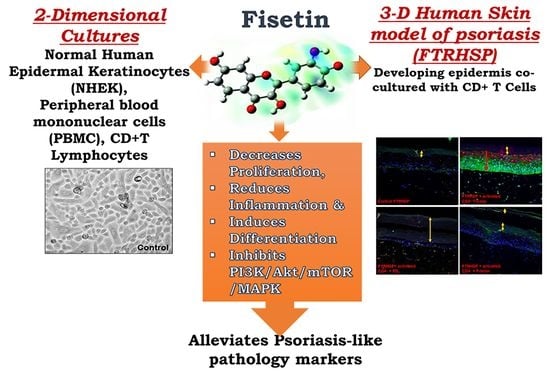
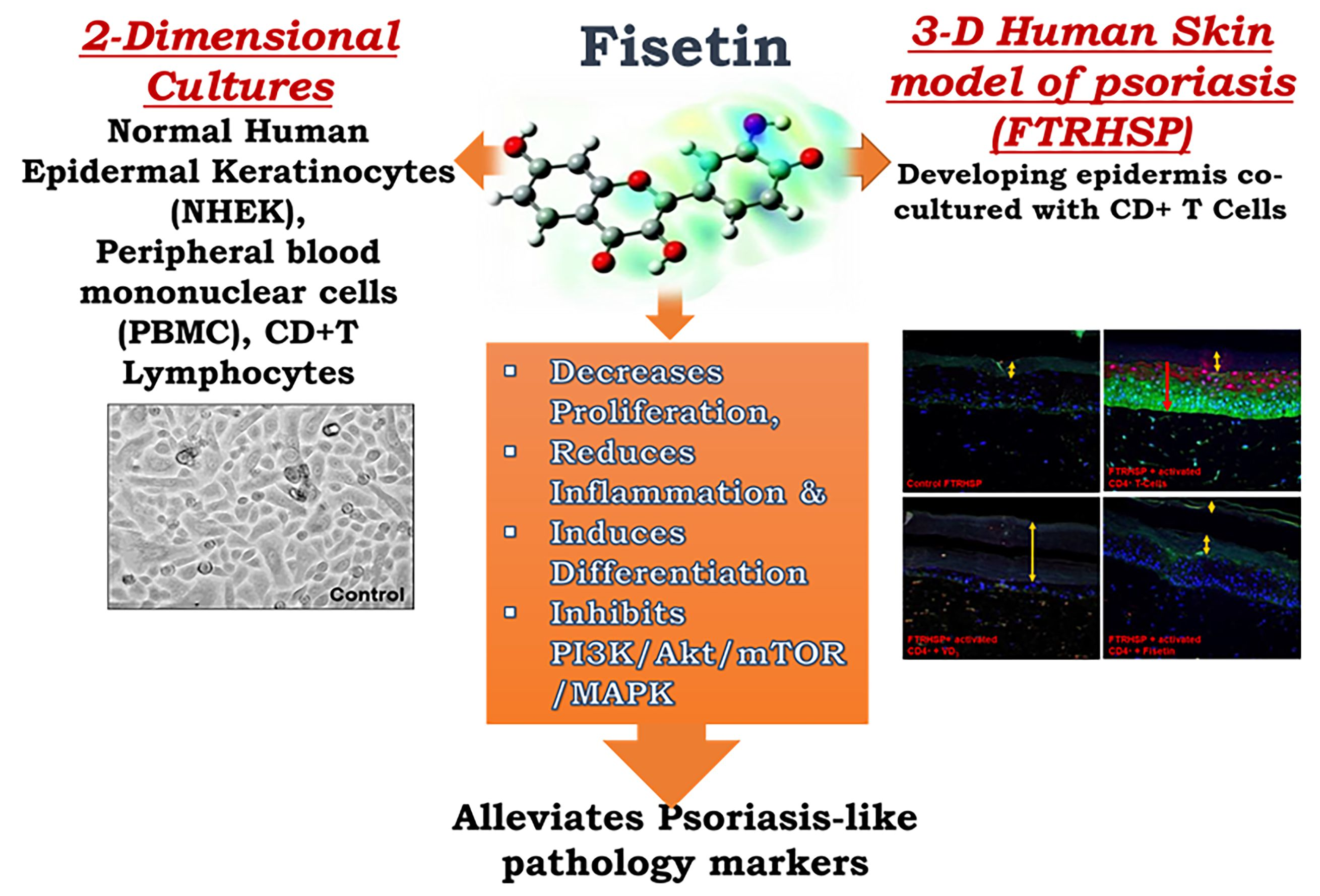
Fisetin, a 3,7,3′,4′-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models

 ,
,  ,
,  , ,
, ,  ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Human Subjects
2.3. Keratinocyte Isolation, Culture, Activation with IL-22, TNF-α, TPA, and Treatments
2.4. Determination of Cell Viability by MTT Assay
2.5. Cell Cycle Analysis
2.6. Preparation, Culture, Activation, and Treatment of Peripheral Blood Mononuclear Cells (PBMC)
2.7. Preparation and Activation of Blood CD4+ T Lymphocytes
2.8. ProcartaPlexTM Multiplex Bead-Based Immunoassays for Cytokines and Chemokines
2.9. ELISA Analysis of Th1-Th2-and-Th17 Cytokines, IL-4, IL-17A, and IFN-γ
2.10. RNA Preparation and Real-Time Quantitative (q)-PCR
2.11. Preparation of Protein Lysates from Cultured Cells and Western Blotting
2.12. Generation of a Three-Dimensional (3D) Full-Thickness Reconstituted Human Skin Model of Psoriasis (FTRHSP)
2.13. Fisetin and Vit-D3 Treatment of the 3D FTRHSP Model System
2.14. Histology, Morphometry, and Immunostaining Analyses of the FTRHSP
2.15. Statistical Analysis
3. Results
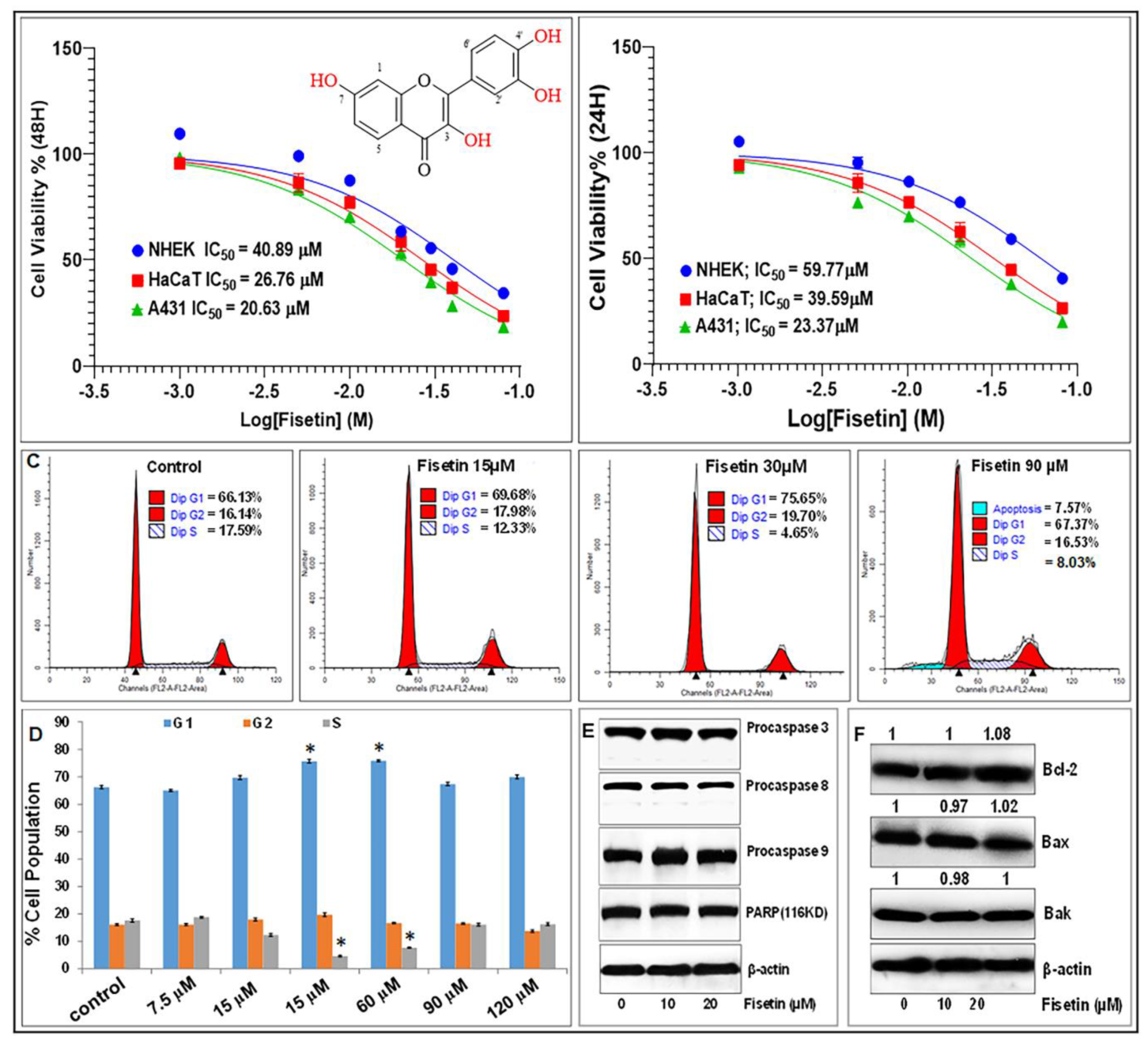
3.1. Fisetin Inhibits Cell Proliferation and Viability, but Does Not Affect Apoptosis of Keratinocytes at Doses ≤20 µM
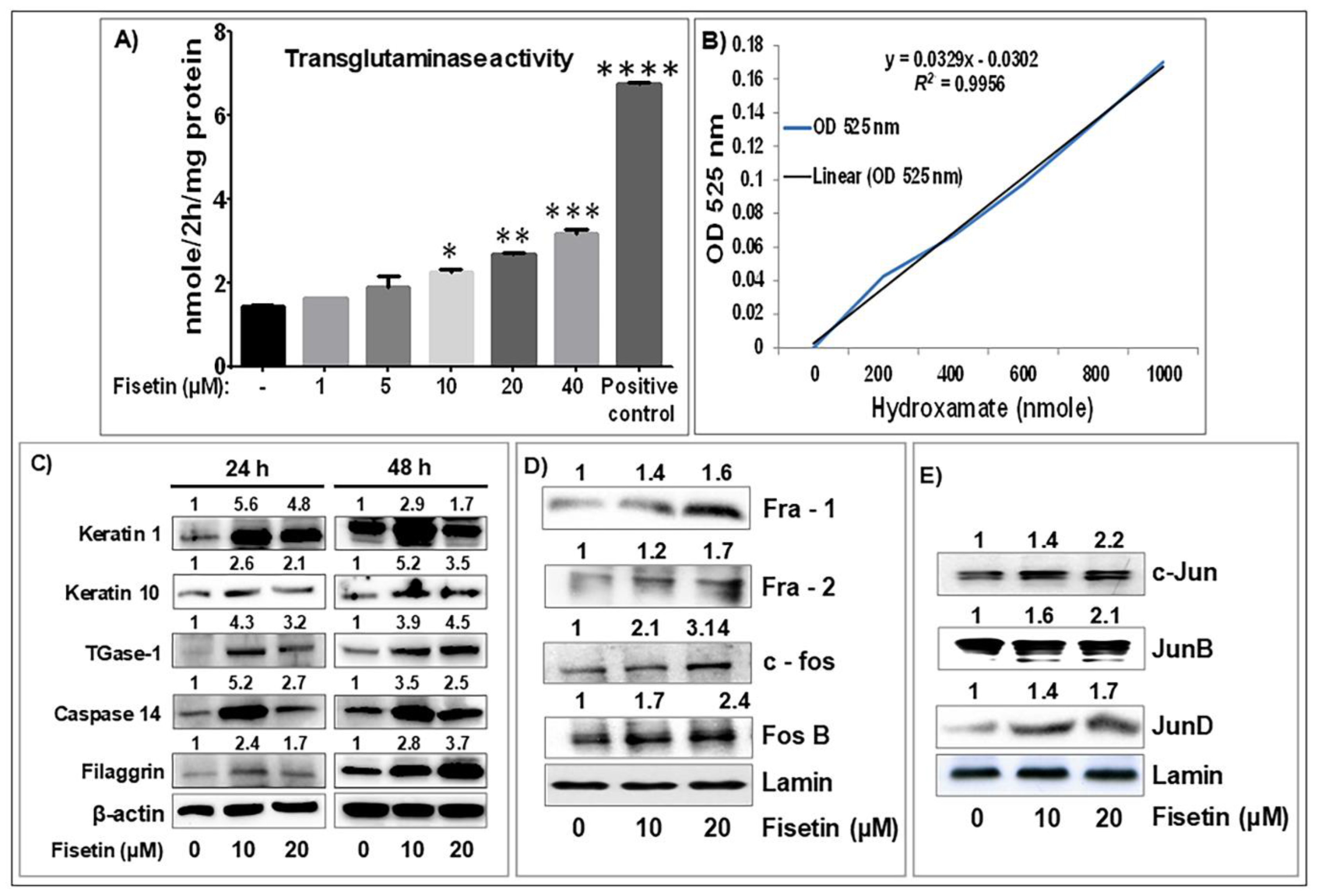
3.2. Fisetin Treatment Promotes Human Primary Epidermal Keratinocyte Differentiation and Upregulates the Expression of AP-1 Transcription Factor Proteins
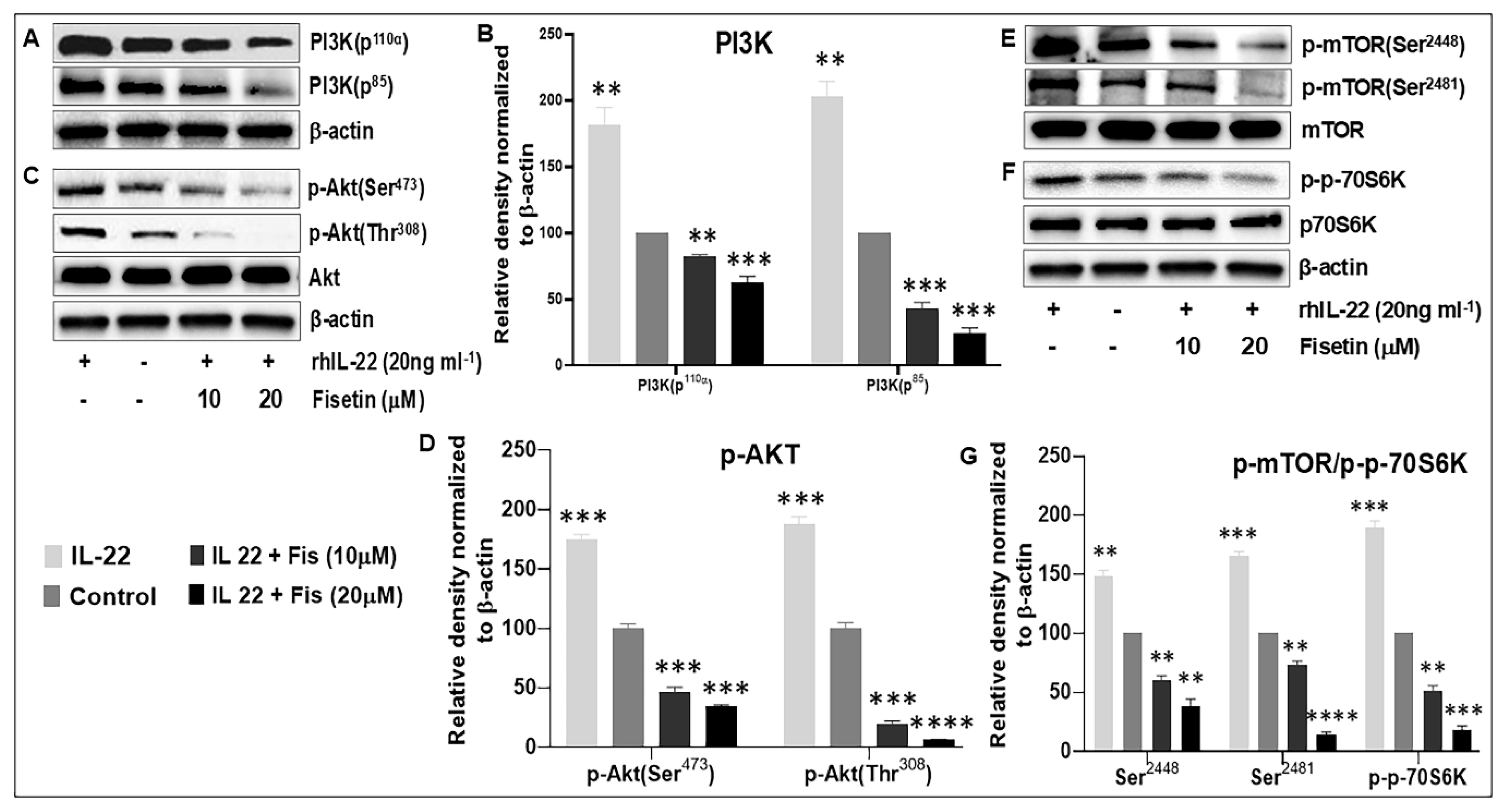
3.3. Fisetin Regulates IL-22-Induced Keratinocyte Proliferation by Inhibiting the PI3K/AKT and mTOR Pathway Components
3.4. Fisetin Regulates TNF-α-Induced Activation of the PI3K/Akt/mTOR and MAPK Signaling Pathways in NHEK
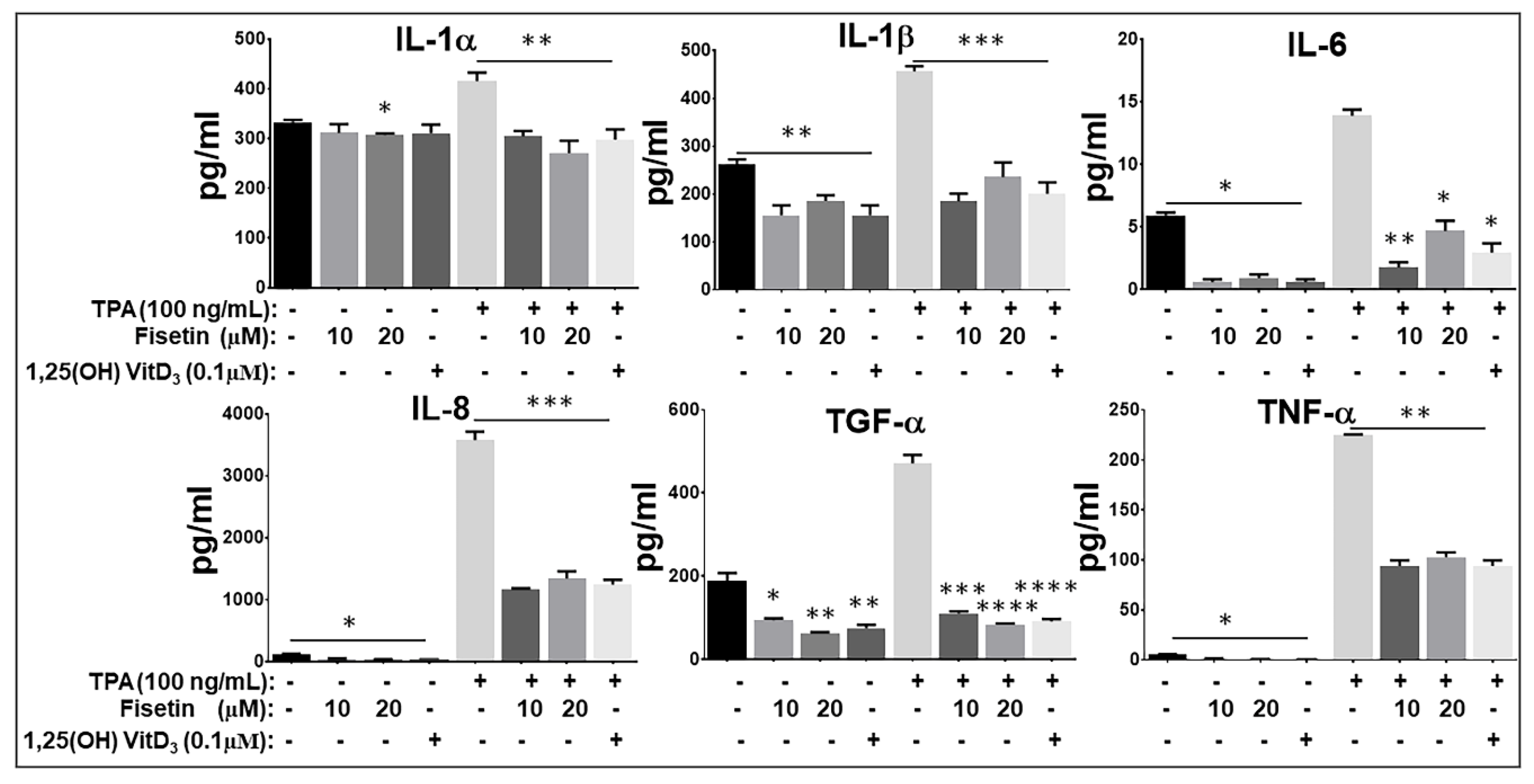
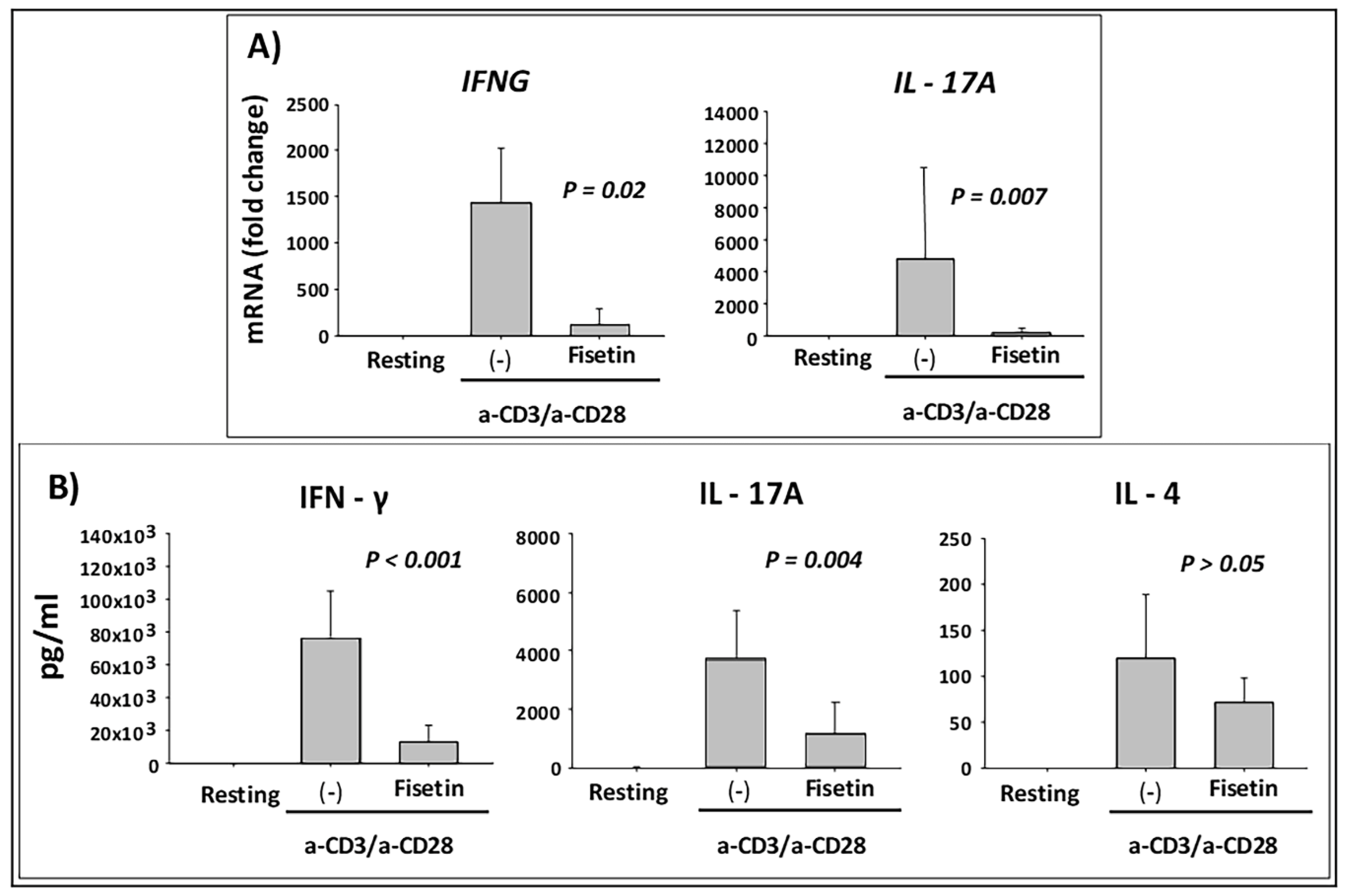
3.5. Fisetin Pretreatment Inhibits Human Epidermal Keratinocytes, Peripheral Blood Mononuclear Cells (PBMC), and CD4+ T-Lymphocytes-Induced Inflammatory Responses
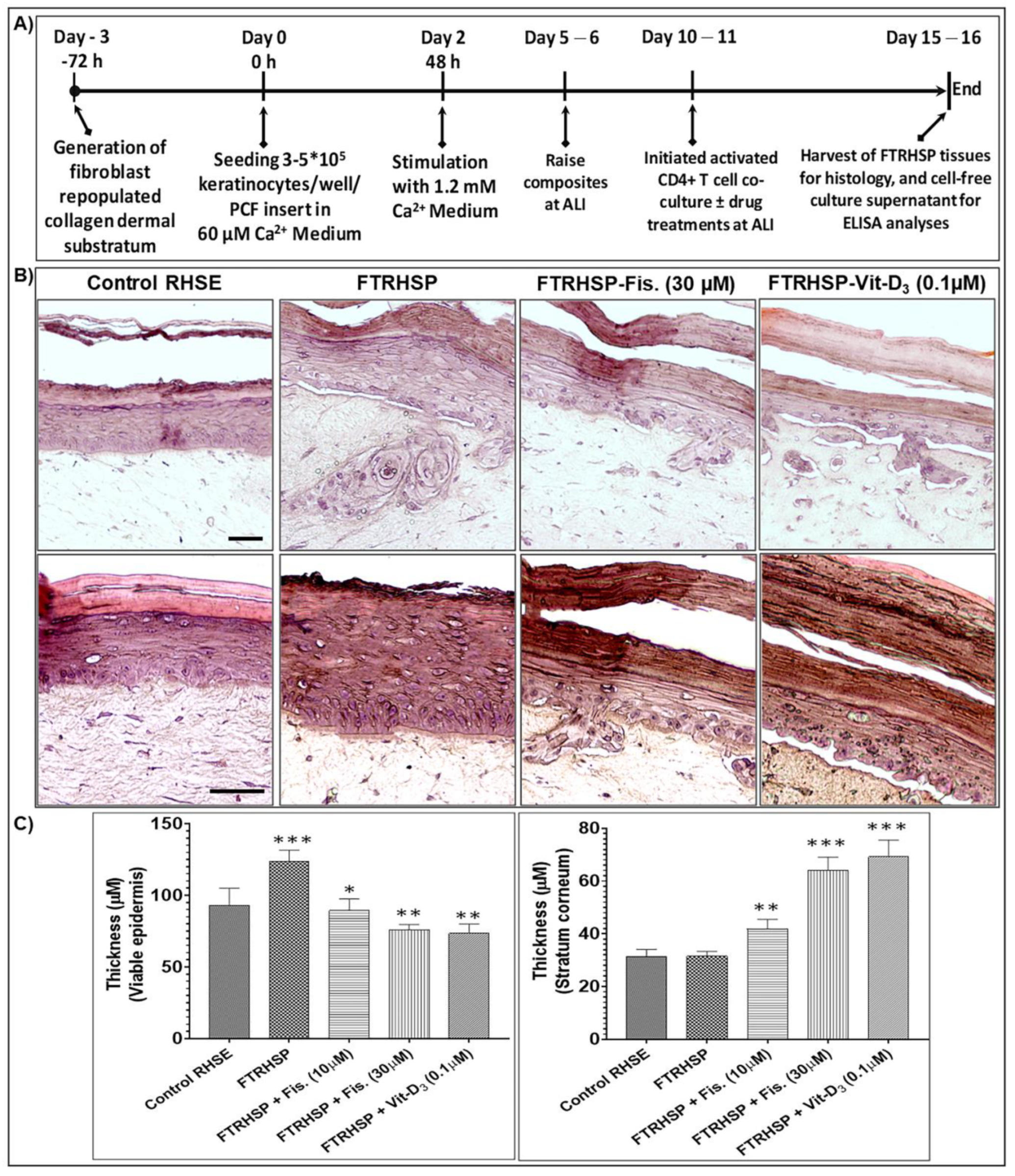
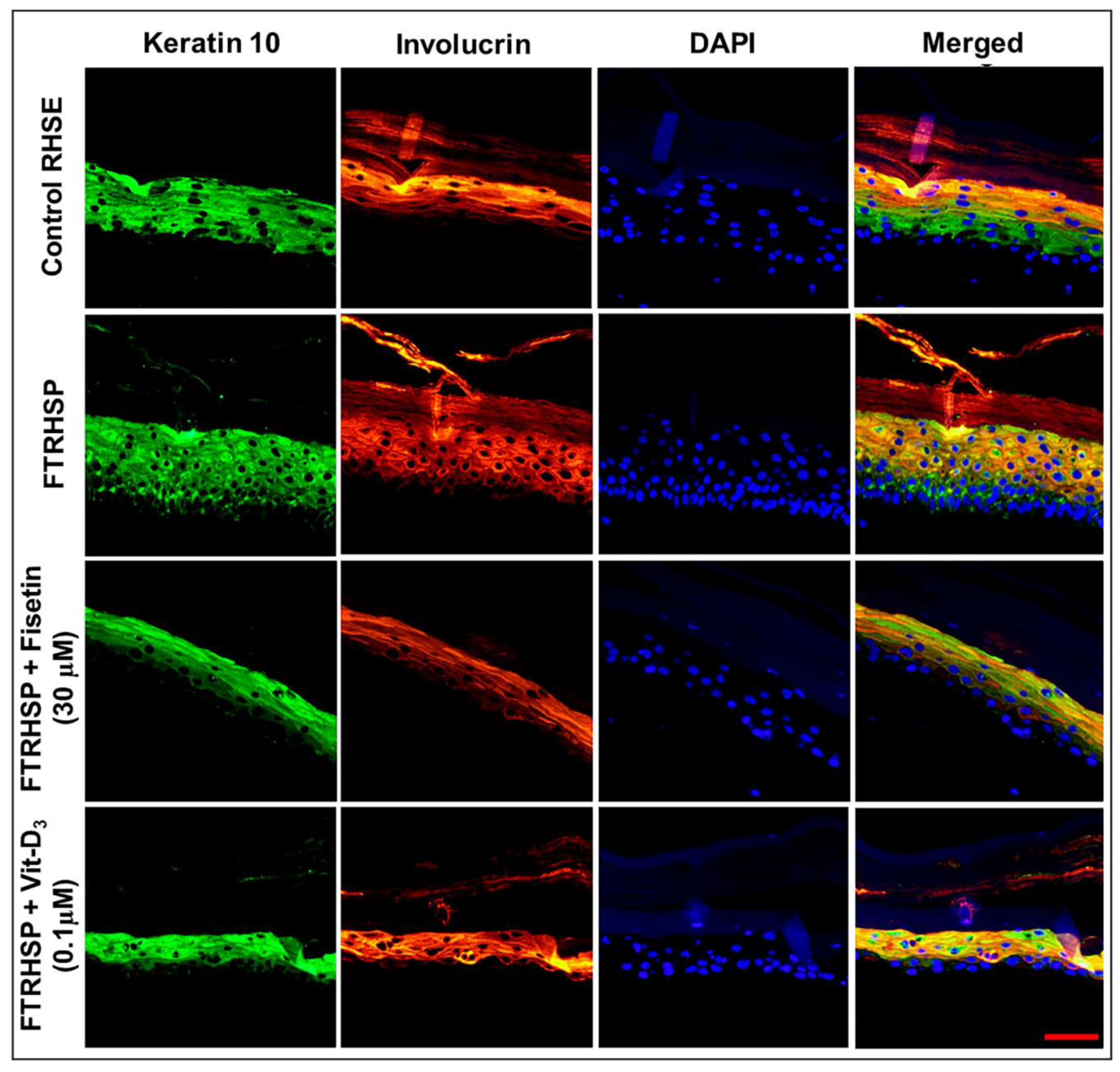
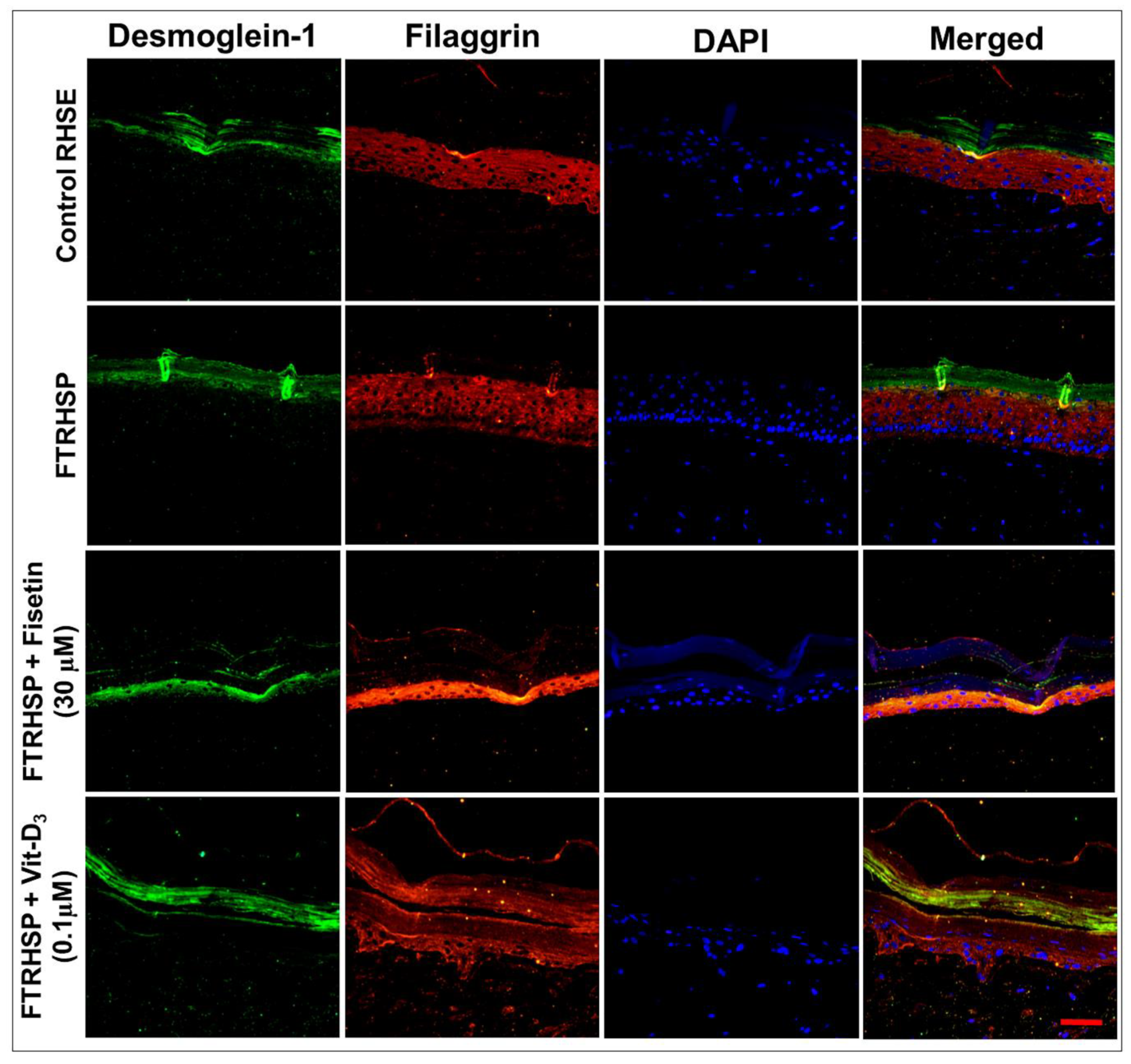
3.6. Topical Application of Fisetin Modulates Psoriasis-Like Features, Suppresses Proliferation, and Modulates Differentiation in a T Cell-Induced Three-Dimensional (3D) Full-Thickness Reconstituted Human Skin Model of Psoriasis (FTRHSP)
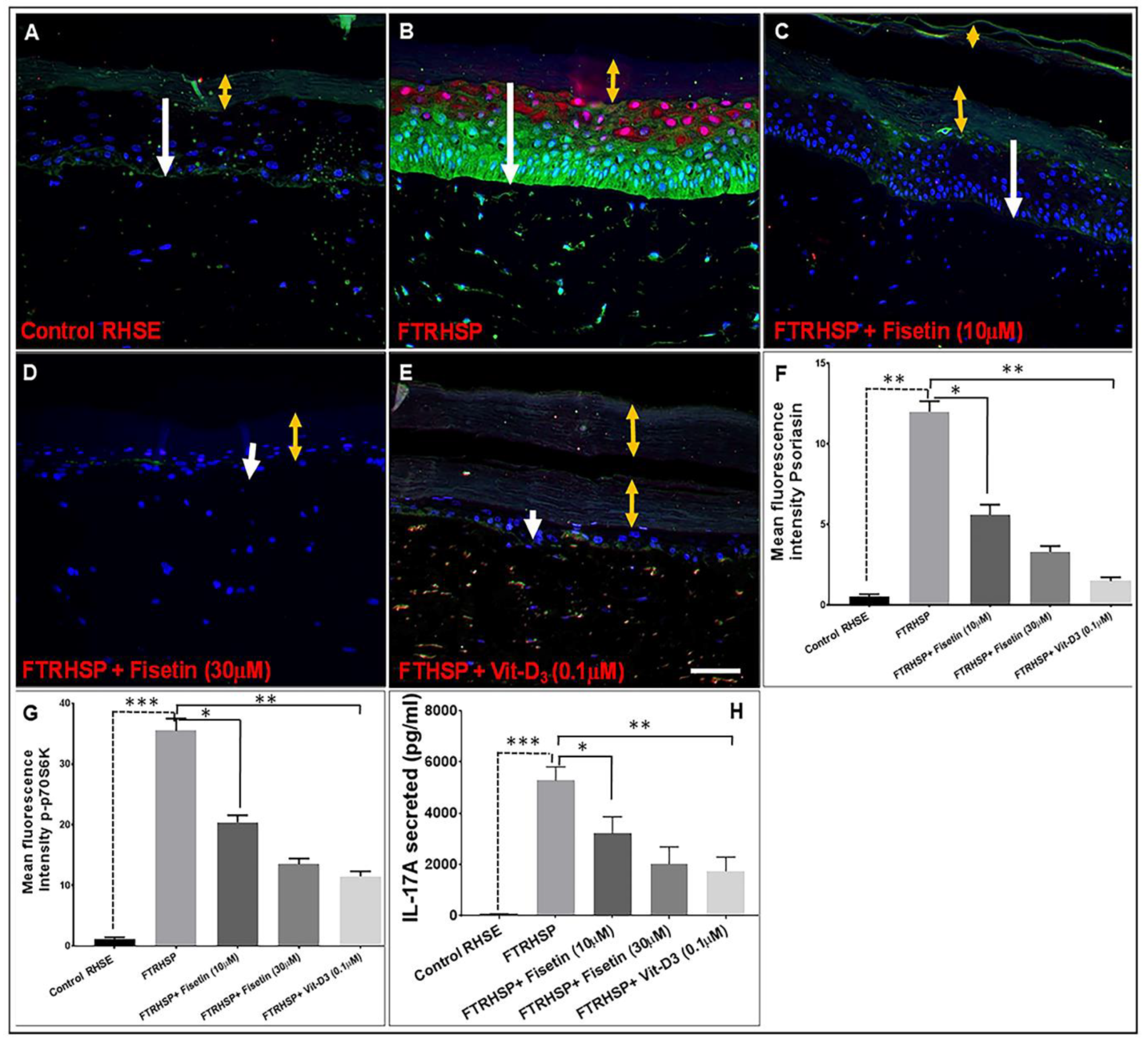
3.7. Topical Application of Fisetin Suppresses mTOR Activation and Inflammation in a T Cell-Induced 3D FTRHSP Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H. Filaggrin in the frontline: role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Siddiqui, I.A.; Syed, D.N.; Adhami, V.M.; Liovic, M.; Mukhtar, H. Keratin gene mutations in disorders of human skin and its appendages. Arch. Biochem. Biophys. 