COUP-TFII in Health and Disease


Abstract
1. Introduction
2. COUP-TFII: An Overview on Structure, Mechanism of Action and Expression
2.1. COUP-TFI and COUP-TFII: Similarities and Differences
2.2. COUP-TFII Signaling: Activation, Repression and Transrepression
2.3. COUP-TFII Protein Structure: Isoforms, the LBD and the Ligands
3. Regulation of Cell Differentiation and Cell Function in Health
3.1. Mesenchymal/Stromal Cells Identity
3.2. COUP-TFII in Metabolism and Metabolic Active Organs
3.2.1. COUP-TFII, Energy Expenditure and the White Adipose Tissue
3.2.2. COUP-TFII and Liver Metabolism
3.2.3. COUP-TFII and Insulin
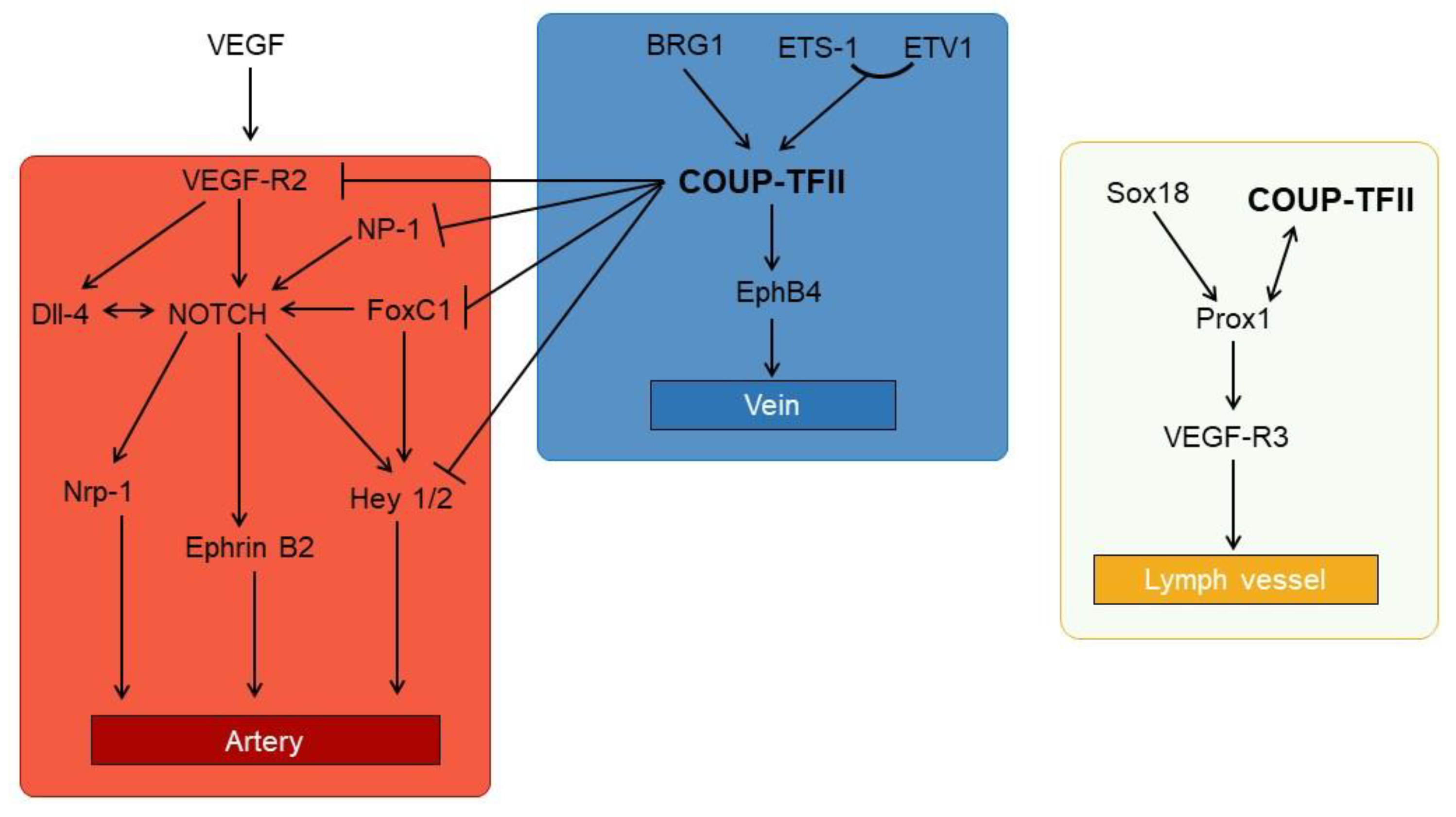
3.3. The Vein Identity and the Link to NOTCH
4. COUP-TFII in Human Pathology
4.1. COUP-TFII in Non-Cancer-Related Pathologies
4.1.1. Cardiovascular Diseases
4.1.2. COUP-TFII and Diabetes
4.1.3. Infertility and Alterations of Mesenchymal Commitment
4.2. COUP-TFII in Cancer: The Good Guy and the Bad Guy
4.2.1. The Gastrointestinal Cancers: Mostly an Oncogene
4.2.2. The Uncertainty of COUP-TFII in the Breast Cancer
4.2.3. Prostate and Renal Cancers
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of Nomenclature of Nuclear Receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Galli, A. Nuclear receptors and pathogenesis of pancreatic cancer. World J. Gastroenterol. WJG 2014, 20, 12062–12081. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Orphan Nuclear Receptors: Shifting Endocrinology into Reverse. Science 1999, 284, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J.; Baulieu, E.; Beato, M.; Becker-Andre, M.; Burbach, P.H.; Camerino, G.; Chambon, P.; Cooney, A.; Dejean, A.; Dreyer, C.; et al. A Unified Nomenclature System for the Nuclear Receptor Superfamily. Cell 1999, 97, 161–163. [Google Scholar]
- Wang, L.H.; Tsai, S.Y.; Sagami, I.; Tsai, M.J.; O’Malley, B.W. Purification and characterization of chicken ovalbumin upstream promoter transcription factor from HeLa cells. J. Biol. Chem. 1987, 262, 16080–16086. [Google Scholar]
- Miyajima, N.; Kadowaki, Y.; Fukushige, S.; Shimizu, S.; Semba, K.; Yamanashi, Y.; Matsubara, K.; Toyoshima, K.; Yamamoto, T. Identification of two novel members of erbA superfamily by molecular cloning: The gene products of the two are highly related to each other. Nucleic Acids Res. 1988, 16, 11057–11074. [Google Scholar] [CrossRef]
- Ladias, J.A.; Karathanasis, S.K. Regulation of the apolipoprotein AI gene by ARP-1, a novel member of the steroid receptor superfamily. Science 1991, 251, 561–565. [Google Scholar] [CrossRef]
- Wang, L.H.; Ing, N.H.; Tsai, S.Y.; O’Malley, B.W.; Tsai, M.J. The COUP-TFs compose a family of functionally related transcription factors. Gene Expr. 1991, 1, 207–216. [Google Scholar]
- Klepsch, V.; Hermann-Kleiter, N.; Baier, G. Beyond CTLA-4 and PD-1: Orphan nuclear receptor NR2F6 as T cell signaling switch and emerging target in cancer immunotherapy. Immunol. Lett. 2016, 178, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Bertacchi, M.; Parisot, J.; Studer, M. The pleiotropic transcriptional regulator COUP-TFI plays multiple roles in neural development and disease. Brain Res. 2019, 1705, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Krishnan, V.; Zeng, Z.; Gilbert, D.J.; Copeland, N.G.; Gibson, L.; Yang-Feng, T.; Jenkins, N.A.; Tsai, M.J.; Tsai, S.Y. Isolation, characterization, and chromosomal localization of mouse and human COUP-TF I and II genes. Genomics 1995, 29, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.A.; Tsai, M.J.; Tsai, S.Y. COUP-TF orphan nuclear receptors in development and differentiation. Cell. Mol. Life Sci. CMLS 2000, 57, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Qin, J.; Tsai, S.Y.; Tsai, M. The role of the orphan nuclear receptor COUP-TFII in tumorigenesis. Acta Pharmacol. Sin. 2015, 36, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.A.; Qiu, Y.; Zhou, G.; Tsai, M.J.; Tsai, S.Y. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev. 1999, 13, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-P.; Kao, C.-Y.; Wang, L.; Creighton, C.J.; Yang, J.; Donti, T.R.; Harmancey, R.; Vasquez, H.G.; Graham, B.H.; Bellen, H.J.; et al. Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat. Commun. 2015, 6, 8245. [Google Scholar] [CrossRef]
- Qin, J.; Chen, X.; Yu-Lee, L.-Y.; Tsai, M.-J.; Tsai, S.Y. Nuclear receptor COUP-TFII controls pancreatic islet tumor angiogenesis by regulating vascular endothelial growth factor/vascular endothelial growth factor receptor-2 signaling. Cancer Res. 2010, 70, 8812–8821. [Google Scholar] [CrossRef]
- Tang, K.; Tsai, S.Y.; Tsai, M.-J. COUP-TFs and Eye Development. Biochim. Biophys. Acta 2015, 1849, 201–209. [Google Scholar] [CrossRef]
- Xie, X.; Wu, S.-P.; Tsai, M.-J.; Tsai, S. The Role of COUP-TFII in Striated Muscle Development and Disease. Curr. Top. Dev. Biol. 2017, 125, 375–403. [Google Scholar]
- Lin, F.-J.; Qin, J.; Tang, K.; Tsai, S.Y.; Tsai, M.-J. Coup d’Etat: An orphan takes control. Endocr. Rev. 2011, 32, 404–421. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Feng, S.; Tang, K. COUP-TF Genes, Human Diseases, and the Development of the Central Nervous System in Murine Models. Curr. Top. Dev. Biol. 2017, 125, 275–301. [Google Scholar] [PubMed]
- Kliewer, S.A.; Umesono, K.; Noonan, D.J.; Heyman, R.A.; Evans, R.M. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992, 358, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.J.; Tsai, S.Y.; O’Malley, B.W.; Tsai, M.J. Chicken ovalbumin upstream promoter transcription factor (COUP-TF) dimers bind to different GGTCA response elements, allowing COUP-TF to repress hormonal induction of the vitamin D3, thyroid hormone, and retinoic acid receptors. Mol. Cell. Biol. 1992, 12, 4153–4163. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Umesono, K.; Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Evans, R.M. Retinoid X receptor-COUP-TF interactions modulate retinoic acid signaling. Proc. Natl. Acad. Sci. USA 1992, 89, 1448–1452. [Google Scholar] [CrossRef]
- Tran, P.; Zhang, X.K.; Salbert, G.; Hermann, T.; Lehmann, J.M.; Pfahl, M. COUP orphan receptors are negative regulators of retinoic acid response pathways. Mol. Cell. Biol. 1992, 12, 4666–4676. [Google Scholar] [CrossRef]
