; Pomytkin, I.; Lyundup, A.; Strekalova, T.; Barteneva, N.S.; Ponomarev, E.D. Cyclic AMP Pathway Suppress Autoimmune Neuroinflammation by Inhibiting Functions of Encephalitogenic CD4 T Cells and Enhancing M2 Macrophage Polarization at the Site of Inflammation. Front. Immunol. 2018, 9, 50. [Google Scholar] [CrossRef]
- Sheldon, K.E.; Shandilya, H.; Kepka-Lenhart, D.; Poljakovic, M.; Ghosh, A.; Morris, S.M. Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. J. Immunol. 2013, 191, 2290–2298. [Google Scholar] [CrossRef] [PubMed]
- Lima, K.M.; Vago, J.P.; Caux, T.R.; Negreiros-Lima, G.L.; Sugimoto, M.A.; Tavares, L.P.; Arribada, R.G.; Carmo, A.A.F.; Galvão, I.; Costa, B.R.C.; et al. The resolution of acute inflammation induced by cyclic AMP is dependent on annexin A1. J. Biol. Chem. 2017, 292, 13758–13773. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.P.; Carmo, A.F.; Rezende, B.M.; Lopes, F.; Silva, D.M.; Alessandri, A.L.; Bonjardim, C.A.; Rossi, A.G.; Teixeira, M.M.; Pinho, V. Cyclic AMP enhances resolution of allergic pleurisy by promoting inflammatory cell apoptosis via inhibition of PI3K/Akt and NF-kappaB. Biochem. Pharmacol. 2009, 78, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.P.; Lopes, F.; Silva, D.M.; Tavares, L.P.; Vieira, A.T.; Rezende, B.M.; Carmo, A.F.; Russo, R.C.; Garcia, C.C.; Bonjardim, C.A.; et al. PDE4 inhibition drives resolution of neutrophilic inflammation by inducing apoptosis in a PKA-PI3K/Akt-dependent and NF-kappaB-independent manner. J. Leukoc. Biol. 2010, 87, 895–904. [Google Scholar] [CrossRef]
- Chiang, N.; Dalli, J.; Colas, R.A.; Serhan, C.N. Identification of resolvin D2 receptor mediating resolution of infections and organ protection. J. Exp. Med. 2015, 212, 1203–1217. [Google Scholar] [CrossRef]
- Lee, H.N.; Surh, Y.J. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem. Pharmacol. 2013, 86, 759–769. [Google Scholar] [CrossRef]
- Montero-Melendez, T.; Patel, H.B.; Seed, M.; Nielsen, S.; Jonassen, T.E.; Perretti, M. The melanocortin agonist AP214 exerts anti-inflammatory and proresolving properties. Am. J. Pathol. 2011, 179, 259–269. [Google Scholar] [CrossRef]
- Patruno, S.; Garrido-Mesa, J.; Romano, M.; Perretti, M.; Montero-Melendez, T. Ligand Bias and Its Association With Pro-resolving Actions of Melanocortin Drugs. Front. Pharmacol. 2018, 9, 919. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Teixeira, M.M.; Sousa, L.P. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J. Immunol. Res. 2016, 2016, 8239258. [Google Scholar] [CrossRef] [PubMed]
- Perucci, L.O.; Sugimoto, M.A.; Gomes, K.B.; Dusse, L.M.; Teixeira, M.M.; Sousa, L.P. Annexin A1 and specialized proresolving lipid mediators: Promoting resolution as a therapeutic strategy in human inflammatory diseases. Expert Opin. Ther. Targets 2017, 21, 879–896. [Google Scholar] [CrossRef]