2011, 508, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Denecker, G.; Hoste, E.; Gilbert, B.; Hochepied, T.; Ovaere, P.; Lippens, S.; van den Broecke, C.; van Damme, P.; D’Herde, K.; Hachem, J.P.; et al. Caspase-14 protects against epidermal UVB photodamage and water loss. Nat. Cell Biol. 2007, 9, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol 2005, 6, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Hoste, E.; Kemperman, P.; Devos, M.; Denecker, G.; Kezic, S.; Yau, N.; Gilbert, B.; Lippens, S.; de Groote, P.; Roelandt, R.; et al. Caspase-14 is required for filaggrin degradation to natural moisturizing factors in the skin. J. Investig. Dermatol. 2011, 131, 2233–2241. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Bowcock, A.M. Psoriasis genetics: breaking the barrier. Trends Genet. TIG 2010, 26, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Abdou, A.G.; Marae, A.H.; Shoeib, M.; Dawood, G.; Abouelfath, E. C-Jun expression in lichen planus, psoriasis, and cutaneous squamous cell carcinoma, an immunohistochemical study. J. Immunoass. Immunochem. 2018, 39, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Grone, A. Keratinocytes and cytokines. Vet. Immunol. Immunopathol. 2002, 88, 1–12. [Google Scholar] [CrossRef]
- Alappatt, C.; Johnson, C.A.; Clay, K.L.; Travers, J.B. Acute keratinocyte damage stimulates platelet-activating factor production. Arch. Dermatol. Res. 2000, 292, 256–259. [Google Scholar] [CrossRef]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar] [CrossRef]
- Declercq, S.D.; Pouliot, R. Promising new treatments for psoriasis. Sci. World J. 2013, 2013, 980419. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; Hsu, L.; Liao, W. Phototherapy in psoriasis: A review of mechanisms of action. J. Cutan. Med. Surg. 2013, 17, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Villasenor-Park, J.; Wheeler, D.; Grandinetti, L. Psoriasis: Evolving treatment for a complex disease. Cleve. Clin. J. Med. 2012, 79, 413–423. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, R.; Rostami-Yazdi, M.; Gerdes, S.; Mrowietz, U. Triptolide in the treatment of psoriasis and other immune-mediated inflammatory diseases. Br. J. Clin. Pharmacol. 2012, 74, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Gniadecki, R.; Bang, B.; Bryld, L.E.; Iversen, L.; Lasthein, S.; Skov, L. Comparison of long-term drug survival and safety of biologic agents in patients with psoriasis vulgaris. Br. J. Dermatol. 2015, 172, 244–252. [Google Scholar] [CrossRef]
- Syed, D.N.; Adhami, V.M.; Khan, N.; Khan, M.I.; Mukhtar, H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin. Cancer Biol. 2016, 40–41, 130–140. [Google Scholar] [CrossRef]
- Khan, N.; Syed, D.N.; Ahmad, N.; Mukhtar, H. Fisetin: A dietary antioxidant for health promotion. Antioxid. Redox Signal. 2013, 19, 151–162. [Google Scholar] [CrossRef]
- Arai, Y.; Watanabe, S.; Kimira, M.; Shimoi, K.; Mochizuki, R.; Kinae, N. Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. J. Nutr. 2000, 130, 2243–2250. [Google Scholar] [CrossRef]
- Vale, P.F.; McNally, L.; Doeschl-Wilson, A.; King, K.C.; Popat, R.; Domingo-Sananes, M.R.; Allen, J.E.; Soares, M.P.; Kummerli, R. Beyond killing: Can we find new ways to manage infection? Evol. Med. Public Health 2016, 2016, 148–157. [Google Scholar] [CrossRef]
