Mesenchymal Stromal Cells from Fetal and Maternal Placenta Possess Key Similarities and Differences: Potential Implications for Their Applications in Regenerative Medicine


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statements
2.2. Isolation of Mesenchymal Stromal Cells from the Amniotic Membrane
2.3. Placental Expanded (PLX) Cells
2.4. Analysis of PLX Cells and hAMSC Phenotype
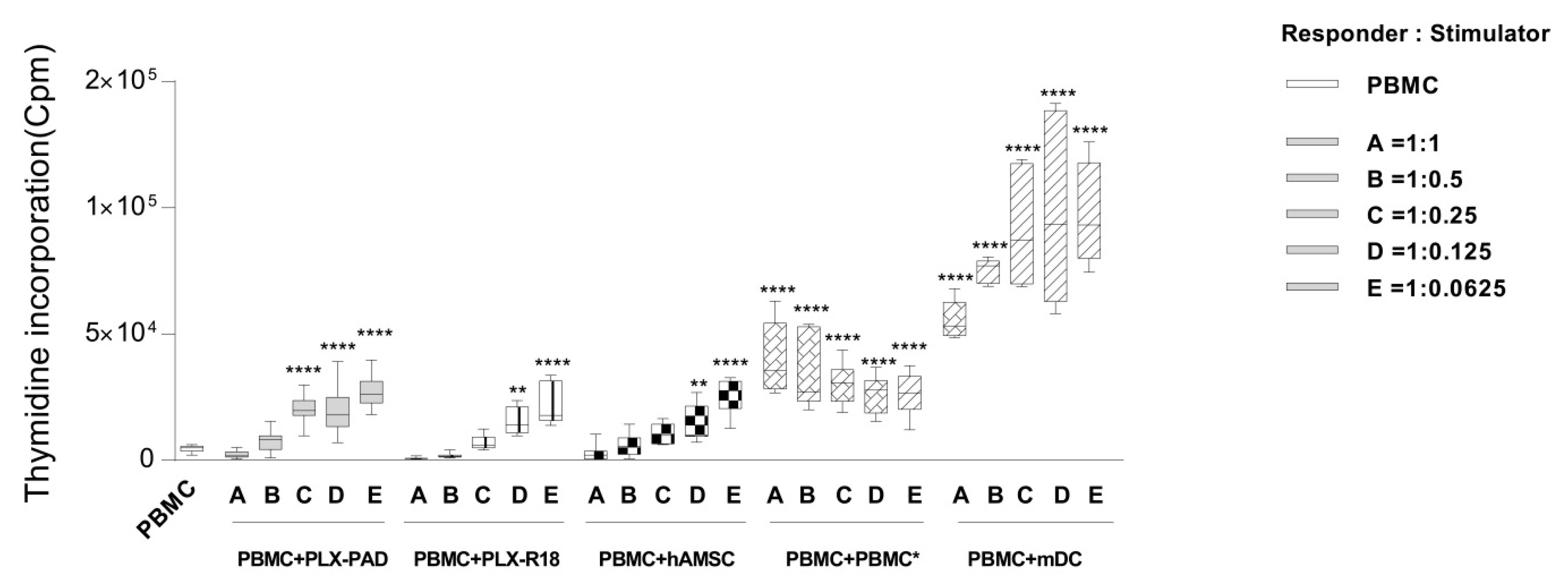
2.5. Analysis of T Cell Proliferation
2.6. Degranulation and Cytotoxic Marker Expression
2.7. Phenotype of CD4+ T Helper (Th) and T Regulatory (Treg) Subsets
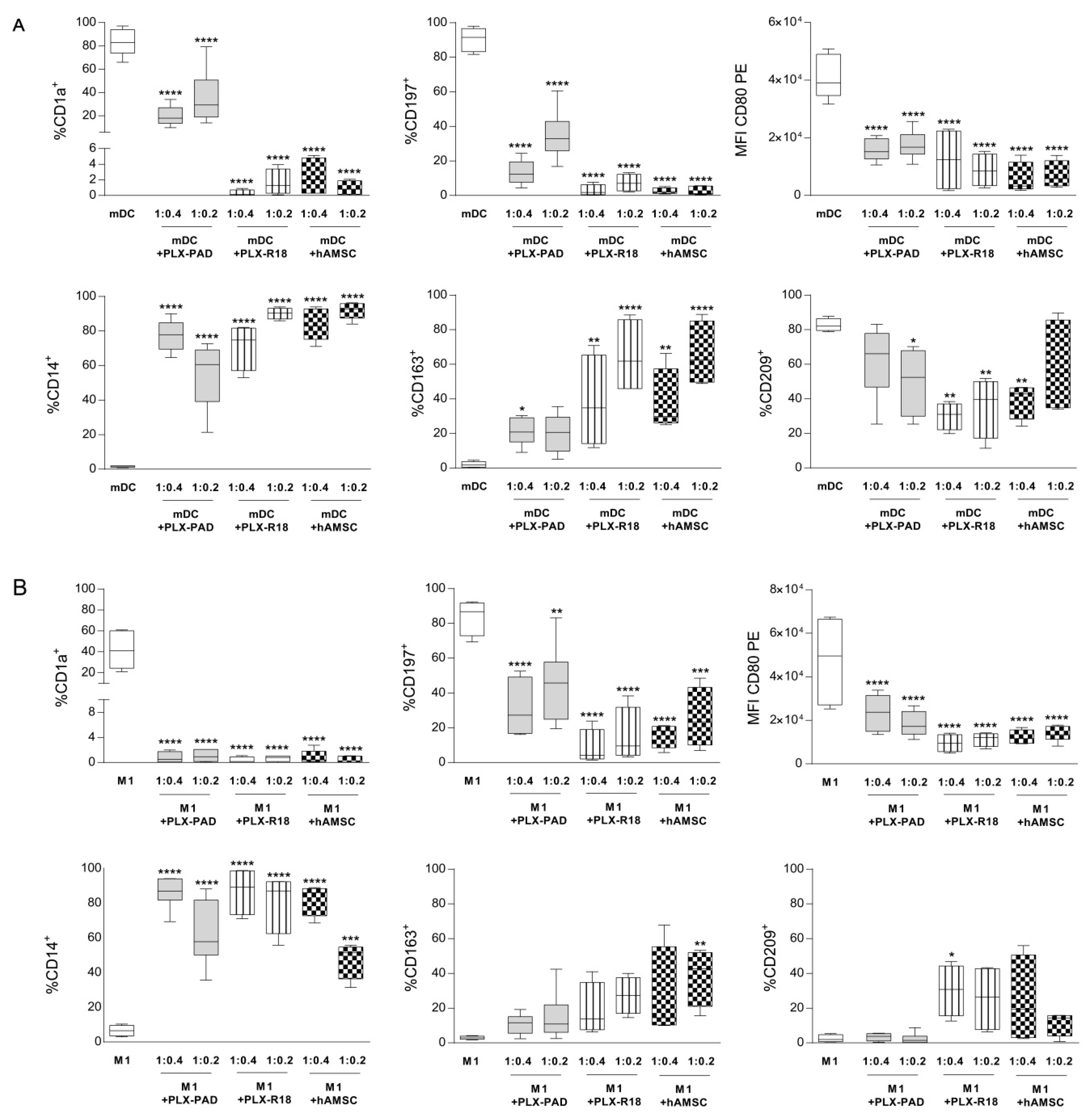
2.8. Analysis of Monocyte Differentiation toward Antigen Presenting Cells
2.9. Cytokine/Chemokine Analysis
2.10. Analysis of Immunogenicity
2.11. Statistical Analysis
3. Results
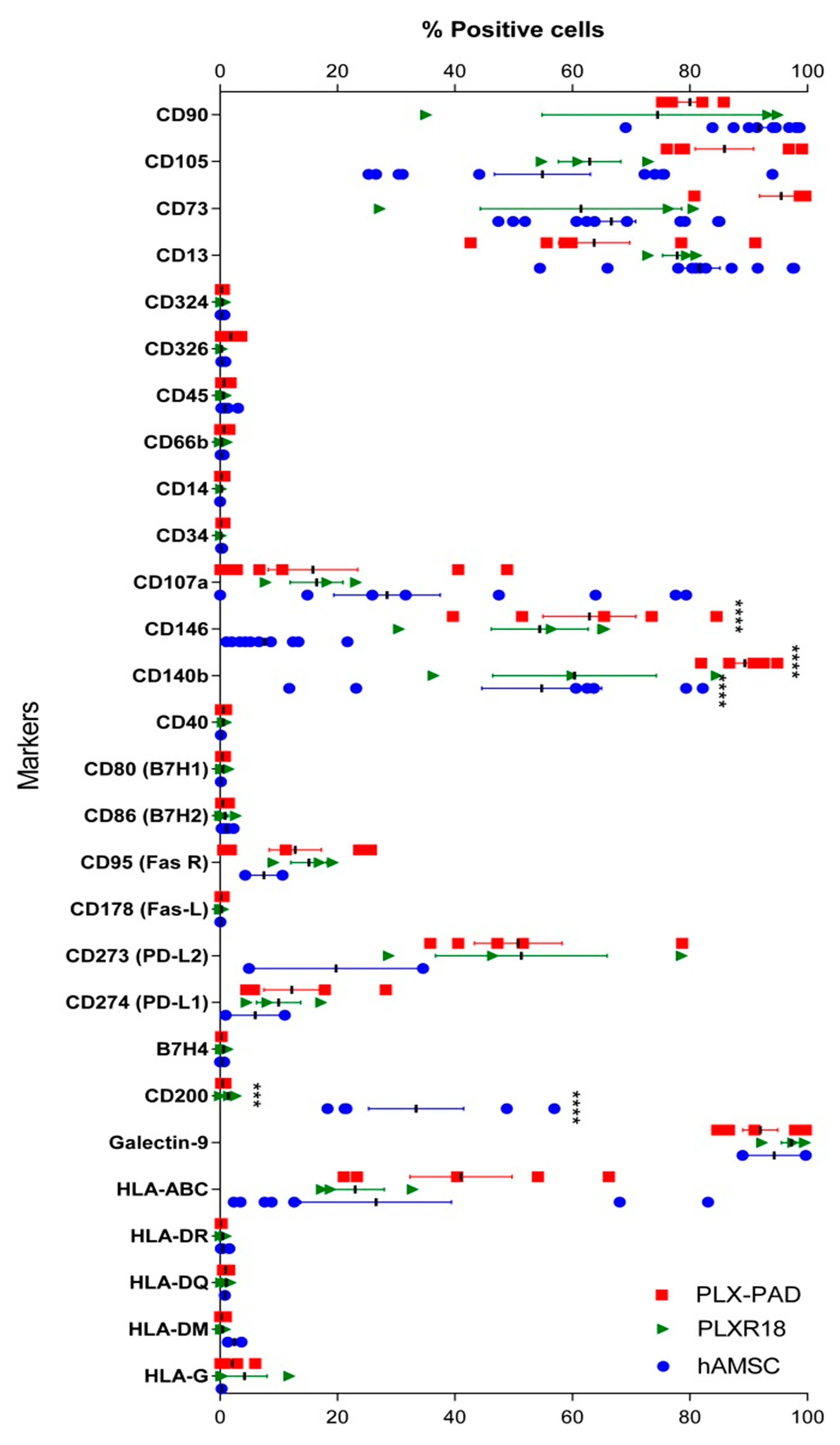
3.1. Immunophenotype of Maternal and Fetal Cells
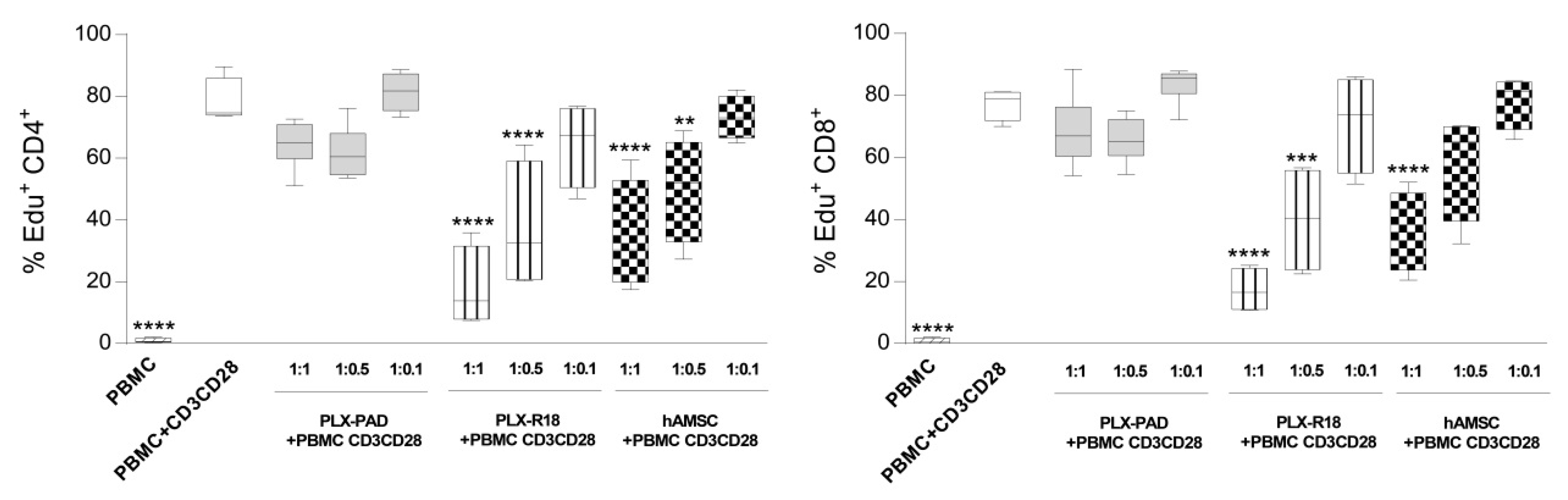
3.2. Maternal and Fetal Cells Differently Impact the Proliferation of T Lymphocytes
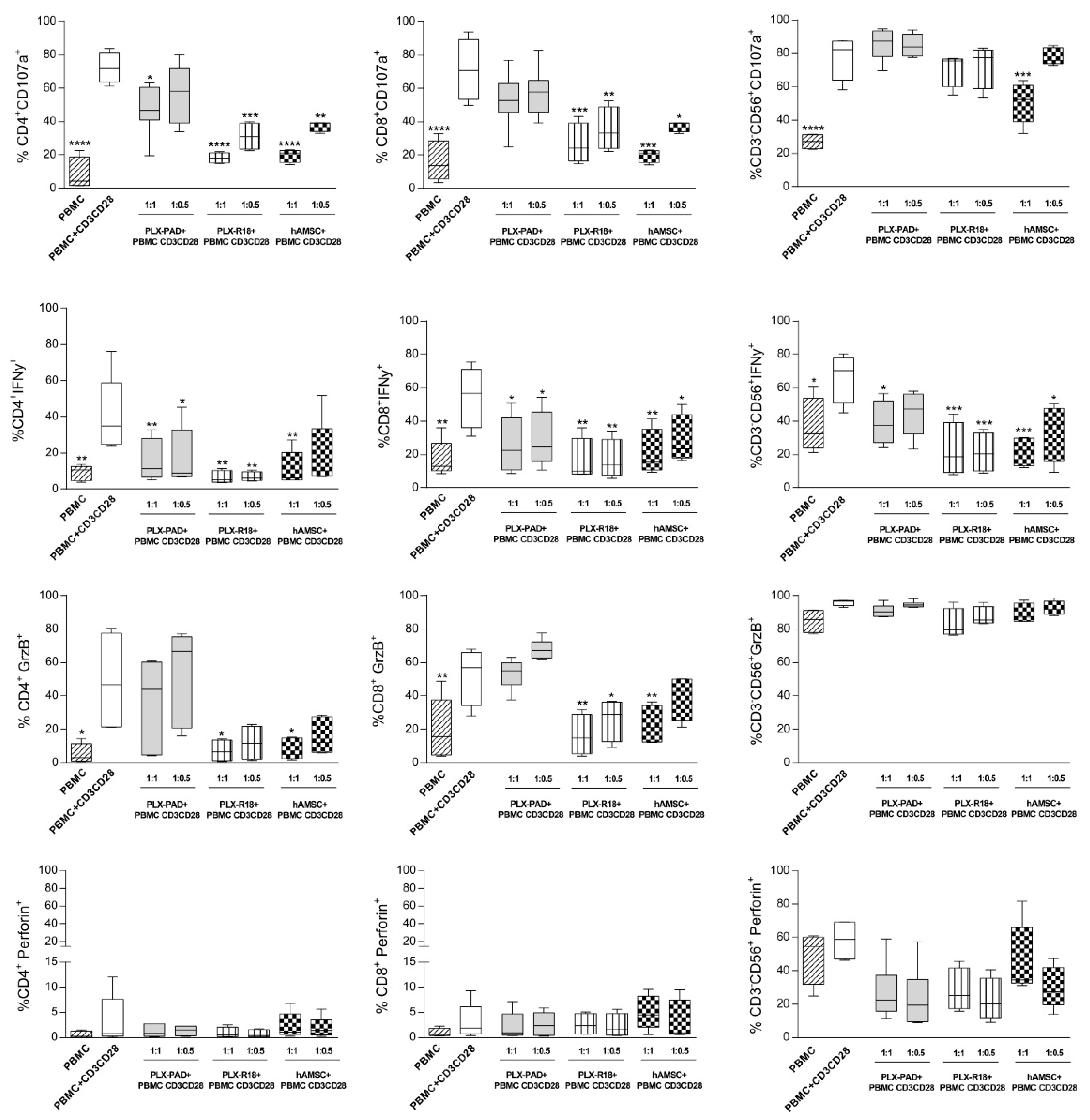
3.3. Maternal and Fetal Cells Affect T Lymphocyte Functions and Reduce the Expression of Cytotoxicity Markers
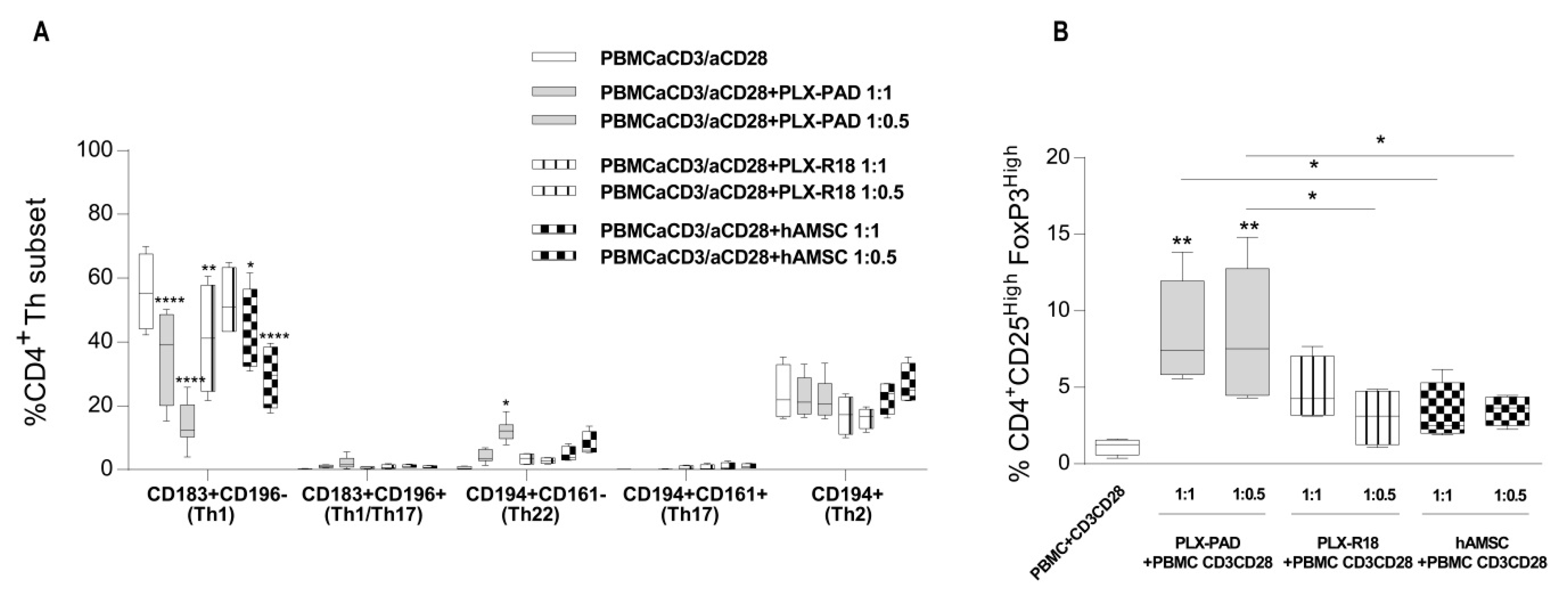
3.4. Maternal and Fetal Cells Inhibit Th1 Priming and Strongly Induce Pro-Regenerative Th22 and T Regulatory Cell Subsets
3.5. Maternal and Fetal MSC Affect the Expression of Th-Cytokines
3.6. Maternal and Fetal Cells Inhibit Monocyte-Derived Antigen Presenting Cell (APC) Differentiation
3.7. Immunogenicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parolini, O.; Alviano, F.; Bagnara, G.P.; Bilic, G.; Buhring, H.J.; Evangelista, M.; Hennerbichler, S.; Liu, B.; Magatti, M.; Mao, N.; et al. Concise review: Isolation and characterization of cells from human term placenta: Outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells 2008, 26, 300–311. [Google Scholar] [CrossRef]
- Soncini, M.; Vertua, E.; Gibelli, L.; Zorzi, F.; Denegri, M.; Albertini, A.; Wengler, G.S.; Parolini, O. Isolation and characterization of mesenchymal cells from human fetal membranes. J. Tissue Eng. Regen. Med. 2007, 1, 296–305. [Google Scholar] [CrossRef]
- Bailo, M.; Soncini, M.; Vertua, E.; Signoroni, P.B.; Sanzone, S.; Lombardi, G.; Arienti, D.; Calamani, F.; Zatti, D.; Paul, P.; et al. Engraftment potential of human amnion and chorion cells derived from term placenta. Transplantation 2004, 78, 1439–1448. [Google Scholar] [CrossRef]
- In’t Anker, P.S.; Scherjon, S.A.; Kleijburg-van der Keur, C.; de Groot-Swings, G.M.; Claas, F.H.; Fibbe, W.E.; Kanhai, H.H. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells 2004, 22, 1338–1345. [Google Scholar] [CrossRef]
- Ilancheran, S.; Michalska, A.; Peh, G.; Wallace, E.M.; Pera, M.; Manuelpillai, U. Stem cells derived from human fetal membranes display multilineage differentiation potential. Biol. Reprod. 2007, 77, 577–588. [Google Scholar] [CrossRef]
- Portmann-Lanz, C.B.; Schoeberlein, A.; Huber, A.; Sager, R.; Malek, A.; Holzgreve, W.; Surbek, D.V. Placental mesenchymal stem cells as potential autologous graft for pre- and perinatal neuroregeneration. Am. J. Obstet. Gynecol. 2006, 194, 664–673. [Google Scholar] [CrossRef]