- García-Osta, A.; Cuadrado-Tejedor, M.; García-Barroso, C.; Oyarzábal, J.; Franco, R. Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem. Neurosci. 2012, 3, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P.H.; Day, R.M. Novel systemic drugs for psoriasis: Mechanism of action for apremilast, a specific inhibitor of PDE4. J. Am. Acad. Dermatol. 2013, 68, 1041–1042. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, M.; Helliwell, P.S. Phosphodiesterase 4 inhibition in the treatment of psoriasis, psoriatic arthritis and other chronic inflammatory diseases. Dermatol Ther. (Heidelb.) 2013, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vago, J.P.; Nogueira, C.R.; Tavares, L.P.; Soriani, F.M.; Lopes, F.; Russo, R.C.; Pinho, V.; Teixeira, M.M.; Sousa, L.P. Annexin A1 modulates natural and glucocorticoid-induced resolution of inflammation by enhancing neutrophil apoptosis. J. Leukoc. Biol. 2012, 92, 249–258. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Ribeiro, A.L.C.; Costa, B.R.C.; Vago, J.P.; Lima, K.M.; Carneiro, F.S.; Ortiz, M.M.O.; Lima, G.L.N.; Carmo, A.A.F.; Rocha, R.M.; et al. Plasmin and plasminogen induce macrophage reprogramming and regulate key steps of inflammation resolution via annexin A1. Blood 2017, 129, 2896–2907. [Google Scholar] [CrossRef]
- Vago, J.P.; Sugimoto, M.A.; Lima, K.M.; Negreiros-Lima, G.L.; Baik, N.; Teixeira, M.M.; Perretti, M.; Parmer, R.J.; Miles, L.A.; Sousa, L.P. Plasminogen and the Plasminogen Receptor, Plg-R. Front. Immunol. 2019, 10, 1458. [Google Scholar] [CrossRef]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular circuits of resolution: Formation and actions of resolvins and protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar] [CrossRef]
- Vago, J.P.; Tavares, L.P.; Sugimoto, M.A.; Lima, G.L.; Galvão, I.; de Caux, T.R.; Lima, K.M.; Ribeiro, A.L.; Carneiro, F.S.; Nunes, F.F.; et al. Proresolving Actions of Synthetic and Natural Protease Inhibitors Are Mediated by Annexin A1. J. Immunol. 2016, 196, 1922–1932. [Google Scholar] [CrossRef]
- Newson, J.; Stables, M.; Karra, E.; Arce-Vargas, F.; Quezada, S.; Motwani, M.; Mack, M.; Yona, S.; Audzevich, T.; Gilroy, D.W. Resolution of acute inflammation bridges the gap between innate and adaptive immunity. Blood 2014, 124, 1748–1764. [Google Scholar] [CrossRef]
- Perez, D.A.; Galvão, I.; Athayde, R.M.; Rezende, B.M.; Vago, J.P.; Silva, J.D.; Reis, A.C.; Ribeiro, L.S.; Gomes, J.H.S.; Pádua, R.M.; et al. Inhibition of the sphingosine-1-phosphate pathway promotes the resolution of neutrophilic inflammation. Eur. J. Immunol. 2019. [Google Scholar] [CrossRef]
- Ward, C.; Chilvers, E.R.; Lawson, M.F.; Pryde, J.G.; Fujihara, S.; Farrow, S.N.; Haslett, C.; Rossi, A.G. NF-kappaB activation is a critical regulator of human granulocyte apoptosis in vitro. J. Biol. Chem. 1999, 274, 4309–4318. [Google Scholar] [CrossRef] [PubMed]
- Marim, F.M.; Silveira, T.N.; Lima, D.S.; Zamboni, D.S. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE 2010, 5, e15263. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kuziel, W.A.; Morgan, S.J.; Dawson, T.C.; Griffin, S.; Smithies, O.; Ley, K.; Maeda, N. Severe reduction in leukocyte adhesion and monocyte extravasation in mice deficient in CC chemokine receptor 2. Proc. Natl. Acad. Sci. USA 1997, 94, 12053–12058. [Google Scholar] [CrossRef]
- Malyshev, I.; Malyshev, Y. Current Concept and Update of the Macrophage Plasticity Concept: Intracellular Mechanisms of Reprogramming and M3 Macrophage “Switch” Phenotype. Biomed. Res. Int 2015, 2015, 341308. [Google Scholar] [CrossRef]