- Cooney, A.J.; Leng, X.; Tsai, S.Y.; O’Malley, B.W.; Tsai, M.J. Multiple mechanisms of chicken ovalbumin upstream promoter transcription factor-dependent repression of transactivation by the vitamin D, thyroid hormone, and retinoic acid receptors. J. Biol. Chem. 1993, 268, 4152–4160. [Google Scholar]
- Kojetin, D.J.; Matta-Camacho, E.; Hughes, T.S.; Srinivasan, S.; Nwachukwu, J.C.; Cavett, V.; Nowak, J.; Chalmers, M.J.; Marciano, D.P.; Kamenecka, T.M.; et al. Structural mechanism for signal transduction in RXR nuclear receptor heterodimers. Nat. Commun. 2015, 6, 8013. [Google Scholar] [CrossRef]
- Lee, S.; Kang, J.; Yoo, J.; Ganesan, S.K.; Cook, S.C.; Aguilar, B.; Ramu, S.; Lee, J.; Hong, Y.-K. Prox1 physically and functionally interacts with COUP-TFII to specify lymphatic endothelial cell fate. Blood 2009, 113, 1856–1859. [Google Scholar] [CrossRef]
- Okamura, M.; Kudo, H.; Wakabayashi, K.; Tanaka, T.; Nonaka, A.; Uchida, A.; Tsutsumi, S.; Sakakibara, I.; Naito, M.; Osborne, T.F.; et al. COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 5819–5824. [Google Scholar] [CrossRef]
- Leng, X.; Cooney, A.J.; Tsai, S.Y.; Tsai, M.J. Molecular mechanisms of COUP-TF-mediated transcriptional repression: Evidence for transrepression and active repression. Mol. Cell. Biol. 1996, 16, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Kruse, S.W.; Suino-Powell, K.; Zhou, X.E.; Kretschman, J.E.; Reynolds, R.; Vonrhein, C.; Xu, Y.; Wang, L.; Tsai, S.Y.; Tsai, M.-J.; et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 2008, 6, e227. [Google Scholar] [CrossRef] [PubMed]
- Le Guével, R.; Oger, F.; Martinez-Jimenez, C.P.; Bizot, M.; Gheeraert, C.; Firmin, F.; Ploton, M.; Kretova, M.; Palierne, G.; Staels, B.; et al. Inactivation of the Nuclear Orphan Receptor COUP-TFII by Small Chemicals. ACS Chem. Biol. 2017, 12, 654–663. [Google Scholar] [CrossRef]
- Wang, L.H.; Tsai, S.Y.; Cook, R.G.; Beattie, W.G.; Tsai, M.J.; O’Malley, B.W. COUP transcription factor is a member of the steroid receptor superfamily. Nature 1989, 340, 163–166. [Google Scholar] [CrossRef]
- Mello, T.; Polvani, S.; Galli, A. Peroxisome proliferator-activated receptor and retinoic x receptor in alcoholic liver disease. PPAR Res. 2009, 2009, 748174. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Stroup, D.; Chiang, J.Y. HNF4 and COUP-TFII interact to modulate transcription of the cholesterol 7alpha-hydroxylase gene (CYP7A1). J. Lipid Res. 2000, 41, 1–11. [Google Scholar]
- Ktistaki, E.; Talianidis, I. Chicken ovalbumin upstream promoter transcription factors act as auxiliary cofactors for hepatocyte nuclear factor 4 and enhance hepatic gene expression. Mol. Cell. Biol. 1997, 17, 2790–2797. [Google Scholar] [CrossRef]
- Schaeffer, E.; Guillou, F.; Part, D.; Zakin, M.M. A different combination of transcription factors modulates the expression of the human transferrin promoter in liver and Sertoli cells. J. Biol. Chem. 1993, 268, 23399–23408. [Google Scholar]
- Yamazaki, T.; Suehiro, J.; Miyazaki, H.; Minami, T.; Kodama, T.; Miyazono, K.; Watabe, T. The COUP-TFII variant lacking a DNA-binding domain inhibits the activation of the Cyp7a1 promoter through physical interaction with COUP-TFII. Biochem. J. 2013, 452, 345–357. [Google Scholar] [CrossRef]
- Rosa, A.; Brivanlou, A.H. A regulatory circuitry comprised of miR-302 and the transcription factors OCT4 and NR2F2 regulates human embryonic stem cell differentiation. EMBO J. 2011, 30, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lambert, M.H.; Xu, H.E. Activation of nuclear receptors: A perspective from structural genomics. Struct. Lond. Engl. 1993 2003, 11, 741–746. [Google Scholar]
- Berry, M.; Metzger, D.; Chambon, P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J. 1990, 9, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Naka-Kaneda, H.; Nakamura, S.; Igarashi, M.; Aoi, H.; Kanki, H.; Tsuyama, J.; Tsutsumi, S.; Aburatani, H.; Shimazaki, T.; Okano, H. The miR-17/106-p38 axis is a key regulator of the neurogenic-to-gliogenic transition in developing neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1604–1609. [Google Scholar] [CrossRef]
- Xie, X.; Tsai, S.Y.; Tsai, M.-J. COUP-TFII regulates satellite cell function and muscular dystrophy. J. Clin. Investig. 2016, 126, 3929–3941. [Google Scholar] [CrossRef]
- Yu, C.-T.; Tang, K.; Suh, J.M.; Jiang, R.; Tsai, S.Y.; Tsai, M.-J. COUP-TFII is essential for metanephric mesenchyme formation and kidney precursor cell survival. Dev. Camb. Engl. 2012, 139, 2330–2339. [Google Scholar] [CrossRef]
- Lee, H.-J.; Kao, C.-Y.; Lin, S.-C.; Xu, M.; Xie, X.; Tsai, S.Y.; Tsai, M.-J. Dysregulation of nuclear receptor COUP-TFII impairs skeletal muscle development. Sci. Rep. 2017, 7, 3136. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Friedman, S.L. Stellate cells: A moving target in hepatic fibrogenesis. Hepatol. Baltim. Md 2004, 40, 1041–1043. [Google Scholar] [CrossRef]
- Apte, M.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cell: Physiologic role, role in fibrosis and cancer. Curr. Opin. Gastroenterol. 2015, 31, 416–423. [Google Scholar] [CrossRef]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Bencini, L.; Galli, A. Peroxisome proliferator activated receptors at the crossroad of obesity, diabetes, and pancreatic cancer. World J. Gastroenterol. 2016, 22, 2441–2459. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Transcriptional regulation of stellate cell activation. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. 3), S79–S83. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.-L.; Gabbiani, G. The Myofibroblast: One Function, Multiple Origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Polvani, S.; Vasseur-Cognet, M.; Tarocchi, M.; Tempesti, S.; Cavalieri, D.; Beltrame, L.; Marroncini, G.; Pinzani, M.; et al. The orphan nuclear receptor COUP-TFII coordinates hypoxia-independent proangiogenic responses in hepatic stellate cells. J. Hepatol. 2017, 66, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Qin, J.; Lin, S.-H.; Tsai, S.Y.; Tsai, M.-J. Nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) modulates mesenchymal cell commitment and differentiation. Proc. Natl. Acad. Sci. USA 2011, 108, 14843–14848. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-N.; Jang, W.-G.; Kim, E.-J.; Oh, S.-H.; Son, H.-J.; Kim, S.-H.; Franceschi, R.; Zhang, X.-K.; Lee, S.-E.; Koh, J.-T. Orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) protein negatively regulates bone morphogenetic protein 2-induced osteoblast differentiation through suppressing runt-related gene 2 (Runx2) activity. J. Biol. Chem. 2012, 287, 18888–18899. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.-C.; Kang, I.-H.; Hwang, Y.-C.; Kim, S.-H.; Koh, J.-T. MicroRNA-194 reciprocally stimulates osteogenesis and inhibits adipogenesis via regulating COUP-TFII expression. Cell Death Dis. 2014, 5, e1532. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.N.; Kim, J.-W.; Oh, S.-H.; Jeong, B.-C.; Hwang, Y.-C.; Koh, J.-T. FGF2 Stimulates COUP-TFII Expression via the MEK1/2 Pathway to Inhibit Osteoblast Differentiation in C3H10T1/2 Cells. PLoS ONE 2016, 11, e0159234. [Google Scholar] [CrossRef]