- Kim, J.A.; Lee, S.; Kim, D.E.; Kim, M.; Kwon, B.M.; Han, D.C. Fisetin, a dietary flavonoid, induces apoptosis of cancer cells by inhibiting HSF1 activity through blocking its binding to the hsp70 promoter. Carcinogenesis 2015, 36, 696–706. [Google Scholar] [CrossRef]
- Syed, D.N.; Chamcheu, J.C.; Khan, M.I.; Sechi, M.; Lall, R.K.; Adhami, V.M.; Mukhtar, H. Fisetin inhibits human melanoma cell growth through direct binding to p70S6K and mTOR: findings from 3-D melanoma skin equivalents and computational modeling. Biochem. Pharmacol. 2014, 89, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.C.; Sharma, S.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin inhibits growth, induces G(2) /M arrest and apoptosis of human epidermoid carcinoma A431 cells: Role of mitochondrial membrane potential disruption and consequent caspases activation. Exp. Dermatol. 2013, 22, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Adhami, V.M.; Khan, M.I.; Mukhtar, H. Inhibition of Akt/mTOR signaling by the dietary flavonoid fisetin. Anti-Cancer Agents Med. Chem. 2013, 13, 995–1001. [Google Scholar] [CrossRef]
- Adhami, V.M.; Syed, D.N.; Khan, N.; Mukhtar, H. Dietary flavonoid fisetin: a novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem. Pharmacol. 2012, 84, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Afaq, F.; Khusro, F.H.; Adhami, V.M.; Suh, Y.; Mukhtar, H. Dual inhibition of phosphatidylinositol 3-kinase/Akt and mammalian target of rapamycin signaling in human nonsmall cell lung cancer cells by a dietary flavonoid fisetin. Int. J. Cancer 2012, 130, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Afaq, F.; Syed, D.N.; Mukhtar, H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis 2008, 29, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.C.; Athar, M.; Elmets, C.A.; Afaq, F. Fisetin inhibits UVB-induced cutaneous inflammation and activation of PI3K/AKT/NFkappaB signaling pathways in SKH-1 hairless mice. Photochem. Photobiol. 2015, 91, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Nakahara, T.; Takahara, M.; Kido, M.; Dugu, L.; Uchi, H.; Takeuchi, S.; Tu, Y.T.; Moroi, Y.; Furue, M. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br. J. Dermatol. 2009, 160, 442–445. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Chaves-Rodriquez, M.I.; Adhami, V.M.; Siddiqui, I.A.; Wood, G.S.; Longley, B.J.; Mukhtar, H. Upregulation of PI3K/AKT/mTOR, FABP5 and PPARbeta/delta in Human Psoriasis and Imiquimod-induced Murine Psoriasiform Dermatitis Model. Acta Derm. Venereol. 2016, 96, 854–856. [Google Scholar]
- Huang, T.; Lin, X.; Meng, X.; Lin, M. Phosphoinositide-3 kinase/protein kinase-B/mammalian target of rapamycin pathway in psoriasis pathogenesis. A potential therapeutic target? Acta Derm. Venereol. 2014, 94, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Higa, S.; Hirano, T.; Kotani, M.; Matsumoto, M.A.; Fujita, M.; Suemura, I.; Kawase, T. Tanaka, Fisetin, a flavonol, inhibits TH2-type cytokine production by activated human basophils. J. Allergy Clin. Immunol. 