- Vago, J.P.; Tavares, L.P.; Garcia, C.C.; Lima, K.M.; Perucci, L.O.; Vieira, É.; Nogueira, C.R.; Soriani, F.M.; Martins, J.O.; Silva, P.M.; et al. The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J. Immunol. 2015, 194, 4940–4950. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized phosphatidylserine-CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006, 203, 2613–2625. [Google Scholar] [CrossRef]
- Yona, S.; Heinsbroek, S.E.; Peiser, L.; Gordon, S.; Perretti, M.; Flower, R.J. Impaired phagocytic mechanism in annexin 1 null macrophages. Br. J. Pharmacol. 2006, 148, 469–477. [Google Scholar] [CrossRef]
- Scannell, M.; Flanagan, M.B.; deStefani, A.; Wynne, K.J.; Cagney, G.; Godson, C.; Maderna, P. Annexin-1 and peptide derivatives are released by apoptotic cells and stimulate phagocytosis of apoptotic neutrophils by macrophages. J. Immunol. 2007, 178, 4595–4605. [Google Scholar] [CrossRef]
- McArthur, S.; Cristante, E.; Paterno, M.; Christian, H.; Roncaroli, F.; Gillies, G.E.; Solito, E. Annexin A1: A central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol. 2010, 185, 6317–6328. [Google Scholar] [CrossRef]
- Maderna, P.; Cottell, D.C.; Toivonen, T.; Dufton, N.; Dalli, J.; Perretti, M.; Godson, C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010, 24, 4240–4249. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Montero-Melendez, T.; McArthur, S.; Perretti, M. Annexin A1 N-terminal derived Peptide ac2-26 exerts chemokinetic effects on human neutrophils. Front. Pharmacol. 2012, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, W.S.; Vaisar, T.; Tang, J.; Wilson, C.L.; Raines, E.W. Macrophage ADAM17 deficiency augments CD36-dependent apoptotic cell uptake and the linked anti-inflammatory phenotype. Circ. Res. 2013, 113, 52–61. [Google Scholar] [CrossRef]
- Novak, M.L.; Thorp, E.B. Shedding light on impaired efferocytosis and nonresolving inflammation. Circ. Res. 2013, 113, 9–12. [Google Scholar] [CrossRef]
- Parks, B.W.; Black, L.L.; Zimmerman, K.A.; Metz, A.E.; Steele, C.; Murphy-Ullrich, J.E.; Kabarowski, J.H. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J. Lipid Res. 2013, 54, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Rajakariar, R.; Newson, J.; Jackson, E.K.; Sawmynaden, P.; Smith, A.; Rahman, F.; Yaqoob, M.M.; Gilroy, D.W. Nonresolving inflammation in gp91phox-/- mice, a model of human chronic granulomatous disease, has lower adenosine and cyclic adenosine 5’-monophosphate. J. Immunol. 2009, 182, 3262–3269. [Google Scholar] [CrossRef] [PubMed]
- Pederzoli-Ribeil, M.; Maione, F.; Cooper, D.; Al-Kashi, A.; Dalli, J.; Perretti, M.; D’Acquisto, F. Design and characterization of a cleavage-resistant Annexin A1 mutant to control inflammation in the microvasculature. Blood 2010, 116, 4288–4296. [Google Scholar] [CrossRef]
- Chiang, N.; Shinohara, M.; Dalli, J.; Mirakaj, V.; Kibi, M.; Choi, A.M.; Serhan, C.N. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J. Immunol. 2013, 190, 6378–6388. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef]