- Hu, S.; Wilson, K.D.; Ghosh, Z.; Han, L.; Wang, Y.; Lan, F.; Ransohoff, K.J.; Burridge, P.; Wu, J.C. MicroRNA-302 increases reprogramming efficiency via repression of NR2F2. Stem Cells Dayt. Ohio 2013, 31, 259–268. [Google Scholar] [CrossRef]
- Zhang, P.; Bennoun, M.; Gogard, C.; Bossard, P.; Leclerc, I.; Kahn, A.; Vasseur-Cognet, M. Expression of COUP-TFII in metabolic tissues during development. Mech. Dev. 2002, 119, 109–114. [Google Scholar] [CrossRef]
- Li, L.; Xie, X.; Qin, J.; Jeha, G.S.; Saha, P.K.; Yan, J.; Haueter, C.M.; Chan, L.; Tsai, S.Y.; Tsai, M.-J. The nuclear orphan receptor COUP-TFII plays an essential role in adipogenesis, glucose homeostasis, and energy metabolism. Cell Metab. 2009, 9, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Neuzillet, C.; Tijeras-Raballand, A.; Faivre, S.; de Gramont, A.; Raymond, E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 2015, 6, 16832–16847. [Google Scholar] [CrossRef] [PubMed]
- Pineda Torra, I.; Jamshidi, Y.; Flavell, D.M.; Fruchart, J.-C.; Staels, B. Characterization of the human PPARalpha promoter: Identification of a functional nuclear receptor response element. Mol. Endocrinol. Baltim. Md 2002, 16, 1013–1028. [Google Scholar]
- Boutant, M.; Ramos, O.H.P.; Lecoeur, C.; Vaillant, E.; Philippe, J.; Zhang, P.; Perilhou, A.; Valcarcel, B.; Sebert, S.; Jarvelin, M.-R.; et al. Glucose-dependent regulation of NR2F2 promoter and influence of SNP-rs3743462 on whole body insulin sensitivity. PLoS ONE 2012, 7, e35810. [Google Scholar] [CrossRef]
- De Martino, M.U.; Bhattachryya, N.; Alesci, S.; Ichijo, T.; Chrousos, G.P.; Kino, T. The glucocorticoid receptor and the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II interact with and mutually affect each other’s transcriptional activities: Implications for intermediary metabolism. Mol. Endocrinol. 2004, 18, 820–833. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.U.; Alesci, S.; Chrousos, G.P.; Kino, T. Interaction of the glucocorticoid receptor and the chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII): Implications for the actions of glucocorticoids on glucose, lipoprotein, and xenobiotic metabolism. Ann. N. Y. Acad. Sci. 2004, 1024, 72–85. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chiang, J.Y.L. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Perilhou, A.; Tourrel-Cuzin, C.; Kharroubi, I.; Henique, C.; Fauveau, V.; Kitamura, T.; Magnan, C.; Postic, C.; Prip-Buus, C.; Vasseur-Cognet, M. The transcription factor COUP-TFII is negatively regulated by insulin and glucose via Foxo1- and ChREBP-controlled pathways. Mol. Cell. Biol. 2008, 28, 6568–6579. [Google Scholar] [CrossRef]
- Planchais, J.; Boutant, M.; Fauveau, V.; Qing, L.D.; Sabra-Makke, L.; Bossard, P.; Vasseur-Cognet, M.; Pégorier, J.-P. The role of chicken ovalbumin upstream promoter transcription factor II in the regulation of hepatic fatty acid oxidation and gluconeogenesis in newborn mice. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E868–E878. [Google Scholar] [CrossRef]
- Li, R.; Zhang, R.; Li, Y.; Zhu, B.; Chen, W.; Zhang, Y.; Chen, G. A RARE of hepatic Gck promoter interacts with RARα, HNF4α and COUP-TFII that affect retinoic acid- and insulin-induced Gck expression. J. Nutr. Biochem. 2014, 25, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Hwung, Y.P.; Crowe, D.T.; Wang, L.H.; Tsai, S.Y.; Tsai, M.J. The COUP transcription factor binds to an upstream promoter element of the rat insulin II gene. Mol. Cell. Biol. 1988, 8, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Bardoux, P.; Zhang, P.; Flamez, D.; Perilhou, A.; Lavin, T.A.; Tanti, J.-F.; Hellemans, K.; Gomas, E.; Godard, C.; Andreelli, F.; et al. Essential role of chicken ovalbumin upstream promoter-transcription factor II in insulin secretion and insulin sensitivity revealed by conditional gene knockout. Diabetes 2005, 54, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Perilhou, A.; Tourrel-Cuzin, C.; Zhang, P.; Kharroubi, I.; Wang, H.; Fauveau, V.; Scott, D.K.; Wollheim, C.B.; Vasseur-Cognet, M. The MODY1 gene for hepatocyte nuclear factor 4alpha and a feedback loop control COUP-TFII expression in pancreatic beta cells. Mol. Cell. Biol. 2008, 28, 4588–4597. [Google Scholar] [CrossRef]
- Navas, M.A.; Munoz-Elias, E.J.; Kim, J.; Shih, D.; Stoffel, M. Functional characterization of the MODY1 gene mutations HNF4(R127W), HNF4(V255M), and HNF4(E276Q). Diabetes 1999, 48, 1459–1465. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Jun, H.-S. Anti-diabetic actions of glucagon-like peptide-1 on pancreatic beta-cells. Metabolism 2014, 63, 9–19. [Google Scholar] [CrossRef]
- Boutant, M.; Ramos, O.H.P.; Tourrel-Cuzin, C.; Movassat, J.; Ilias, A.; Vallois, D.; Planchais, J.; Pégorier, J.-P.; Schuit, F.; Petit, P.X.; et al. COUP-TFII controls mouse pancreatic β-cell mass through GLP-1-β-catenin signaling pathways. PLoS ONE 2012, 7, e30847. [Google Scholar] [CrossRef]
- Fuentealba, P.; Klausberger, T.; Karayannis, T.; Suen, W.Y.; Huck, J.; Tomioka, R.; Rockland, K.; Capogna, M.; Studer, M.; Morales, M.; et al. Expression of COUP-TFII nuclear receptor in restricted GABAergic neuronal populations in the adult rat hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 1595–1609. [Google Scholar] [CrossRef]
- Reinchisi, G.; Ijichi, K.; Glidden, N.; Jakovcevski, I.; Zecevic, N. COUP-TFII expressing interneurons in human fetal forebrain. Cereb. Cortex 2012, 22, 2820–2830. [Google Scholar] [CrossRef]
- Tang, K.; Rubenstein, J.L.R.; Tsai, S.Y.; Tsai, M.-J. COUP-TFII controls amygdala patterning by regulating neuropilin expression. Dev. Camb. Engl. 2012, 139, 1630–1639. [Google Scholar] [CrossRef]