2003, 111, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Guan, S.; Lu, J.; Chen, Z.; Huang, G.; Li, G.; Xiong, Y.; Zhang, S.; Yue, Z.; Deng, X. Suppressive effects of fisetin on mice T lymphocytes in vitro and in vivo. J. Surg. Res. 2013, 185, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Goh, F.Y.; Upton, N.; Guan, S.; Cheng, C.; Shanmugam, M.K.; Sethi, G.; Leung, B.P.; Wong, W.S. Fisetin, a bioactive flavonol, attenuates allergic airway inflammation through negative regulation of NF-kappaB. Eur. J. Pharmacol. 2012, 679, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Krueger, J.G.; Bowcock, A.M. The immunogenetics of Psoriasis: A comprehensive review. J. Autoimmun. 2015, 64, 66–73. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Afaq, F.; Syed, D.N.; Siddiqui, I.A.; Adhami, V.M.; Khan, N.; Singh, S.; Boylan, B.T.; Wood, G.S.; Mukhtar, H. Delphinidin, a dietary antioxidant, induces human epidermal keratinocyte differentiation but not apoptosis: studies in submerged and three-dimensional epidermal equivalent models. Exp. Dermatol. 2013, 22, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Pal, H.C.; Siddiqui, I.A.; Adhami, V.M.; Ayehunie, S.; Boylan, B.T.; Noubissi, F.K.; Khan, N.; Syed, D.N.; Elmets, C.A.; et al. Prodifferentiation, anti-inflammatory and antiproliferative effects of delphinidin, a dietary anthocyanidin, in a full-thickness three-dimensional reconstituted human skin model of psoriasis. Skin Pharmacol. Physiol. 2015, 28, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Adhami, V.M.; Esnault, S.; Sechi, M.; Siddiqui, I.A.; Satyshur, K.A.; Syed, D.N.; Dodwad, S.M.; Chaves-Rodriquez, M.I.; Longley, B.J.; et al. Dual Inhibition of PI3K/Akt and mTOR by the Dietary Antioxidant, Delphinidin, Ameliorates Psoriatic Features In Vitro and in an Imiquimod-Induced Psoriasis-Like Disease in Mice. Antioxid. Redox Signal. 2017, 26, 49–69. [Google Scholar] [CrossRef]
- Esnault, S.; Shen, Z.J.; Whitesel, E.; Malter, J.S. The peptidyl-prolyl isomerase Pin1 regulates granulocyte-macrophage colony-stimulating factor mRNA stability in T lymphocytes. J. Immunol. 2006, 177, 6999–7006. [Google Scholar] [CrossRef]
- Esnault, S.; Kelly, E.A.; Nettenstrom, L.M.; Cook, E.B.; Seroogy, C.M.; Jarjour, N.N. Human eosinophils release IL-1ss and increase expression of IL-17A in activated CD4+ T lymphocytes. Clin. Exp. Allergy 2012, 42, 1756–1764. [Google Scholar] [CrossRef]
- Kelly, E.A.; Rodriguez, R.R.; Busse, W.W.; Jarjour, N.N. The effect of segmental bronchoprovocation with allergen on airway lymphocyte function. Am. J. Respir. Crit. Care Med. 1997, 156, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Navsaria, H.; Pihl-Lundin, I.; Liovic, M.; Vahlquist, A.; Torma, H. Chemical chaperones protect epidermolysis bullosa simplex keratinocytes from heat stress-induced keratin aggregation: involvement of heat shock proteins and MAP kinases. J. Invest. Dermatol. 2011, 131, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Castro, L.; Syed, D.N.; Chamcheu, J.C.; Vilela, F.M.; Perez, A.M.; Vaillant, F.; Rojas, M.; Mukhtar, H. Protective effect of tropical highland blackberry juice (Rubus adenotrichos Schltdl.) against UVB-mediated damage in human epidermal keratinocytes and in a reconstituted skin equivalent model. Photochem. Photobiol. 2013, 89, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Chamcheu, J.C.; Siddiqui, I.A.; Adhami, V.M.; Esnault, S.; Bharali, D.J.; Babatunde, A.S.; Adame, S.; Massey, R.J.; Wood, G.S.; Longley, B.J.; et al. Chitosan-based nanoformulated (-)-epigallocatechin-3-gallate (EGCG) modulates human keratinocyte-induced responses and alleviates imiquimod-induced murine psoriasiform dermatitis. Int. J. Nanomed. 2018, 13, 4189–4206. [Google Scholar] [CrossRef] [PubMed]