- Igura, K.; Zhang, X.; Takahashi, K.; Mitsuru, A.; Yamaguchi, S.; Takashi, T.A. Isolation and characterization of mesenchymal progenitor cells from chorionic villi of human placenta. Cytotherapy 2004, 6, 543–553. [Google Scholar] [CrossRef]
- Fukuchi, Y.; Nakajima, H.; Sugiyama, D.; Hirose, I.; Kitamura, T.; Tsuji, K. Human placenta-derived cells have mesenchymal stem/progenitor cell potential. Stem Cells 2004, 22, 649–658. [Google Scholar] [CrossRef]
- Castrechini, N.M.; Murthi, P.; Gude, N.M.; Erwich, J.J.; Gronthos, S.; Zannettino, A.; Brennecke, S.P.; Kalionis, B. Mesenchymal stem cells in human placental chorionic villi reside in a vascular Niche. Placenta 2010, 31, 203–212. [Google Scholar] [CrossRef]
- Abumaree, M.H.; Al Jumah, M.A.; Kalionis, B.; Jawdat, D.; Al Khaldi, A.; AlTalabani, A.A.; Knawy, B.A. Phenotypic and functional characterization of mesenchymal stem cells from chorionic villi of human term placenta. Stem Cell Rev. 2013, 9, 16–31. [Google Scholar] [CrossRef]
- Roselli, E.A.; Lazzati, S.; Iseppon, F.; Manganini, M.; Marcato, L.; Gariboldi, M.B.; Maggi, F.; Grati, F.R.; Simoni, G. Fetal mesenchymal stromal cells from cryopreserved human chorionic villi: Cytogenetic and molecular analysis of genome stability in long-term cultures. Cytotherapy 2013, 15, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Abomaray, F.M.; Al Jumah, M.A.; Alsaad, K.O.; Jawdat, D.; Al Khaldi, A.; AlAskar, A.S.; Al Harthy, S.; Al Subayyil, A.M.; Khatlani, T.; Alawad, A.O.; et al. Phenotypic and Functional Characterization of Mesenchymal Stem/Multipotent Stromal Cells from Decidua Basalis of Human Term Placenta. Stem Cells Int. 2016, 2016, 5184601. [Google Scholar] [CrossRef] [PubMed]
- Macias, M.I.; Grande, J.; Moreno, A.; Dominguez, I.; Bornstein, R.; Flores, A.I. Isolation and characterization of true mesenchymal stem cells derived from human term decidua capable of multilineage differentiation into all 3 embryonic layers. Am. J. Obstet. Gynecol. 2010, 203, 495.e9–495.e23. [Google Scholar] [CrossRef]
- Troyer, D.L.; Weiss, M.L. Wharton’s jelly-derived cells are a primitive stromal cell population. Stem Cells 2008, 26, 591–599. [Google Scholar] [CrossRef]
- La Rocca, G.; Anzalone, R.; Corrao, S.; Magno, F.; Loria, T.; Lo Iacono, M.; Di Stefano, A.; Giannuzzi, P.; Marasà, L.; Cappello, F.; et al. Isolation and characterization of Oct-4+/HLA-G+ mesenchymal stem cells from human umbilical cord matrix: Differentiation potential and detection of new markers. Histochem. Cell Biol. 2009, 131, 267–282. [Google Scholar] [CrossRef]
- La Rocca, G.; Anzalone, R.; Farina, F. The expression of CD68 in human umbilical cord mesenchymal stem cells: New evidences of presence in non-myeloid cell types. Scand. J. Immunol. 2009, 70, 161–162. [Google Scholar] [CrossRef]
- Saito, S.; Nakashima, A.; Shima, T.; Ito, M. Th1/Th2/Th17 and regulatory T-cell paradigm in pregnancy. Am. J. Reprod. Immunol. 2010, 63, 601–610. [Google Scholar] [CrossRef]
- Heikkinen, J.; Mottonen, M.; Alanen, A.; Lassila, O. Phenotypic characterization of regulatory T cells in the human decidua. Clin. Exp. Immunol. 2004, 136, 373–378. [Google Scholar] [CrossRef]
- Mjosberg, J.; Berg, G.; Jenmalm, M.C.; Ernerudh, J. FOXP3+ regulatory T cells and T helper 1, T helper 2, and T helper 17 cells in human early pregnancy decidua. Biol. Reprod. 2010, 82, 698–705. [Google Scholar] [CrossRef]