- Sabra-Makke, L.; Tourrel-Cuzin, C.; Denis, R.G.P.; Moldes, M.; Pégorier, J.-P.; Luquet, S.; Vasseur-Cognet, M.; Bossard, P. The nutritional induction of COUP-TFII gene expression in ventromedial hypothalamic neurons is mediated by the melanocortin pathway. PLoS ONE 2010, 5, e13464. [Google Scholar] [CrossRef] [PubMed]
- Sabra-Makke, L.; Maritan, M.; Planchais, J.; Boutant, M.; Pégorier, J.-P.; Even, P.C.; Vasseur-Cognet, M.; Bossard, P. Hypothalamic ventromedial COUP-TFII protects against hypoglycemia-associated autonomic failure. Proc. Natl. Acad. Sci. USA 2013, 110, 4333–4338. [Google Scholar] [CrossRef] [PubMed]
- Patan, S. Vasculogenesis and angiogenesis. Cancer Treat. Res. 2004, 117, 3–32. [Google Scholar] [PubMed]
- Fang, J.; Hirschi, K. Molecular regulation of arteriovenous endothelial cell specification. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Hu, H.; Isaji, T.; Dardik, A. Molecular identity of arteries, veins, and lymphatics. J. Vasc. Surg. 2019, 69, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Sancho, R.; Cremona, C.A.; Behrens, A. Stem cell and progenitor fate in the mammalian intestine: Notch and lateral inhibition in homeostasis and disease. EMBO Rep. 2015, 16, 571–581. [Google Scholar] [CrossRef]
- Demitrack, E.S.; Samuelson, L.C. Notch regulation of gastrointestinal stem cells. J. Physiol. 2016, 594, 4791–4803. [Google Scholar] [CrossRef]
- Espinoza, I.; Miele, L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013, 341, 41–45. [Google Scholar] [CrossRef]
- Park, J.K.; Lee, T.W.; Do, E.K.; Moon, H.J.; Kim, J.H. Role of Notch1 in the arterial specification and angiogenic potential of mouse embryonic stem cell-derived endothelial cells. Stem Cell Res. Ther. 2018, 9, 197. [Google Scholar] [CrossRef]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar]
- Hrabĕ de Angelis, M.; McIntyre, J.; Gossler, A. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature 1997, 386, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Chen, X.; Xie, X.; Tsai, M.-J.; Tsai, S.Y. COUP-TFII regulates tumor growth and metastasis by modulating tumor angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 3687–3692. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Fujita, H.; Nakano, A.; Kang, M.; Duarte, A.; Kume, T. The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev. Biol. 2006, 294, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-J.; Chen, X.; Qin, J.; Hong, Y.-K.; Tsai, M.-J.; Tsai, S.Y. Direct transcriptional regulation of neuropilin-2 by COUP-TFII modulates multiple steps in murine lymphatic vessel development. J. Clin. Investig. 2010, 120, 1694–1707. [Google Scholar] [CrossRef]
- Nagasaki, S.; Suzuki, T.; Miki, Y.; Akahira, J.; Shibata, H.; Ishida, T.; Ohuchi, N.; Sasano, H. Chicken ovalbumin upstream promoter transcription factor II in human breast carcinoma: Possible regulator of lymphangiogenesis via vascular endothelial growth factor-C expression. Cancer Sci. 2009, 100, 639–645. [Google Scholar] [CrossRef]
- Davis, R.B.; Curtis, C.D.; Griffin, C.T. BRG1 promotes COUP-TFII expression and venous specification during embryonic vascular. Development 2013, 140, 1272–1281. [Google Scholar] [CrossRef]
- Takada, Y.; Fukuda, A.; Chiba, T.; Seno, H. Brg1 plays an essential role in development and homeostasis of the duodenum through regulation of Notch signaling. Dev. Camb. Engl. 2016, 143, 3532–3539. [Google Scholar]
- Zhang, X.; Lu, Y.; Wang, J.; He, N. Overexpression of Brg1 alleviates high glucose-induced retinal ganglion cell apoptosis though regulating Notch/Hes1 signaling. Biochem. Biophys. Res. Commun. 2019, 514, 1160–1166. [Google Scholar] [CrossRef]
- Petit, F.G.; Salas, R.; Tsai, M.-J.; Tsai, S.Y. The regulation of COUP-TFII gene expression by Ets-1 is enhanced by the steroid receptor co-activators. Mech. Ageing Dev. 2004, 125, 719–732. [Google Scholar] [CrossRef]
- Chen, X.; Qin, J.; Cheng, C.-M.; Tsai, M.-J.; Tsai, S.Y. COUP-TFII is a major regulator of cell cycle and Notch signaling pathways. Mol. Endocrinol. 2012, 26, 1268–1277. [Google Scholar] [CrossRef]
- You, L.-R.; Lin, F.-J.; Lee, C.T.; DeMayo, F.J.; Tsai, M.-J.; Tsai, S.Y. Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 2005, 435, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Hu, H.; Guo, J.; Ige, M.; Wang, T.; Isaji, T.; Kudze, T.; Liu, H.; Yatsula, B.; Hashimoto, T.; et al. Polyester vascular patches acquire arterial or venous identity depending on their environment. J. Biomed. Mater. Res. A 2017, 105, 3422–3431. [Google Scholar] [CrossRef] [PubMed]
- Diez, H.; Fischer, A.; Winkler, A.; Hu, C.-J.; Hatzopoulos, A.K.; Breier, G.; Gessler, M. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp. Cell Res. 2007, 313, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.A.; Yamakuchi, M.; Kondo, M.; Oettgen, P.; Lowenstein, C.J. Ets-1 and Ets-2 regulate the expression of microRNA-126 in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, G.M.; Pitarresi, J.R.; Balakrishnan, S.; Ostrowski, M.C. The ETS family of oncogenic transcription factors in solid tumours. Nat. Rev. Cancer 2017, 17, 337–351. [Google Scholar] [CrossRef]
- Furlan, A.; Vercamer, C.; Heliot, L.; Wernert, N.; Desbiens, X.; Pourtier, A. Ets-1 drives breast cancer cell angiogenic potential and interactions between breast cancer and endothelial cells. Int. J. Oncol. 2018, 54, 29–40. [Google Scholar] [CrossRef]
- van Impel, A.; Zhao, Z.; Hermkens, D.M.A.; Roukens, M.G.; Fischer, J.C.; Peterson-Maduro, J.; Duckers, H.; Ober, E.A.; Ingham, P.W.; Schulte-Merker, S. Divergence of zebrafish and mouse lymphatic cell fate specification pathways. Dev. Camb. Engl. 2014, 141, 1228–1238. [Google Scholar] [CrossRef]
- Srinivasan, R.S.; Geng, X.; Yang, Y.; Wang, Y.; Mukatira, S.; Studer, M.; Porto, M.P.R.; Lagutin, O.; Oliver, G. The nuclear hormone receptor Coup-TFII is required for the initiation and early maintenance of Prox1 expression in lymphatic endothelial cells. Genes Dev. 2010, 24, 696–707. [Google Scholar] [CrossRef]
- Aranguren, X.L.; Beerens, M.; Vandevelde, W.; Dewerchin, M.; Carmeliet, P.; Luttun, A. Transcription factor COUP-TFII is indispensable for venous and lymphatic development in zebrafish and Xenopus laevis. Biochem. Biophys. Res. Commun. 2011, 410, 121–126. [Google Scholar] [CrossRef]
- Jha, S.K.; Rauniyar, K.; Jeltsch, M. Key molecules in lymphatic development, function, and identification. Ann. Anat. Anat. Anz. Off. Organ Anat. Ges. 2018, 219, 25–34. [Google Scholar] [CrossRef]