- Yang, J.X.; Hsiung, T.C.; Weng, F.C.; Ding, S.L.; Wu, C.P.; Conti, M.; Chuang, T.H.; Catherine Jin, S.L. Synergistic effect of phosphodiesterase 4 inhibitor and serum on migration of endotoxin-stimulated macrophages. Innate Immun. 2018, 24, 501–512. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.; Gobbetti, T.; Kusters, D.H.; Reutelingsperger, C.P.; Flower, R.J.; Perretti, M. Definition of a Novel Pathway Centered on Lysophosphatidic Acid To Recruit Monocytes during the Resolution Phase of Tissue Inflammation. J. Immunol. 2015, 195, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J.F.; Serhan, C.N. Lipoxin A4 and B4 are potent stimuli for human monocyte migration and adhesion: Selective inactivation by dehydrogenation and reduction. J. Exp. Med. 1996, 183, 137–146. [Google Scholar] [CrossRef]
- Maddox, J.F.; Hachicha, M.; Takano, T.; Petasis, N.A.; Fokin, V.V.; Serhan, C.N. Lipoxin A4 stable analogs are potent mimetics that stimulate human monocytes and THP-1 cells via a G-protein-linked lipoxin A4 receptor. J. Biol. Chem. 1997, 272, 6972–6978. [Google Scholar] [CrossRef] [PubMed]
- Carmo, A.A.; Costa, B.R.; Vago, J.P.; de Oliveira, L.C.; Tavares, L.P.; Nogueira, C.R.; Ribeiro, A.L.; Garcia, C.C.; Barbosa, A.S.; Brasil, B.S.; et al. Plasmin induces in vivo monocyte recruitment through protease-activated receptor-1-, MEK/ERK-, and CCR2-mediated signaling. J. Immunol. 2014, 193, 3654–3663. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, T.; Warr, G.; Loy, J.; Bravo, R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997, 186, 1757–1762. [Google Scholar] [CrossRef]
- Cushing, S.D.; Berliner, J.A.; Valente, A.J.; Territo, M.C.; Navab, M.; Parhami, F.; Gerrity, R.; Schwartz, C.J.; Fogelman, A.M. Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc. Natl. Acad. Sci. USA 1990, 87, 5134–5138. [Google Scholar] [CrossRef]
- Standiford, T.J.; Kunkel, S.L.; Phan, S.H.; Rollins, B.J.; Strieter, R.M. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J. Biol. Chem. 1991, 266, 9912–9918. [Google Scholar]
- Brown, Z.; Strieter, R.M.; Neild, G.H.; Thompson, R.C.; Kunkel, S.L.; Westwick, J. IL-1 receptor antagonist inhibits monocyte chemotactic peptide 1 generation by human mesangial cells. Kidney Int. 1992, 42, 95–101. [Google Scholar] [CrossRef]
- Barna, B.P.; Pettay, J.; Barnett, G.H.; Zhou, P.; Iwasaki, K.; Estes, M.L. Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J. Neuroimmunol. 1994, 50, 101–107. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, Y.G.; Shaw, M.; Kanneganti, T.D.; Fujimoto, Y.; Fukase, K.; Inohara, N.; Núñez, G. Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J. Immunol. 2007, 179, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Rollins, B.J. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: Therapeutic opportunities and controversies. Microcirculation 2003, 10, 247–257. [Google Scholar] [CrossRef] [PubMed]
- França, C.N.; Izar, M.C.O.; Hortêncio, M.N.S.; do Amaral, J.B.; Ferreira, C.E.S.; Tuleta, I.D.; Fonseca, F.A.H. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin. Sci. (Lond.) 2017, 131, 1215–1224. [Google Scholar] [CrossRef]
- Cailhier, J.F.; Partolina, M.; Vuthoori, S.; Wu, S.; Ko, K.; Watson, S.; Savill, J.; Hughes, J.; Lang, R.A. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 2005, 174, 2336–2342. [Google Scholar] [CrossRef]
- Motoyoshi, S.; Yamamoto, Y.; Munesue, S.; Igawa, H.; Harashima, A.; Saito, H.; Han, D.; Watanabe, T.; Sato, H.; Yamamoto, H. cAMP ameliorates inflammation by modulation of macrophage receptor for advanced glycation end-products. Biochem. J. 2014, 463, 75–82. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 130. [Google Scholar] [CrossRef]