- Nagamatsu, T.; Schust, D.J. The contribution of macrophages to normal and pathological pregnancies. Am. J. Reprod. Immunol. 2010, 63, 460–471. [Google Scholar] [CrossRef]
- Moffett, A.; Loke, C. Immunology of placentation in eutherian mammals. Nat. Rev. Immunol. 2006, 6, 584–594. [Google Scholar] [CrossRef]
- Hanna, J.; Mandelboim, O. When killers become helpers. Trends Immunol. 2007, 28, 201–206. [Google Scholar] [CrossRef]
- Trowsdale, J.; Betz, A.G. Mother’s little helpers: Mechanisms of maternal-fetal tolerance. Nat. Immunol. 2006, 7, 241–246. [Google Scholar] [CrossRef]
- Chen, S.J.; Liu, Y.L.; Sytwu, H.K. Immunologic regulation in pregnancy: From mechanism to therapeutic strategy for immunomodulation. Clin. Dev. Immunol. 2012, 2012, 258391. [Google Scholar] [CrossRef]
- Magatti, M.; Abumaree, M.H.; Silini, A.R.; Anzalone, R.; Saieva, S.; Russo, E.; Trapani, M.E.; La Rocca, G.; Parolini, O. The Immunomodulatory Features of Mesenchymal Stromal Cells Derived from Wharton’s Jelly, Amniotic Membrane and Chorionic Villi: In Vitro and In Vivo Data. In Placenta the Tree of Life; Parolini, O., Ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 91–128. [Google Scholar]
- Silini, A.R.; Magatti, M.; Cargnoni, A.; Parolini, O. Is Immune Modulation the Mechanism Underlying the Beneficial Effects of Amniotic Cells and Their Derivatives in Regenerative Medicine? Cell Transplant. 2017, 26, 531–539. [Google Scholar] [CrossRef]
- Weiss, M.L.; Anderson, C.; Medicetty, S.; Seshareddy, K.B.; Weiss, R.J.; VanderWerff, I.; Troyer, D.; McIntosh, K.R. Immune properties of human umbilical cord Wharton’s jelly-derived cells. Stem Cells 2008, 26, 2865–2874. [Google Scholar] [CrossRef]
- Abomaray, F.M.; Al Jumah, M.A.; Kalionis, B.; AlAskar, A.S.; Al Harthy, S.; Jawdat, D.; Al Khaldi, A.; Alkushi, A.; Knawy, B.A.; Abumaree, M.H. Human Chorionic Villous Mesenchymal Stem Cells Modify the Functions of Human Dendritic Cells, and Induce an Anti-Inflammatory Phenotype in CD1+ Dendritic Cells. Stem Cell Rev. Rep. 2015, 11, 423–441. [Google Scholar] [CrossRef]
- Abumaree, M.H.; Abomaray, F.M.; Alshehri, N.A.; Almutairi, A.; AlAskar, A.S.; Kalionis, B.; Al Jumah, M.A. Phenotypic and Functional Characterization of Mesenchymal Stem/Multipotent Stromal Cells From Decidua Parietalis of Human Term Placenta. Reprod. Sci. 2016, 23, 1193–1207. [Google Scholar] [CrossRef]
- Centurione, L.; Passaretta, F.; Centurione, M.A.; Munari, S.; Vertua, E.; Silini, A.; Liberati, M.; Parolini, O.; Di Pietro, R. Mapping of the Human Placenta: Experimental Evidence of Amniotic Epithelial Cell Heterogeneity. Cell Transplant. 2018, 27, 12–22. [Google Scholar] [CrossRef]
- Hass, R.; Kasper, C.; Bohm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. CCS 2011, 9, 12. [Google Scholar] [CrossRef]
- Roelen, D.L.; van der Mast, B.J.; in’t Anker, P.S.; Kleijburg, C.; Eikmans, M.; van Beelen, E.; de Groot-Swings, G.M.; Fibbe, W.E.; Kanhai, H.H.; Scherjon, S.A.; et al. Differential immunomodulatory effects of fetal versus maternal multipotent stromal cells. Hum. Immunol. 2009, 70, 16–23. [Google Scholar] [CrossRef]
- Gonzalez, P.L.; Carvajal, C.; Cuenca, J.; Alcayaga-Miranda, F.; Figueroa, F.E.; Bartolucci, J.; Salazar-Aravena, L.; Khoury, M. Chorion Mesenchymal Stem Cells Show Superior Differentiation, Immunosuppressive, and Angiogenic Potentials in Comparison with Haploidentical Maternal Placental Cells. Stem Cells Transl. Med. 2015, 4, 1109–1121. [Google Scholar] [CrossRef]