- Yamazaki, T.; Yoshimatsu, Y.; Morishita, Y.; Miyazono, K.; Watabe, T. COUP-TFII regulates the functions of Prox1 in lymphatic endothelial cells through direct interaction. Genes Cells Devoted Mol. Cell. Mech. 2009, 14, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Yoo, J.; Lee, S.; Tang, W.; Aguilar, B.; Ramu, S.; Choi, I.; Otu, H.H.; Shin, J.W.; Dotto, G.P.; et al. An exquisite cross-control mechanism among endothelial cell fate regulators directs the plasticity and heterogeneity of lymphatic endothelial cells. Blood 2010, 116, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Geenen, I.L.A.; Molin, D.G.M.; van den Akker, N.M.S.; Jeukens, F.; Spronk, H.M.; Schurink, G.W.H.; Post, M.J. Endothelial cells (ECs) for vascular tissue engineering: Venous ECs are less thrombogenic than arterial ECs. J. Tissue Eng. Regen. Med. 2015, 9, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Lu, Y.W.; Lee, V.; Kim, D.; Dorsey, T.; Wang, Q.; Lee, Y.; Vincent, P.; Schwarz, J.; Dai, G. Venous Endothelial Marker COUP-TFII Regulates the Distinct Pathologic Potentials of Adult Arteries and Veins. Sci. Rep. 2015, 5, 16193. [Google Scholar] [CrossRef]
- Wu, X.; Zou, Y.; Liang, Y.; Zhou, Q.; Gong, H.; Sun, A.; Yuan, L.; Wang, K.; Ge, J. COUP-TFII switches responses of venous endothelium to atherosclerotic factors through controlling the profile of various inherent genes expression. J. Cell. Biochem. 2011, 112, 256–264. [Google Scholar] [CrossRef]
- Lehoux, S.; Jones, E.A. Shear stress, arterial identity and atherosclerosis. Thromb. Haemost. 2015, 115, 467–473. [Google Scholar]
- Nakamura, E.; Makita, Y.; Okamoto, T.; Nagaya, K.; Hayashi, T.; Sugimoto, M.; Manabe, H.; Taketazu, G.; Kajino, H.; Fujieda, K. 5.78 Mb terminal deletion of chromosome 15q in a girl, evaluation of NR2F2 as candidate gene for congenital heart defects. Eur. J. Med. Genet. 2011, 54, 354–356. [Google Scholar] [CrossRef]
- Al Turki, S.; Manickaraj, A.K.; Mercer, C.L.; Gerety, S.S.; Hitz, M.-P.; Lindsay, S.; D’Alessandro, L.C.A.; Swaminathan, G.J.; Bentham, J.; Arndt, A.-K.; et al. Rare Variants in NR2F2 Cause Congenital Heart Defects in Humans. Am. J. Hum. Genet. 2014, 94, 574–585. [Google Scholar] [CrossRef]
- Lyu, G.; Zhang, C.; Ling, T.; Liu, R.; Zong, L.; Guan, Y.; Huang, X.; Sun, L.; Zhang, L.; Li, C.; et al. Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genom. 2018, 19, 428. [Google Scholar] [CrossRef]
- Qiao, X.-H.; Wang, Q.; Wang, J.; Liu, X.-Y.; Xu, Y.-J.; Huang, R.-T.; Xue, S.; Li, Y.-J.; Zhang, M.; Qu, X.-K.; et al. A novel NR2F2 loss-of-function mutation predisposes to congenital heart defect. Eur. J. Med. Genet. 2017. [Google Scholar] [CrossRef]
- Upadia, J.; Gonzales, P.R.; Robin, N.H. Novel de novo pathogenic variant in the NR2F2 gene in a boy with congenital heart defect and dysmorphic features. Am. J. Med. Genet. A 2018, 176, 1423–1426. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Abhinav, P.; Xu, Y.-J.; Li, R.-G.; Zhang, M.; Qiu, X.-B.; Di, R.-M.; Qiao, Q.; Li, X.-M.; Huang, R.-T.; et al. NR2F2 loss-of-function mutation is responsible for congenital bicuspid aortic valve. Int. J. Mol. Med. 2019, 43, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.; Girijamma, A.; Nair, R.; Ramegowda, K.S.; Nair, D.H.; Thulaseedharan, J.V.; Lakkappa, R.B.; Kamalapurkar, G.; Kartha, C.C. Forkhead box C2 promoter variant c.-512C>T is associated with increased susceptibility to chronic venous diseases. PLoS ONE 2014, 9, e90682. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Redondo, V.; Pettersson, A.T.; Ruas, J.L. The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 2015, 58, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Brunssen, C.; Korten, S.; Brux, M.; Seifert, S.; Roesler, J.; Bornstein, S.R.; Morawietz, H.; Goettsch, W. COUP-TFII is regulated by high glucose in endothelial cells. Horm. Metab. Res. Horm. Stoffwechselforschung Horm. Metab. 2010, 42, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Galli, A. PPARγ and Oxidative Stress: Con(β) Catenating NRF2 and FOXO. PPAR Res. 2012, 2012, 641087. [Google Scholar] [CrossRef] [PubMed]
- Balkau, B. [An epidemiologic survey from a network of French Health Examination Centres, (D.E.S.I.R.): Epidemiologic data on the insulin resistance syndrome]. Rev. Epidemiol. Sante Publique 1996, 44, 373–375. [Google Scholar]
- Shoemaker, L.D.; Fuentes, L.F.; Santiago, S.M.; Allen, B.M.; Cook, D.J.; Steinberg, G.K.; Chang, S.D. Human brain arteriovenous malformations express lymphatic-associated genes. Ann. Clin. Transl. Neurol. 2014, 1, 982–995. [Google Scholar] [CrossRef]
- Thomas, J.M.; Surendran, S.; Abraham, M.; Sasankan, D.; Bhaadri, S.; Rajavelu, A.; Kartha, C.C. Gene expression analysis of nidus of cerebral arteriovenous malformations reveals vascular structures with deficient differentiation and maturation. PLoS ONE 2018, 13, e0198617. [Google Scholar] [CrossRef]
- Mancini, M.L.; Terzic, A.; Conley, B.A.; Oxburgh, L.H.; Nicola, T.; Vary, C.P.H. Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 2479–2493. [Google Scholar] [CrossRef]
- Kumarasamy, S.; Waghulde, H.; Gopalakrishnan, K.; Mell, B.; Morgan, E.; Joe, B. Mutation within the hinge region of the transcription factor Nr2f2 attenuates salt-sensitive hypertension. Nat. Commun. 2015, 6, 6252. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Tsai, M.-J.; Tsai, S.Y. Essential roles of COUP-TFII in Leydig cell differentiation and male fertility. PLoS ONE 2008, 3, e3285. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Y.; Ge, R.; Zirkin, B.R. Leydig cell stem cells: Identification, proliferation and differentiation. Mol. Cell. Endocrinol. 2017, 445, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Villarroel, R.E.; Di-Luoffo, M.; Camiré, E.; Giner, X.C.; Brousseau, C.; Tremblay, J.J. The Insl3 gene is a direct target for the orphan nuclear receptor COUP-TFII in Leydig cells. J. Mol. Endocrinol. 2014, 53, 43–55. [Google Scholar] [CrossRef]
- Mendoza-Villarroel, R.E.; Robert, N.M.; Martin, L.J.; Brousseau, C.; Tremblay, J.J. The nuclear receptor NR2F2 activates star expression and steroidogenesis in mouse MA-10 and MLTC-1 Leydig cells. Biol. Reprod. 2014, 91, 26. [Google Scholar] [CrossRef]