- Chakravortty, D.; Hensel, M. Inducible nitric oxide synthase and control of intracellular bacterial pathogens. Microbes Infect. 2003, 5, 621–627. [Google Scholar] [CrossRef]
- Tavares, L.P.; Garcia, C.C.; Vago, J.P.; Queiroz-Junior, C.M.; Galvão, I.; David, B.A.; Rachid, M.A.; Silva, P.M.; Russo, R.C.; Teixeira, M.M.; et al. Inhibition of Phosphodiesterase-4 during Pneumococcal Pneumonia Reduces Inflammation and Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2016, 55, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Koscsó, B.; Csóka, B.; Kókai, E.; Németh, Z.H.; Pacher, P.; Virág, L.; Leibovich, S.J.; Haskó, G. Adenosine augments IL-10-induced STAT3 signaling in M2c macrophages. J. Leukoc. Biol. 2013, 94, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Özeş, A.R.; Pulliam, N.; Ertosun, M.G.; Yılmaz, Ö.; Tang, J.; Çopuroğlu, E.; Matei, D.; Özeş, O.N.; Nephew, K.P. Protein kinase A-mediated phosphorylation regulates STAT3 activation and oncogenic EZH2 activity. Oncogene 2018, 37, 3589–3600. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Feng, P.P.; Zhao, Z.B.; Zhu, W.; Gong, J.P.; Du, H.M. Liraglutide protects against inflammatory stress in non-alcoholic fatty liver by modulating Kupffer cells M2 polarization via cAMP-PKA-STAT3 signaling pathway. Biochem. Biophys. Res. Commun 2019, 510, 20–26. [Google Scholar] [CrossRef]
- MacKenzie, K.F.; Clark, K.; Naqvi, S.; McGuire, V.A.; Nöehren, G.; Kristariyanto, Y.; van den Bosch, M.; Mudaliar, M.; McCarthy, P.C.; Pattison, M.J.; et al. PGE(2) induces macrophage IL-10 production and a regulatory-like phenotype via a protein kinase A-SIK-CRTC3 pathway. J. Immunol. 2013, 190, 565–577. [Google Scholar] [CrossRef]
- Wein, M.N.; Foretz, M.; Fisher, D.E.; Xavier, R.J.; Kronenberg, H.M. Salt-Inducible Kinases: Physiology, Regulation by cAMP, and Therapeutic Potential. Trends Endocrinol. Metab. 2018, 29, 723–735. [Google Scholar] [CrossRef]
- Ozanne, J.; Prescott, A.R.; Clark, K. The clinically approved drugs dasatinib and bosutinib induce anti-inflammatory macrophages by inhibiting the salt-inducible kinases. Biochem J. 2015, 465, 271–279. [Google Scholar] [CrossRef]
- Clark, K.; MacKenzie, K.F.; Petkevicius, K.; Kristariyanto, Y.; Zhang, J.; Choi, H.G.; Peggie, M.; Plater, L.; Pedrioli, P.G.; McIver, E.; et al. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, 16986–16991. [Google Scholar] [CrossRef]
- Gilroy, D.W.; Lawrence, T.; Perretti, M.; Rossi, A.G. Inflammatory resolution: New opportunities for drug discovery. Nat. Rev. Drug Discov. 2004, 3, 401–416. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef]
- Patel, H.B.; Montero-Melendez, T.; Greco, K.V.; Perretti, M. Melanocortin receptors as novel effectors of macrophage responses in inflammation. Front. Immunol. 2011, 2, 41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frasch, S.C.; Bratton, D.L. Emerging roles for lysophosphatidylserine in resolution of inflammation. Prog. Lipid Res. 2012, 51, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Frasch, S.C.; Fernandez-Boyanapalli, R.F.; Berry, K.Z.; Leslie, C.C.; Bonventre, J.V.; Murphy, R.C.; Henson, P.M.; Bratton, D.L. Signaling via macrophage G2A enhances efferocytosis of dying neutrophils by augmentation of Rac activity. J. Biol. Chem. 2011, 286, 12108–12122. [Google Scholar] [CrossRef]