- Malak, T.M.; Bell, S.C. Structural characteristics of term human fetal membranes: A novel zone of extreme morphological alteration within the rupture site. Br. J. Obstet. Gynaecol. 1994, 101, 375–386. [Google Scholar] [CrossRef]
- Banerjee, A.; Weidinger, A.; Hofer, M.; Steinborn, R.; Lindenmair, A.; Hennerbichler-Lugscheider, S.; Eibl, J.; Redl, H.; Kozlov, A.V.; Wolbank, S. Different metabolic activity in placental and reflected regions of the human amniotic membrane. Placenta 2015, 36, 1329–1332. [Google Scholar] [CrossRef]
- Allen, H.; Shraga-Heled, N.; Blumenfeld, M.; Dego-Ashto, T.; Fuchs-Telem, D.; Gilert, A.; Aberman, Z.; Ofir, R. Human Placental-Derived Adherent Stromal Cells Co-Induced with TNF-alpha and IFN-gamma Inhibit Triple-Negative Breast Cancer in Nude Mouse Xenograft Models. Sci. Rep. 2018, 8, 670. [Google Scholar] [CrossRef]
- Chatterjee, P.; Chiasson, V.L.; Pinzur, L.; Raveh, S.; Abraham, E.; Jones, K.A.; Bounds, K.R.; Ofir, R.; Flaishon, L.; Chajut, A.; et al. Human placenta-derived stromal cells decrease inflammation, placental injury and blood pressure in hypertensive pregnant mice. Clin. Sci. 2016, 130, 513–523. [Google Scholar] [CrossRef]
- Metheny, L.; Eid, S.; Lingas, K.; Ofir, R.; Pinzur, L.; Meyerson, H.; Hillard, M.L.; Huang, Y.A. Posttransplant Intramuscular Injection of PLX-R18 Mesenchymal-Like Adherent Stromal Cells Improves Human Hematopoietic Engraftment in A Murine Transplant Model. Front. Med. 2018, 2, 5–37. [Google Scholar]
- Lahiani, A.; Zahavi, E.; Netzer, N.; Ofir, R.; Pinzur, L.; Raveh, S.; Arien-Zakay, H.; Yavin, E.; Lazarovici, P. Human PLacental eXpanded (PLX) mesenchymal-like adherent stromal cells confer neuroprotection to nerve growth factor (NGF)-differentiated PC12 cells exposed to ischemia by secretion of IL-6 and VEGF. Biochim. Biophys. Acta 2015, 1853, 422–430. [Google Scholar] [CrossRef]
- Zahavi-Goldstein, E.; Blumenfeld, M.; Fuchs-Telem, D.; Pinzur, L.; Rubin, S.; Aberman, Z.; Sher, N.; Ofir, R. Placenta-derived PLX-PAD mesenchymal-like stromal cells are efficacious in rescuing blood flow in hind limb ischemia mouse model by a dose- and site-dependent mechanism of action. Cytotherapy 2017, 19, 1438–1446. [Google Scholar] [CrossRef]
- Magatti, M.; Pianta, S.; Silini, A.; Parolini, O. Isolation, Culture, and Phenotypic Characterization of Mesenchymal Stromal Cells from the Amniotic Membrane of the Human Term Placenta. Methods Mol. Biol. 2016, 1416, 233–244. [Google Scholar]
- Winkler, T.; Perka, C.; von Roth, P.; Agres, A.N.; Plage, H.; Preininger, B.; Pumberger, M.; Geissler, S.; Hagai, E.L.; Ofir, R.; et al. Immunomodulatory placental-expanded, mesenchymal stromal cells improve muscle function following hip arthroplasty. J. Cachexia Sarcopenia Muscle 2018, 9, 880–897. [Google Scholar] [CrossRef]
- Prather, W.R.; Toren, A.; Meiron, M.; Ofir, R.; Tschope, C.; Horwitz, E.M. The role of placental-derived adherent stromal cell (PLX-PAD) in the treatment of critical limb ischemia. Cytotherapy 2009, 11, 427–434. [Google Scholar] [CrossRef]