- Kawamura, K.; Kumagai, J.; Sudo, S.; Chun, S.-Y.; Pisarska, M.; Morita, H.; Toppari, J.; Fu, P.; Wade, J.D.; Bathgate, R.A.D.; et al. Paracrine regulation of mammalian oocyte maturation and male germ cell survival. Proc. Natl. Acad. Sci. USA 2004, 101, 7323–7328. [Google Scholar] [CrossRef]
- Lottrup, G.; Nielsen, J.E.; Maroun, L.L.; Møller, L.M.A.; Yassin, M.; Leffers, H.; Skakkebæk, N.E.; Rajpert-De Meyts, E. Expression patterns of DLK1 and INSL3 identify stages of Leydig cell differentiation during normal development and in testicular pathologies, including testicular cancer and Klinefelter syndrome. Hum. Reprod. Oxf. Engl. 2014, 29, 1637–1650. [Google Scholar] [CrossRef]
- Carvalheira, G.; Malinverni, A.M.; Moysés-Oliveira, M.; Ueta, R.; Cardili, L.; Monteagudo, P.; Mathez, A.L.G.; Verreschi, I.T.; Maluf, M.A.; Shida, M.E.F.; et al. The Natural History of a Man With Ovotesticular 46,XX DSD Caused by a Novel 3-Mb 15q26.2 Deletion Containing NR2F2 Gene. J. Endocr. Soc. 2019, 3, 2107–2113. [Google Scholar] [CrossRef]
- Ferrero, S.; Remorgida, V.; Maganza, C.; Venturini, P.L.; Salvatore, S.; Papaleo, E.; Candiani, M.; Leone Roberti Maggiore, U. Aromatase and endometriosis: Estrogens play a role. Ann. N. Y. Acad. Sci. 2014, 1317, 17–23. [Google Scholar] [CrossRef]
- Zeitoun, K.; Takayama, K.; Michael, M.D.; Bulun, S.E. Stimulation of aromatase P450 promoter (II) activity in endometriosis and its inhibition in endometrium are regulated by competitive binding of steroidogenic factor-1 and chicken ovalbumin upstream promoter transcription factor to the same cis-acting element. Mol. Endocrinol. 1999, 13, 239–253. [Google Scholar]
- Li, X.; Large, M.J.; Creighton, C.J.; Lanz, R.B.; Jeong, J.-W.; Young, S.L.; Lessey, B.A.; Palomino, W.A.; Tsai, S.Y.; Demayo, F.J. COUP-TFII regulates human endometrial stromal genes involved in inflammation. Mol. Endocrinol. 2013, 27, 2041–2054. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, Y.M.; Wu, S.-P.; Anderson, M.L.; Hawkins, S.M.; Creighton, C.J.; Ray, M.; Tsai, S.Y.; Tsai, M.-J.; Lydon, J.P.; DeMayo, F.J. Endometrial Expression of Steroidogenic Factor 1 Promotes Cystic Glandular Morphogenesis. Mol. Endocrinol. 2016, 30, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Li, Y.-H.; Wu, M.-H.; Chang, Y.-F.; Lee, D.-K.; Tsai, S.Y.; Tsai, M.-J.; Tsai, S.-J. Suppression of COUP-TFII by proinflammatory cytokines contributes to the pathogenesis of endometriosis. J. Clin. Endocrinol. Metab. 2014, 99, E427–E437. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.-L.; Hsiao, K.-Y.; Lee, H.-C.; Li, W.-N.; Chang, N.; Wu, M.-H.; Tsai, S.-J. Suppression of COUP-TFII upregulates angiogenin and promotes angiogenesis in endometriosis. Hum. Reprod. Oxf. Engl. 2018, 33, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, I.; Lee, D.-K.; Petit, F.G.; Jeong, J.; Lee, K.; Lydon, J.P.; DeMayo, F.J.; Tsai, M.-J.; Tsai, S.Y. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007, 3, e102. [Google Scholar] [CrossRef] [PubMed]
- Petit, F.G.; Jamin, S.P.; Kurihara, I.; Behringer, R.R.; DeMayo, F.J.; Tsai, M.-J.; Tsai, S.Y. Deletion of the orphan nuclear receptor COUP-TFII in uterus leads to placental deficiency. Proc. Natl. Acad. Sci. USA 2007, 104, 6293–6298. [Google Scholar] [CrossRef]
- You, L.-R.; Takamoto, N.; Yu, C.-T.; Tanaka, T.; Kodama, T.; Demayo, F.J.; Tsai, S.Y.; Tsai, M.-J. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia. Proc. Natl. Acad. Sci. USA 2005, 102, 16351–16356. [Google Scholar] [CrossRef]
- High, F.A.; Bhayani, P.; Wilson, J.M.; Bult, C.J.; Donahoe, P.K.; Longoni, M. De novo frameshift mutation in COUP-TFII (NR2F2) in human congenital diaphragmatic hernia. Am. J. Med. Genet. A. 2016, 170, 2457–2461. [Google Scholar] [CrossRef]
- van den Brink, G.R.; Hardwick, J.C.H.; Nielsen, C.; Xu, C.; ten Kate, F.J.; Glickman, J.; van Deventer, S.J.H.; Roberts, D.J.; Peppelenbosch, M.P. Sonic hedgehog expression correlates with fundic gland differentiation in the adult gastrointestinal tract. Gut 2002, 51, 628–633. [Google Scholar] [CrossRef]
- Shiotani, A.; Kamada, T.; Yamanaka, Y.; Manabe, N.; Kusunoki, H.; Hata, J.; Haruma, K. Sonic hedgehog and CDX2 expression in the stomach. J. Gastroenterol. Hepatol. 2008, 23 (Suppl. 2), S161–S166. [Google Scholar] [CrossRef]
- Lees, C.; Howie, S.; Sartor, R.B.; Satsangi, J. The hedgehog signalling pathway in the gastrointestinal tract: Implications for development, homeostasis, and disease. Gastroenterology 2005, 129, 1696–1710. [Google Scholar] [CrossRef]
- Takamoto, N.; You, L.-R.; Moses, K.; Chiang, C.; Zimmer, W.E.; Schwartz, R.J.; DeMayo, F.J.; Tsai, M.-J.; Tsai, S.Y. COUP-TFII is essential for radial and anteroposterior patterning of the stomach. Dev. Camb. Engl. 2005, 132, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Elberg, G.; Tsai, M.J.; Tsai, S.Y. Identification of a novel sonic hedgehog response element in the chicken ovalbumin upstream promoter-transcription factor II promoter. Mol. Endocrinol. 1997, 11, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Todisco, A. Regulation of Gastric Metaplasia, Dysplasia, and Neoplasia by Bone Morphogenetic Protein Signaling. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 339–347. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shinohara, M.; Mao, M.; Keeley, T.M.; El-Zaatari, M.; Lee, H.-J.; Eaton, K.A.; Samuelson, L.C.; Merchant, J.L.; Goldenring, J.R.; Todisco, A. Bone morphogenetic protein signaling regulates gastric epithelial cell development and proliferation in mice. Gastroenterology 2010, 139, 2050–2060.e2. [Google Scholar] [CrossRef] [PubMed]
- Holbeck, S.; Chang, J.; Best, A.M.; Bookout, A.L.; Mangelsdorf, D.J.; Martinez, E.D. Expression profiling of nuclear receptors in the NCI60 cancer cell panel reveals receptor-drug and receptor-gene interactions. Mol. Endocrinol. 2010, 24, 1287–1296. [Google Scholar] [CrossRef]
- Shin, S.-W.; Kwon, H.-C.; Rho, M.-S.; Choi, H.-J.; Kwak, J.-Y.; Park, J.-I. Clinical significance of chicken ovalbumin upstream promoter-transcription factor II expression in human colorectal cancer. Oncol. Rep. 2009, 21, 101–106. [Google Scholar]
- Yun, S.-H.; Park, M.-G.; Kim, Y.-M.; Roh, M.-S.; Park, J.-I. Expression of chicken ovalbumin upstream promoter-transcription factor II and liver X receptor as prognostic indicators for human colorectal cancer. Oncol. Lett. 2017, 14, 4011–4020. [Google Scholar] [CrossRef]