- Brock, T.G.; McNish, R.W.; Mancuso, P.; Coffey, M.J.; Peters-Golden, M. Prolonged lipopolysaccharide inhibits leukotriene synthesis in peritoneal macrophages: Mediation by nitric oxide and prostaglandins. Prostaglandins Other Lipid Mediat. 2003, 71, 131–145. [Google Scholar] [CrossRef]
- Maderna, P.; Yona, S.; Perretti, M.; Godson, C. Modulation of phagocytosis of apoptotic neutrophils by supernatant from dexamethasone-treated macrophages and annexin-derived peptide Ac(2-26). J. Immunol. 2005, 174, 3727–3733. [Google Scholar] [CrossRef]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Gugiu, B.; Fox, P.L.; et al. Identification of a novel family of oxidized phospholipids that serve as ligands for the macrophage scavenger receptor CD36. J. Biol. Chem. 2002, 277, 38503–38516. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.G.; McCutcheon, J.C.; Roy, N.; Chilvers, E.R.; Haslett, C.; Dransfield, I. Regulation of macrophage phagocytosis of apoptotic cells by cAMP. J. Immunol. 1998, 160, 3562–3568. [Google Scholar]
- Aronoff, D.M.; Carstens, J.K.; Chen, G.H.; Toews, G.B.; Peters-Golden, M. Short communication: Differences between macrophages and dendritic cells in the cyclic AMP-dependent regulation of lipopolysaccharide-induced cytokine and chemokine synthesis. J. Interferon Cytokine Res. 2006, 26, 827–833. [Google Scholar] [CrossRef]
- Kumaran Satyanarayanan, S.; El Kebir, D.; Soboh, S.; Butenko, S.; Sekheri, M.; Saadi, J.; Peled, N.; Assi, S.; Othman, A.; Schif-Zuck, S.; et al. IFN-β is a macrophage-derived effector cytokine facilitating the resolution of bacterial inflammation. Nat. Commun. 2019, 10, 3471. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negreiros-Lima, G.L.; Lima, K.M.; Moreira, I.Z.; Jardim, B.L.O.; Vago, J.P.; Galvão, I.; Teixeira, L.C.R.; Pinho, V.; Teixeira, M.M.; Sugimoto, M.A.; et al. Cyclic AMP Regulates Key Features of Macrophages via PKA: Recruitment, Reprogramming and Efferocytosis. Cells 2020, 9, 128. https://doi.org/10.3390/cells9010128
Negreiros-Lima GL, Lima KM, Moreira IZ, Jardim BLO, Vago JP, Galvão I, Teixeira LCR, Pinho V, Teixeira MM, Sugimoto MA, et al. Cyclic AMP Regulates Key Features of Macrophages via PKA: Recruitment, Reprogramming and Efferocytosis. Cells. 2020; 9(1):128. https://doi.org/10.3390/cells9010128
Chicago/Turabian StyleNegreiros-Lima, Graziele L., Kátia M. Lima, Isabella Z. Moreira, Bruna Lorrayne O. Jardim, Juliana P. Vago, Izabela Galvão, Lívia Cristina R. Teixeira, Vanessa Pinho, Mauro M. Teixeira, Michelle A. Sugimoto, and et al. 2020. "Cyclic AMP Regulates Key Features of Macrophages via PKA: Recruitment, Reprogramming and Efferocytosis" Cells 9, no. 1: 128. https://doi.org/10.3390/cells9010128
APA StyleNegreiros-Lima, G. L., Lima, K. M., Moreira, I. Z., Jardim, B. L. O., Vago, J. P., Galvão, I., Teixeira, L. C. R., Pinho, V., Teixeira, M. M., Sugimoto, M. A., & Sousa, L. P. (2020). Cyclic AMP Regulates Key Features of Macrophages via PKA: Recruitment, Reprogramming and Efferocytosis. Cells, 9(1), 128. https://doi.org/10.3390/cells9010128