- Karrys, A.; Rady, I.; Chamcheu, R.N.; Sabir, M.S.; Mallick, S.; Chamcheu, J.C.; Jurutka, P.W.; Haussler, M.R.; Whitfield, G.K. Bioactive Dietary VDR Ligands Regulate Genes Encoding Biomarkers of Skin Repair That Are Associated with Risk for Psoriasis. Nutrients 2018, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Syed, D.N.; Afaq, F.; Maddodi, N.; Johnson, J.J.; Sarfaraz, S.; Ahmad, A.; Setaluri, V.; Mukhtar, H. Inhibition of human melanoma cell growth by the dietary flavonoid fisetin is associated with disruption of Wnt/beta-catenin signaling and decreased Mitf levels. J. Invest. Dermatol. 2011, 131, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Nograles, K.E.; Davidovici, B.; Krueger, J.G. New insights in the immunologic basis of psoriasis. Semin. Cutan. Med. Surg. 2010, 29, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Sturniolo, M.T.; Broome, A.M.; Ruse, M.; Rorke, E.A. Transglutaminase function in epidermis. J. Invest. Dermatol. 2005, 124, 481–492. [Google Scholar] [CrossRef]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef]
- Eckert, R.L.; Adhikary, G.; Young, C.A.; Jans, R.; Crish, J.F.; Xu, W.; Rorke, E.A. AP1 transcription factors in epidermal differentiation and skin cancer. J. Skin Cancer 2013, 2013, 537028. [Google Scholar] [CrossRef]
- Zenz, R.; Eferl, R.; Kenner, L.; Florin, L.; Hummerich, L.; Mehic, D.; Scheuch, H.; Angel, P.; Tschachler, E.; Wagner, E.F. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 2005, 437, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Elmets, C.A. An animal model of psoriasis in mice deficient in epidermal Jun proteins. Arch. Dermatol. 2006, 142, 1499–1500. [Google Scholar] [PubMed]
- Zenz, R.; Eferl, R.; Scheinecker, C.; Redlich, K.; Smolen, J.; Schonthaler, H.B.; Kenner, L.; Tschachler, E.; Wagner, E.F. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 2008, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, T.; Takekoshi, T.; Hwang, S.T. Epidermal CCR6+ gammadelta T cells are major producers of IL-22 and IL-17 in a murine model of psoriasiform dermatitis. J. Immunol. 2011, 187, 5026–5031. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, M.N.; Lonsdorf, A.S.; Shirakawa, A.K.; Lee, C.C.R.; Liao, F.; Singh, S.P.; Zhang, H.H.; Grinberg, A.; Love, P.E.; Hwang, S.T.; et al. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J. Clin. Invest. 2009, 119, 2317–2329. [Google Scholar] [CrossRef]
- Mitra, A.; Raychaudhuri, S.K.; Raychaudhuri, S.P. IL-22 induced cell proliferation is regulated by PI3K/Akt/mTOR signaling cascade. Cytokine 2012, 60, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Haugen, H.S.; Xu, W.; Witte, E.; Waggie, K.; Anderson, M.; Baur, E.V.; Witte, K.; Warszawska, K.; Philipp, S.; et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J. Mol. Med. (Berl) 2009, 87, 523–536. [Google Scholar] [CrossRef]