- Bao, Y.; Gu, D.; Feng, W.; Sun, X.; Wang, X.; Zhang, X.; Shi, Q.; Cui, G.; Yu, H.; Tang, C.; et al. COUP-TFII regulates metastasis of colorectal adenocarcinoma cells by modulating Snail1. Br. J. Cancer 2014, 111, 933–943. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, Y.; Ruan, R.; Zheng, M.; Han, W.; Liao, L. High expression of COUP-TF II cooperated with negative Smad4 expression predicts poor prognosis in patients with colorectal cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 7112–7121. [Google Scholar]
- Wang, H.; Nie, L.; Wu, L.; Liu, Q.; Guo, X. NR2F2 inhibits Smad7 expression and promotes TGF-β-dependent epithelial-mesenchymal transition of CRC via transactivation of miR-21. Biochem. Biophys. Res. Commun. 2017, 485, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Lu, Y.; Feng, W.; Yu, H.; Guo, H.; Tao, Y.; Shi, Q.; Chen, W.; Wang, X. COUP-TFII promotes epithelial-mesenchymal transition by inhibiting miR-34a expression in colorectal cancer. Int. J. Oncol. 2019, 54, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Mello, T.; Ceni, E.; Buccoliero, F.; D’Amico, M.; Boddi, V.; Farsi, M.; Nesi, S.; et al. COUP-TFII in pancreatic adenocarcinoma: Clinical implication for patient survival and tumor progression. Int. J. Cancer 2014, 134, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.F.; Nam, K.T.; Petersen, C.P.; Lee, H.-J.; Yang, H.-K.; Kim, W.H.; Goldenring, J.R. miR-30-HNF4γ and miR-194-NR2F2 regulatory networks contribute to the upregulation of metaplasia markers in the stomach. Gut 2016, 65, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Zhang, Y.; Cai, H.; Liu, G.; Ye, Y.; Xu, G.; Wang, H.; Xiong, D.; Zhang, C.; Huang, Z.; et al. Overexpression of COUP-TFII suppresses proliferation and metastasis of human gastric cancer cells. Mol. Med. Rep. 2018, 17, 2393–2401. [Google Scholar] [CrossRef]
- Feng, Q.; Wu, X.; Li, F.; Ning, B.; Lu, X.; Zhang, Y.; Pan, Y.; Guan, W. miR-27b inhibits gastric cancer metastasis by targeting NR2F2. Protein Cell 2017, 8, 114–122. [Google Scholar] [CrossRef]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of prognostic biomarkers in prostate cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Cao, X.; Wang, L.; Dhanasekaran, S.M.; Kalyana-Sundaram, S.; Wei, J.T.; Rubin, M.A.; Pienta, K.J.; et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007, 39, 41–51. [Google Scholar] [CrossRef]
- Qin, J.; Wu, S.-P.; Creighton, C.J.; Dai, F.; Xie, X.; Cheng, C.-M.; Frolov, A.; Ayala, G.; Lin, X.; Feng, X.-H.; et al. COUP-TFII inhibits TGF-β-induced growth barrier to promote prostate tumorigenesis. Nature 2013, 493, 236–240. [Google Scholar] [CrossRef]
- Lilis, I.; Giopanou, I.; Papadaki, H.; Gyftopoulos, K. The expression of p-mTOR and COUP-TFII correlates with increased lymphangiogenesis and lymph node metastasis in prostate adenocarcinoma. Urol. Oncol. 2018, 36, 311.e27–311.e35. [Google Scholar] [CrossRef]
- Lin, S.-C.; Kao, C.-Y.; Lee, H.-J.; Creighton, C.J.; Ittmann, M.M.; Tsai, S.-J.; Tsai, S.Y.; Tsai, M.-J. Dysregulation of miRNAs-COUP-TFII-FOXM1-CENPF axis contributes to the metastasis of prostate cancer. Nat. Commun. 2016, 7, 11418. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, M.; Qin, J.; Lin, S.-C.; Lee, H.-J.; Tsai, S.Y.; Tsai, M.-J. MPC1, a key gene in cancer metabolism, is regulated by COUPTFII in human prostate cancer. Oncotarget 2016, 7, 14673–14683. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Qin, W.; Jiao, D.; Ren, J.; Wei, M.; Shi, S.; Xi, W.; Wang, H.; Yang, A.-G.; Huan, Y.; et al. Knockdown of COUP-TFII inhibits cell proliferation and induces apoptosis through upregulating BRCA1 in renal cell carcinoma cells. Int. J. Cancer 2016, 139, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Liu, C.-X.; Zeng, X.-R.; Huang, X.-M.; Chen, W.-L.; Wang, Y.; Ai, F. Orphan nuclear receptor COUP-TFII is an oncogenic gene in renal cell carcinoma. Clin. Transl. Oncol. 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, L.M.; Riggs, K.A.; Hockenberry, A.M.; Oliver, L.D.; Barnhart, K.G.; Cai, J.; Pierce, W.M.; Ivanova, M.M.; Bates, P.J.; Appana, S.N.; et al. Identification and characterization of nucleolin as a COUP-TFII coactivator of retinoic acid receptor β transcription in breast cancer cells. PLoS ONE 2012, 7, e38278. [Google Scholar] [CrossRef]
- Al-Rayyan, N.; Litchfield, L.M.; Ivanova, M.M.; Radde, B.N.; Cheng, A.; Elbedewy, A.; Klinge, C.M. 5-aza-2-deoxycytidine and Trichostatin A increase COUP-TFII expression in antiestrogen-resistant breast cancer cell lines. Cancer Lett. 2014, 347, 139–150. [Google Scholar] [CrossRef][Green Version]
- Jiang, G.; Wang, X.; Sheng, D.; Zhou, L.; Liu, Y.; Xu, C.; Liu, S.; Zhang, J. Cooperativity of co-factor NR2F2 with Pioneer Factors GATA3, FOXA1 in promoting ERα function. Theranostics 2019, 9, 6501–6516. [Google Scholar] [CrossRef]
- Muscat, G.E.O.; Eriksson, N.A.; Byth, K.; Loi, S.; Graham, D.; Jindal, S.; Davis, M.J.; Clyne, C.; Funder, J.W.; Simpson, E.R.; et al. Research resource: Nuclear receptors as transcriptome: Discriminant and prognostic value in breast cancer. Mol. Endocrinol. 2013, 27, 350–365. [Google Scholar] [CrossRef]
- Zhang, C.; Han, Y.; Huang, H.; Qu, L.; Shou, C. High NR2F2 transcript level is associated with increased survival and its expression inhibits TGF-β-dependent epithelial-mesenchymal transition in breast cancer. Breast Cancer Res. Treat. 2014, 147, 265–281. [Google Scholar] [CrossRef]
- Nakshatri, H.; Mendonca, M.S.; Bhat-Nakshatri, P.; Patel, N.M.; Goulet, R.J.; Cornetta, K. The orphan receptor COUP-TFII regulates G2/M progression of breast cancer cells by modulating the expression/activity of p21(WAF1/CIP1), cyclin D1, and cdk2. Biochem. Biophys. Res. Commun. 2000, 270, 1144–1153. [Google Scholar] [CrossRef]
- Litchfield, L.M.; Appana, S.N.; Datta, S.; Klinge, C.M. COUP-TFII inhibits NFkappaB activation in endocrine-resistant breast cancer cells. Mol. Cell. Endocrinol. 2014, 382, 358–367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prahalad, P.; Dakshanamurthy, S.; Ressom, H.; Byers, S.W. Retinoic acid mediates regulation of network formation by COUP-TFII and VE-cadherin expression by TGFbeta receptor kinase in breast cancer cells. PLoS ONE 2010, 5, e10023. [Google Scholar] [CrossRef] [PubMed]