- Ramot, Y.; Meiron, M.; Toren, A.; Steiner, M.; Nyska, A. Safety and biodistribution profile of placental-derived mesenchymal stromal cells (PLX-PAD) following intramuscular delivery. Toxicol. Pathol. 2009, 37, 606–616. [Google Scholar] [CrossRef]
- Pianta, S.; Magatti, M.; Vertua, E.; Bonassi Signoroni, P.; Muradore, I.; Nuzzo, A.M.; Rolfo, A.; Silini, A.; Quaglia, F.; Todros, T.; et al. Amniotic mesenchymal cells from pre-eclamptic placentae maintain immunomodulatory features as healthy controls. J. Cell. Mol. Med. 2016, 20, 157–169. [Google Scholar] [CrossRef]
- Magatti, M.; Caruso, M.; De Munari, S.; Vertua, E.; De, D.; Manuelpillai, U.; Parolini, O. Human Amniotic Membrane-Derived Mesenchymal and Epithelial Cells Exert Different Effects on Monocyte-Derived Dendritic Cell Differentiation and Function. Cell Transplant. 2015, 24, 1733–1752. [Google Scholar] [CrossRef]
- Chiossone, L.; Conte, R.; Spaggiari, G.M.; Serra, M.; Romei, C.; Bellora, F.; Becchetti, F.; Andaloro, A.; Moretta, L.; Bottino, C.; et al. Mesenchymal Stromal Cells Induce Peculiar Alternatively Activated Macrophages Capable of Dampening Both Innate and Adaptive Immune Responses. Stem Cells 2016, 34, 1909–1921. [Google Scholar] [CrossRef]
- Spaggiari, G.M.; Abdelrazik, H.; Becchetti, F.; Moretta, L. MSCs inhibit monocyte-derived DC maturation and function by selectively interfering with the generation of immature DCs: Central role of MSC-derived prostaglandin E2. Blood 2009, 113, 6576–6583. [Google Scholar] [CrossRef]
- Magatti, M.; Vertua, E.; De Munari, S.; Caro, M.; Caruso, M.; Silini, A.; Delgado, M.; Parolini, O. Human amnion favours tissue repair by inducing the M1-to-M2 switch and enhancing M2 macrophage features. J. Tissue Eng. Regen. Med. 2017, 11, 2895–2911. [Google Scholar] [CrossRef]
- Ferreira, L.M.R.; Meissner, T.B.; Tilburgs, T.; Strominger, J.L. HLA-G: At the Interface of Maternal-Fetal Tolerance. Trends Immunol. 2017, 38, 272–286. [Google Scholar] [CrossRef]
- Le Gallo, M.; Poissonnier, A.; Blanco, P.; Legembre, P. CD95/Fas, Non-Apoptotic Signaling Pathways, and Kinases. Front. Immunol. 2017, 8, 1216. [Google Scholar] [CrossRef]
- Poissonnier, A.; Sanseau, D.; Le Gallo, M.; Malleter, M.; Levoin, N.; Viel, R.; Morere, L.; Penna, A.; Blanco, P.; Dupuy, A.; et al. CD95-Mediated Calcium Signaling Promotes T Helper 17 Trafficking to Inflamed Organs in Lupus-Prone Mice. Immunity 2016, 45, 209–223. [Google Scholar] [CrossRef]
- Wang, F.; Lu, Z.; Hawkes, M.; Yang, H.; Kain, K.C.; Liles, W.C. Fas (CD95) induces rapid, TLR4/IRAK4-dependent release of pro-inflammatory HMGB1 from macrophages. J. Inflamm. 2010, 7, 30. [Google Scholar] [CrossRef]
- Zhu, Y.; Yang, Y.; Zhang, Y.; Hao, G.; Liu, T.; Wang, L.; Yang, T.; Wang, Q.; Zhang, G.; Wei, J.; et al. Placental mesenchymal stem cells of fetal and maternal origins demonstrate different therapeutic potentials. Stem Cell Res. Ther. 2014, 5, 48. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.J.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–377. [Google Scholar] [CrossRef]
- Samsonraj, R.M.; Raghunath, M.; Nurcombe, V.; Hui, J.H.; van Wijnen, A.J. Concise Review: Multifaceted Characterization of Human Mesenchymal Stem Cells for Use in Regenerative Medicine. Stem Cells Transl. Med. 2017, 6, 2173–2185. [Google Scholar] [CrossRef]