- Johnson-Huang, L.M.; Lowes, M.A.; Krueger, J.G. Putting together the psoriasis puzzle: an update on developing targeted therapies. Dis. Models Mech. 2012, 5, 423–433. [Google Scholar] [CrossRef]
- Zaba, L.C.; Krueger, J.G.; Lowes, M.A. Resident and “inflammatory” dendritic cells in human skin. J. Invest. Dermatol. 2009, 129, 302–308. [Google Scholar] [CrossRef]
- Mitra, A.D.; Raychaudhuri, S.P.; Abria, C.J.; Mitra, A.; Wright, R.; Ray, R.; Kundu-Raychaudhuri, S. 1alpha,25-Dihydroxyvitamin-D3-3-bromoacetate regulates AKT/mTOR signaling cascades: A therapeutic agent for psoriasis. J. Invest. Dermatol. 2013, 133, 1556–1564. [Google Scholar] [CrossRef]
- Kim, B.H.; Lee, J.M.; Jung, Y.G.; Kim, S.; Kim, T.Y. Phytosphingosine derivatives ameliorate skin inflammation by inhibiting NF-kappaB and JAK/STAT signaling in keratinocytes and mice. J. Invest. Dermatol. 2014, 134, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Brembilla, N.C.; Senra, L.; Boehncke, W.H. The IL-17 Family of Cytokines in Psoriasis: IL-17A and Beyond. Front. Immunol. 2018, 9, 1682. [Google Scholar] [CrossRef] [PubMed]
- Ecoeur, F.; Weiss, J.; Kaupmann, K.; Hintermann, S.; Orain, D.; Guntermann, C. Antagonizing Retinoic Acid-Related-Orphan Receptor Gamma Activity Blocks the T Helper 17/Interleukin-17 Pathway Leading to Attenuated Pro-inflammatory Human Keratinocyte and Skin Responses. Front. Immunol. 2019, 10, 577. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Wolk, K.; Loyal, L.; Docke, W.D.; Ghoreschi, K. T cell pathology in skin inflammation. Semin. Immunopathol. 2019, 41, 359–377. [Google Scholar] [CrossRef] [PubMed]
- van den Bogaard, E.H.; Tjabringa, G.S.; Joosten, I.; Vonk-Bergers, M.; van Rijssen, E.; Tijssen, H.J.; Erkens, M.; Schalkwijk, J.; Koenen, H. Crosstalk between keratinocytes and T cells in a 3D microenvironment: a model to study inflammatory skin diseases. J. Invest. Dermatol. 2014, 134, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Howard, O.M.; Dong, H.F.; Voscopoulos, C.; Boeshans, K.; Winston, J.; Divi, R.; Gunsior, M.; Goldsmith, P.; Ahvazi, B.; et al. Chemotactic activity of S100A7 (Psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J. Immunol. 2008, 181, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Ekor, M. The growing use of herbal medicines: issues relating to adverse reactions and challenges in monitoring safety. Front. Pharmacol. 2014, 4, 177. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Conrad, C.; Gilliet, M. Psoriasis: From pathogenesis to targeted therapies. Clin. Rev. Allergy Immunol. 2018, 54, 102–113. [Google Scholar] [CrossRef]
- Candi, E.; Oddi, S.; Paradisi, A.; Terrinoni, A.; Ranalli, M.; Teofoli, P.; Citro, G.; Scarpato, S.; Puddu, P.; Melino, G. Expression of transglutaminase 5 in normal and pathologic human epidermis. J. Invest. Dermatol. 2002, 119, 670–677. [Google Scholar] [CrossRef]