- Brzozowa, M.; Michalski, M.; Wyrobiec, G.; Piecuch, A.; Dittfeld, A.; Harabin-Słowińska, M.; Boroń, D.; Wojnicz, R. The role of Snail1 transcription factor in colorectal cancer progression and metastasis. Contemp. Oncol. 2015, 19, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Freihen, V.; Rönsch, K.; Mastroianni, J.; Frey, P.; Rose, K.; Boerries, M.; Zeiser, R.; Busch, H.; Hecht, A. SNAIL1 employs β-Catenin-LEF1 complexes to control colorectal cancer cell invasion and proliferation. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Zhang, L.; Ye, Y.; Long, X.; Xiao, P.; Ren, X.; Yu, J. BMP signaling and its paradoxical effects in tumorigenesis and dissemination. Oncotarget 2016, 7, 78206. [Google Scholar] [CrossRef]
- Zhou, B.; Song, J.; Han, T.; Huang, M.; Jiang, H.; Qiao, H.; Shi, J.; Wang, Y. MiR-382 inhibits cell growth and invasion by targeting NR2F2 in colorectal cancer. Mol. Carcinog. 2016, 55, 2260–2267. [Google Scholar] [CrossRef]
- Chen, T.; Ren, H.; Thakur, A.; Yang, T.; Li, Y.; Zhang, S.; Wang, T.; Chen, M. miR-382 inhibits tumor progression by targeting SETD8 in non-small cell lung cancer. Biomed. Pharmacother. 2017, 86, 248–253. [Google Scholar] [CrossRef]
- Feng, J.; Qi, B.; Guo, L.; Chen, L.-Y.; Wei, X.-F.; Liu, Y.-Z.; Zhao, B.-S. miR-382 functions as a tumor suppressor against esophageal squamous cell carcinoma. World J. Gastroenterol. 2017, 23, 4243–4251. [Google Scholar] [CrossRef]
- Tan, H.; He, Q.; Gong, G.; Wang, Y.; Li, J.; Wang, J.; Zhu, D.; Wu, X. miR-382 inhibits migration and invasion by targeting ROR1 through regulating EMT in ovarian cancer. Int. J. Oncol. 2016, 48, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, J.; Qiu, J.; Fu, X.; Tang, Q.; Yang, F.; Zhao, Z.; Wang, H. MicroRNA-382 inhibits prostate cancer cell proliferation and metastasis through targeting COUP-TFII. Oncol. Rep. 2016, 36, 3707–3715. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, H.; Jiang, P. MicroRNA-382 inhibits cell growth and migration in colorectal cancer by targeting SP1. Biol. Res. 2018, 51, 51. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Xia, D.; Li, Z.-L.; Ren, L.; Wang, M.-M.; Chen, W.-S.; Hu, Z.-C.; Yi, G.-P.; Xu, L. MiR-382 functions as tumor suppressor and chemosensitizer in colorectal cancer. Biosci. Rep. 2019, 39, BSR20180441. [Google Scholar] [CrossRef]
- Zhang, D.; Qiu, X.; Li, J.; Zheng, S.; Li, L.; Zhao, H. TGF-β secreted by tumor-associated macrophages promotes proliferation and invasion of colorectal cancer via miR-34a-VEGF axis. Cell Cycle Georget. Tex 2018, 17, 2766–2778. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Chen, J.-J.; Lv, Q.; Qin, J.; Huang, Y.-Z.; Yu, M.-H.; Zhong, M. Long non-coding RNA NEAT1 promotes colorectal cancer progression by competitively binding miR-34a with SIRT1 and enhancing the Wnt/β-catenin signaling pathway. Cancer Lett. 2019, 440–441, 11–22. [Google Scholar] [CrossRef]
- Li, Y.; Gong, P.; Hou, J.-X.; Huang, W.; Ma, X.-P.; Wang, Y.-L.; Li, J.; Cui, X.-B.; Li, N. miR-34a Regulates Multidrug Resistance via Positively Modulating OAZ2 Signaling in Colon Cancer Cells. J. Immunol. Res. 2018, 2018, 7498514. [Google Scholar] [CrossRef]
- Wu, Q.-B.; Sheng, X.; Zhang, N.; Yang, M.-W.; Wang, F. Role of microRNAs in the resistance of colorectal cancer to chemoradiotherapy. Mol. Clin. Oncol. 2018, 8, 528–532. [Google Scholar]
- Suzuki, T.; Moriya, T.; Darnel, A.D.; Takeyama, J.; Sasano, H. Immunohistochemical distribution of chicken ovalbumin upstream promoter transcription factor II in human tissues. Mol. Cell. Endocrinol. 2000, 164, 69–75. [Google Scholar] [CrossRef]
- Moré, E.; Fellner, T.; Doppelmayr, H.; Hauser-Kronberger, C.; Dandachi, N.; Obrist, P.; Sandhofer, F.; Paulweber, B. Activation of the MAP kinase pathway induces chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) expression in human breast cancer cell lines. J. Endocrinol. 2003, 176, 83–94. [Google Scholar] [CrossRef]
- Erdős, E.; Bálint, B.L. COUP-TFII is a modulator of cell-type-specific genetic programs based on genomic localization maps. J. Biotechnol. 2019, 301, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cortés, M.; Delgado-Bellido, D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 2019, 9, 803. [Google Scholar]
- Ding, Z.; Wu, C.-J.; Chu, G.C.; Xiao, Y.; Ho, D.; Zhang, J.; Perry, S.R.; Labrot, E.S.; Wu, X.; Lis, R.; et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011, 470, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Aranguren, X.L.; Beerens, M.; Coppiello, G.; Wiese, C.; Vandersmissen, I.; Lo Nigro, A.; Verfaillie, C.M.; Gessler, M.; Luttun, A. COUP-TFII orchestrates venous and lymphatic endothelial identity by homo- or hetero-dimerisation with PROX1. J. Cell Sci. 2013, 126, 1164–1175. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Role | Ref. | Effects |
|---|---|---|---|
| Colorectal cancer | Onco-suppressor (?) | [157,158] | ↑survival rate |
| Oncogene (?) | [159,160,161,162] | ↑risk of metastasis | |
| ↓overall survival | |||
| ↑invasion | |||
| ↑EMT | |||
| Pancreatic cancer | Oncogene | [163] | ↓survival rate |
| ↑invasiveness | |||
| ↑anchorage-independent cells growth | |||
| ↑neoangiogenesis | |||
| Gastric cancer | Onco-suppressor (?) | [164,165] | ↓proliferation |
| ↓migration | |||
| ↓invasiveness | |||
| ↓cell growth | |||
| ↓metastasis | |||
| ↑survival rate | |||
| Oncogene (?) | [166] | negative prognostic role | |
| Prostate cancer | Oncogene | [167,168,169,170,171,172] | ↑tumor cell growth |
| ↑proliferation | |||
| ↑metastatic potential | |||
| ↑invasion | |||
| Renal cancer | Oncogene | [173,174] | ↓survival rate |
| ↑proliferation | |||
| ↓apoptosis | |||
| Breast cancer | Oncogene(?) | [95] | ↓survival rate |
| ↑lymph node metastasis | |||
| Onco-suppressor(?) | [175,176,177,178,179,180,181,182] | ↑effects of Tamoxifen (TAM) | |
| ↓EMT | |||
| ↓proliferation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polvani, S.; Pepe, S.; Milani, S.; Galli, A. COUP-TFII in Health and Disease. Cells 2020, 9, 101. https://doi.org/10.3390/cells9010101
Polvani S, Pepe S, Milani S, Galli A. COUP-TFII in Health and Disease. Cells. 2020; 9(1):101. https://doi.org/10.3390/cells9010101
Chicago/Turabian StylePolvani, Simone, Sara Pepe, Stefano Milani, and Andrea Galli. 2020. "COUP-TFII in Health and Disease" Cells 9, no. 1: 101. https://doi.org/10.3390/cells9010101
APA StylePolvani, S., Pepe, S., Milani, S., & Galli, A. (2020). COUP-TFII in Health and Disease. Cells, 9(1), 101. https://doi.org/10.3390/cells9010101