- Peltzer, J.; Montespan, F.; Thepenier, C.; Boutin, L.; Uzan, G.; Rouas-Freiss, N.; Lataillade, J.J. Heterogeneous functions of perinatal mesenchymal stromal cells require a preselection before their banking for clinical use. Stem Cells Dev. 2015, 24, 329–344. [Google Scholar] [CrossRef]
- Sivasubramaniyan, K.; Harichandan, A.; Schumann, S.; Sobiesiak, M.; Lengerke, C.; Maurer, A.; Kalbacher, H.; Bühring, H.J. Prospective isolation of mesenchymal stem cells from human bone marrow using novel antibodies directed against Sushi domain containing 2. Stem Cells Dev. 2013, 22, 1944–1954. [Google Scholar] [CrossRef]
- Schmelzer, E.; McKeel, D.T.; Gerlach, J.C. Characterization of Human Mesenchymal Stem Cells from Different Tissues and Their Membrane Encasement for Prospective Transplantation Therapies. BioMed Res. Int. 2019, 2019, 6376271. [Google Scholar] [CrossRef]
- Li, J.; Koike-Soko, C.; Sugimoto, J.; Yoshida, T.; Okabe, M.; Nikaido, T. Human Amnion-Derived Stem Cells Have Immunosuppressive Properties on NK Cells and Monocytes. Cell Transplant. 2015, 24, 2065–2076. [Google Scholar] [CrossRef]
- Coles, S.J.; Wang, E.C.; Man, S.; Hills, R.K.; Burnett, A.K.; Tonks, A.; Darley, R.L. CD200 expression suppresses natural killer cell function and directly inhibits patient anti-tumor response in acute myeloid leukemia. Leukemia 2011, 25, 792–799. [Google Scholar] [CrossRef]
- Clark, D.A.; Wong, K.; Banwatt, D.; Chen, Z.; Liu, J.; Lee, L.; Gorczynski, R.M.; Blajchman, M.A. CD200-dependent and nonCD200-dependent pathways of NK cell suppression by human IVIG. J. Assist. Reprod. Genet. 2008, 25, 67–72. [Google Scholar] [CrossRef]
- Croxatto, D.; Vacca, P.; Canegallo, F.; Conte, R.; Venturini, P.L.; Moretta, L.; Mingari, M.C. Stromal cells from human decidua exert a strong inhibitory effect on NK cell function and dendritic cell differentiation. PLoS ONE 2014, 9, e89006. [Google Scholar] [CrossRef]
- Hsu, P.; Santner-Nanan, B.; Hu, M.; Skarratt, K.; Lee, C.H.; Stormon, M.; Wong, M.; Fuller, S.J.; Nanan, R. IL-10 Potentiates Differentiation of Human Induced Regulatory T Cells via STAT3 and Foxo1. J. Immunol. 2015, 195, 3665–3674. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Davidson, T.S.; DiPaolo, R.J.; Andersson, J.; Shevach, E.M. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J. Immunol. 2007, 178, 4022–4026. [Google Scholar] [CrossRef]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 is essential for TGF-beta to convert naive CD4+CD25- cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef]
- Pianta, S.; Bonassi Signoroni, P.; Muradore, I.; Rodrigues, M.F.; Rossi, D.; Silini, A.; Parolini, O. Amniotic membrane mesenchymal cells-derived factors skew T cell polarization toward Treg and downregulate Th1 and Th17 cells subsets. Stem Cell Rev. 2015, 11, 394–407. [Google Scholar] [CrossRef]
- Ohta, A. A Metabolic Immune Checkpoint: Adenosine in Tumor Microenvironment. Front. Immunol. 2016, 7, 109. [Google Scholar] [CrossRef]
- Song, W.J.; Li, Q.; Ryu, M.O.; Ahn, J.O.; Bhang, D.H.; Jung, Y.C.; Youn, H.Y. TSG-6 Secreted by Human Adipose Tissue-derived Mesenchymal Stem Cells Ameliorates DSS-induced colitis by Inducing M2 Macrophage Polarization in Mice. Sci. Rep. 2017, 7, 5187. [Google Scholar] [CrossRef]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles as Mediators of Anti-Inflammatory Effects: Endorsement of Macrophage Polarization. Stem Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef]