- Harrison, C.A.; Layton, C.M.; Hau, Z.; Bullock, A.J.; Johnson, T.S.; MacNeil, S. Transglutaminase inhibitors induce hyperproliferation and parakeratosis in tissue-engineered skin. Br. J. Dermatol. 2007, 156, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Lippens, S.; Kockx, M.; Denecker, G.; Knaapen, M.; Verheyen, A.; Christiaen, R.; Tschachler, E.; Vandenabeele, P.; Declercq, W. Vitamin D3 induces caspase-14 expression in psoriatic lesions and enhances caspase-14 processing in organotypic skin cultures. Am. J. Pathol. 2004, 165, 833–841. [Google Scholar] [CrossRef]
- Hattinger, E.; Zwicker, S.; Ruzicka, T.; Yuspa, S.H.; Wolf, R. Opposing functions of psoriasin (S100A7) and koebnerisin (S100A15) in epithelial carcinogenesis. Curr. Opin. Pharmacol. 2013, 13, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Sabat, R.; Philipp, S.; Hoflich, C.; Kreutzer, S.; Wallace, E.; Asadullah, K.; Volk, H.D.; Sterry, W.; Wolk, K. Immunopathogenesis of psoriasis. Exp. Dermatol. 2007, 16, 779–798. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, Z.; Zwicker, S.; Bureik, D.; Peric, M.; Koglin, S.; Batycka-Baran, A.; Prinz, J.C.; Ruzicka, T.; Schauber, J.; Wolf, R. Vitamin D analog calcipotriol suppresses the Th17 cytokine-induced proinflammatory S100 “alarmins” psoriasin (S100A7) and koebnerisin (S100A15) in psoriasis. J. Invest. Dermatol. 2012, 132, 1416–1424. [Google Scholar] [CrossRef]
- Buerger, C. Epidermal mTORC1 Signaling Contributes to the Pathogenesis of Psoriasis and Could Serve as a Therapeutic Target. Front. Immunol. 2018, 9, 2786. [Google Scholar] [CrossRef]
- Griffiths, C.E. Comparing biological therapies in psoriasis: Implications for clinical practice. J. Eur. Acad. Dermatol. Venereol. JEADV 2010, 24 (Suppl. 6), 10–14. [Google Scholar] [CrossRef]
- Leonardi, C.L.; Romiti, R.; Tebbey, P.W. Ten years on: the impact of biologics on the practice of dermatology. Dermatol. Clin. 2015, 33, 111–125. [Google Scholar] [CrossRef]
- Schmitt, J.; Rosumeck, S.; Thomaschewski, G.; Sporbeck, B.; Haufe, E.; Nast, A. Efficacy and safety of systemic treatments for moderate-to-severe psoriasis: meta-analysis of randomized controlled trials. Br. J. Dermatol. 2014, 170, 274–303. [Google Scholar] [CrossRef]
- Arican, O.; Aral, M.; Sasmaz, S.; Ciragil, P. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005, 2005, 273–279. [Google Scholar] [CrossRef]
- Taniguchi, K.; Arima, K.; Masuoka, M.; Ohta, S.; Shiraishi, H.; Ontsuka, K.; Suzuki, S.; Inamitsu, M.; Yamamoto, K.; Simmons, O.; et al. Periostin controls keratinocyte proliferation and differentiation by interacting with the paracrine IL-1alpha/IL-6 loop. J. Invest. Dermatol. 2014, 134, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Muhr, P.; Renne, J.; Schaefer, V.; Werfel, T.; Wittmann, M. Primary human keratinocytes efficiently induce IL-1-dependent IL-17 in CCR6+ T cells. Exp. Dermatol. 2010, 19, 1105–1107. [Google Scholar] [CrossRef] [PubMed]