- Nemeth, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Doi, K.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M.; et al. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef]
- Enninga, E.A.; Nevala, W.K.; Holtan, S.G.; Leontovich, A.A.; Markovic, S.N. Galectin-9 modulates immunity by promoting Th2/M2 differentiation and impacts survival in patients with metastatic melanoma. Melanoma Res. 2016, 26, 429–441. [Google Scholar] [CrossRef]
- Pietila, M.; Lehtonen, S.; Tuovinen, E.; Lahteenmaki, K.; Laitinen, S.; Leskela, H.V.; Nätynki, A.; Pesälä, J.; Nordström, K.; Lehenkari, P. CD200 positive human mesenchymal stem cells suppress TNF-alpha secretion from CD200 receptor positive macrophage-like cells. PLoS ONE 2012, 7, e31671. [Google Scholar] [CrossRef]
- Pontikoglou, C.; Langonne, A.; Ba, M.A.; Varin, A.; Rosset, P.; Charbord, P.; Sensébé, L.; Deschaseaux, F. CD200 expression in human cultured bone marrow mesenchymal stem cells is induced by pro-osteogenic and pro-inflammatory cues. J. Cell. Mol. Med. 2016, 20, 655–665. [Google Scholar] [CrossRef]
- Lindenmair, A.; Hatlapatka, T.; Kollwig, G.; Hennerbichler, S.; Gabriel, C.; Wolbank, S.; Redl, H.; Kasper, C. Mesenchymal stem or stromal cells from amnion and umbilical cord tissue and their potential for clinical applications. Cells 2012, 1, 1061–1088. [Google Scholar] [CrossRef]
- Insausti, C.L.; Blanquer, M.; Garcia-Hernandez, A.M.; Castellanos, G.; Moraleda, J.M. Amniotic membrane-derived stem cells: Immunomodulatory properties and potential clinical application. Stem Cells Cloning Adv. Appl. 2014, 7, 53–63. [Google Scholar] [CrossRef]
- Le Blanc, K.; Tammik, L.; Sundberg, B.; Haynesworth, S.E.; Ringden, O. Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand. J. Immunol. 2003, 57, 11–20. [Google Scholar] [CrossRef]
- Wolbank, S.; Peterbauer, A.; Fahrner, M.; Hennerbichler, S.; van Griensven, M.; Stadler, G.; Redl, H.; Gabriel, C. Dose-dependent immunomodulatory effect of human stem cells from amniotic membrane: A comparison with human mesenchymal stem cells from adipose tissue. Tissue Eng. 2007, 13, 1173–1183. [Google Scholar] [CrossRef]
- Karlsson, H.; Erkers, T.; Nava, S.; Ruhm, S.; Westgren, M.; Ringden, O. Stromal cells from term fetal membrane are highly suppressive in allogeneic settings in vitro. Clin. Exp. Immunol. 2012, 167, 543–555. [Google Scholar] [CrossRef]
- Kronsteiner, B.; Wolbank, S.; Peterbauer, A.; Hackl, C.; Redl, H.; van Griensven, M.; Gabriel, C. Human mesenchymal stem cells from adipose tissue and amnion influence T-cells depending on stimulation method and presence of other immune cells. Stem Cells Dev. 2011, 20, 2115–2126. [Google Scholar] [CrossRef]
- Chan, J.L.; Tang, K.C.; Patel, A.P.; Bonilla, L.M.; Pierobon, N.; Ponzio, N.M.; Rameshwar, P. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood 2006, 107, 4817–4824. [Google Scholar] [CrossRef]
- van Megen, K.M.; van’t Wout, E.T.; Lages Motta, J.; Dekker, B.; Nikolic, T.; Roep, B.O. Activated Mesenchymal Stromal Cells Process and Present Antigens Regulating Adaptive Immunity. Front. Immunol. 2019, 10, 694. [Google Scholar] [CrossRef]
- Galipeau, J.; Krampera, M.; Barrett, J.; Dazzi, F.; Deans, R.J.; DeBruijn, J.; Dominici, M.; Fibbe, W.E.; Gee, A.P.; Gimble, J.M.; et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 2016, 18, 151–159. [Google Scholar] [CrossRef]
- Krampera, M.; Galipeau, J.; Shi, Y.; Tarte, K.; Sensebe, L. Therapy MSCCotISfC. Immunological characterization of multipotent mesenchymal stromal cells—The International Society for Cellular Therapy (ISCT) working proposal. Cytotherapy 2013, 15, 1054–1061. [Google Scholar]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef]
- Parolini, O.; Souza-Moreira, L.; O’Valle, F.; Magatti, M.; Hernandez-Cortes, P.; Gonzalez-Rey, E.; Delgado, M. Therapeutic effect of human amniotic membrane-derived cells on experimental arthritis and other inflammatory disorders. Arthritis Rheumatol. 2014, 66, 327–339. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBMC CD3CD28 | PBMC CD3CD28+ PLX-PAD | PBMC CD3CD28+ PLX-R18 | PBMC CD3CD28+ hAMSC | ||
|---|---|---|---|---|---|
| Th1 | IFNγ | 2521.9 ± 446.2 | 2021.9 ± 728.2 | 183.6 ± 91.2 | 181.2 ± 111.4 |
| TNFα | 1076.0 ± 319.6 | 168.4 ± 132.2 | 3.8 ± 0.9 | 6.1 ± 4.8 | |
| Th2 | IL-4 | 1.9 ± 0.5 | 6.4 ± 2.4 | 1.9 ± 0.7 | 1.4 ± 0.4 |
| IL-5 | 775.1 ± 456.7 | 205.6 ± 100.7 | 35.5 ± 29.4 | 30.0 ± 20.9 | |
| IL-13 | 893.6 ± 290.3 | 992.4 ± 202.6 | 141.8 ± 79.7 | 195.9 ± 125.0 | |
| Th17 | IL-17A | 164.9 ± 197.5 | 60.8 ± 35.3 | 174.6 ± 111.1 | 227.6 ± 151.6 |
| Treg | IL-10 | 115.9 ± 14.6 | 112.8 ± 26.1 | 96.9 ± 34.6 | 117.9 ± 37.5 |
| TGFβ1 | 359.4 ± 98.9 | 931.0 ± 185.3 | 938.3 ± 61.8 | 933.7 ± 126.7 | |
| Cytotox | GrzB | 5778.0 ± 450.6 | 3490.1 ± 580.6 | 2397.2 ± 1429.2 | 2451.2 ± 528.6 |
| GrzA | 1963.5 ± 662.4 | 1842.7 ± 12.2 | 1122.5 ± 989.0 | 640.4 ± 406.5 | |
| RANTES | 3108.5 ± 822.7 | 3937.9 ± 424.5 | 443.7 ± 143.2 | 377.2 ± 248.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papait, A.; Vertua, E.; Magatti, M.; Ceccariglia, S.; De Munari, S.; Silini, A.R.; Sheleg, M.; Ofir, R.; Parolini, O. Mesenchymal Stromal Cells from Fetal and Maternal Placenta Possess Key Similarities and Differences: Potential Implications for Their Applications in Regenerative Medicine. Cells 2020, 9, 127. https://doi.org/10.3390/cells9010127
Papait A, Vertua E, Magatti M, Ceccariglia S, De Munari S, Silini AR, Sheleg M, Ofir R, Parolini O. Mesenchymal Stromal Cells from Fetal and Maternal Placenta Possess Key Similarities and Differences: Potential Implications for Their Applications in Regenerative Medicine. Cells. 2020; 9(1):127. https://doi.org/10.3390/cells9010127
Chicago/Turabian StylePapait, Andrea, Elsa Vertua, Marta Magatti, Sabrina Ceccariglia, Silvia De Munari, Antonietta Rosa Silini, Michal Sheleg, Racheli Ofir, and Ornella Parolini. 2020. "Mesenchymal Stromal Cells from Fetal and Maternal Placenta Possess Key Similarities and Differences: Potential Implications for Their Applications in Regenerative Medicine" Cells 9, no. 1: 127. https://doi.org/10.3390/cells9010127
APA StylePapait, A., Vertua, E., Magatti, M., Ceccariglia, S., De Munari, S., Silini, A. R., Sheleg, M., Ofir, R., & Parolini, O. (2020). Mesenchymal Stromal Cells from Fetal and Maternal Placenta Possess Key Similarities and Differences: Potential Implications for Their Applications in Regenerative Medicine. Cells, 9(1), 127. https://doi.org/10.3390/cells9010127