- Nograles, K.E.; Zaba, L.C.; Guttman-Yassky, E.; Fuentes-Duculan, J.; Suarez-Farinas, M.; Cardinale, I.; Khatcherian, A.; Gonzalez, J.; Pierson, K.C.; White, T.R.; et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol. 2008, 159, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Brotas, A.M.; Cunha, J.M.; Lago, E.H.; Machado, C.C.; Carneiro, S.C. Tumor necrosis factor-alpha and the cytokine network in psoriasis. An. Bras. Dermatol. 2012, 87, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, J.E.; Johnston, A.; Dyson, M.; Valdimarsson, H.; Elder, J.T. Mouse models of psoriasis. J. Invest. Dermatol. 2007, 127, 1292–1308. [Google Scholar] [CrossRef] [PubMed]
- Groves, R.W.; Mizutani, H.; Kieffer, J.D.; Kupper, T.S. Inflammatory skin disease in transgenic mice that express high levels of interleukin 1 alpha in basal epidermis. Proc. Natl. Acad. Sci. USA 1995, 92, 11874–11878. [Google Scholar] [CrossRef]
- Nestle, F.O.; di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease, Nature reviews. Immunology 2009, 9, 679–691. [Google Scholar]
- Helwa, I.; Patel, R.; Karempelis, P.; Kaddour-Djebbar, I.; Choudhary, V.; Bollag, W.B. The antipsoriatic agent monomethylfumarate has antiproliferative, prodifferentiative, and anti-inflammatory effects on keratinocytes. J. Pharmacol. Exp. Ther. 2015, 352, 90–97. [Google Scholar] [CrossRef]
- Hsieh, M.H.; Tsai, J.P.; Yang, S.F.; Chiou, H.L.; Lin, C.L.; Hsieh, Y.H.; Chang, H.R. Fisetin Suppresses the Proliferation and Metastasis of Renal Cell Carcinoma through Upregulation of MEK/ERK-Targeting CTSS and ADAM9. Cells 2019, 8, 948. [Google Scholar] [CrossRef] [PubMed]
- Sechi, M.; Lall, R.K.; Afolabi, S.O.; Singh, A.; Joshi, D.C.; Chiu, S.-Y.; Mukhtar, H.; Syed, D.N. Fisetin targets YB-1/RSK axis independent of its effect on ERK signaling: insights from in vitro and in vivo melanoma models. Sci. Rep. 2018, 8, 15726. [Google Scholar] [CrossRef] [PubMed]
- Trepicchio, W.L.; Ozawa, M.; Walters, I.B.; Kikuchi, T.; Gilleaudeau, P.; Bliss, J.L.; Schwertschlag, U.; Dorner, A.J.; Krueger, J.G. Interleukin-11 therapy selectively downregulates type I cytokine proinflammatory pathways in psoriasis lesions. J. Clin. Invest. 1999, 104, 1527–1537. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamcheu, J.C.; Esnault, S.; Adhami, V.M.; Noll, A.L.; Banang-Mbeumi, S.; Roy, T.; Singh, S.S.; Huang, S.; Kousoulas, K.G.; Mukhtar, H. Fisetin, a 3,7,3′,4′-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models. Cells 2019, 8, 1089. https://doi.org/10.3390/cells8091089
Chamcheu JC, Esnault S, Adhami VM, Noll AL, Banang-Mbeumi S, Roy T, Singh SS, Huang S, Kousoulas KG, Mukhtar H. Fisetin, a 3,7,3′,4′-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models. Cells. 2019; 8(9):1089. https://doi.org/10.3390/cells8091089
Chicago/Turabian StyleChamcheu, Jean Christopher, Stephane Esnault, Vaqar M. Adhami, Andrea L. Noll, Sergette Banang-Mbeumi, Tithi Roy, Sitanshu S. Singh, Shile Huang, Konstantin G. Kousoulas, and Hasan Mukhtar. 2019. "Fisetin, a 3,7,3′,4′-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models" Cells 8, no. 9: 1089. https://doi.org/10.3390/cells8091089
APA StyleChamcheu, J. C., Esnault, S., Adhami, V. M., Noll, A. L., Banang-Mbeumi, S., Roy, T., Singh, S. S., Huang, S., Kousoulas, K. G., & Mukhtar, H. (2019). Fisetin, a 3,7,3′,4′-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models. Cells, 8(9), 1089. https://doi.org/10.3390/cells8091089