ER-Mitochondria Communication in Cells of the Innate Immune System


Abstract
1. Introduction
2. Basic Considerations towards the ER-Mitochondrial Communication
3. The Innate Immune System
4. ER-Mitochondria in Macrophage Lipid Metabolism
4.1. Introduction to Macrophage Lipid Metabolism
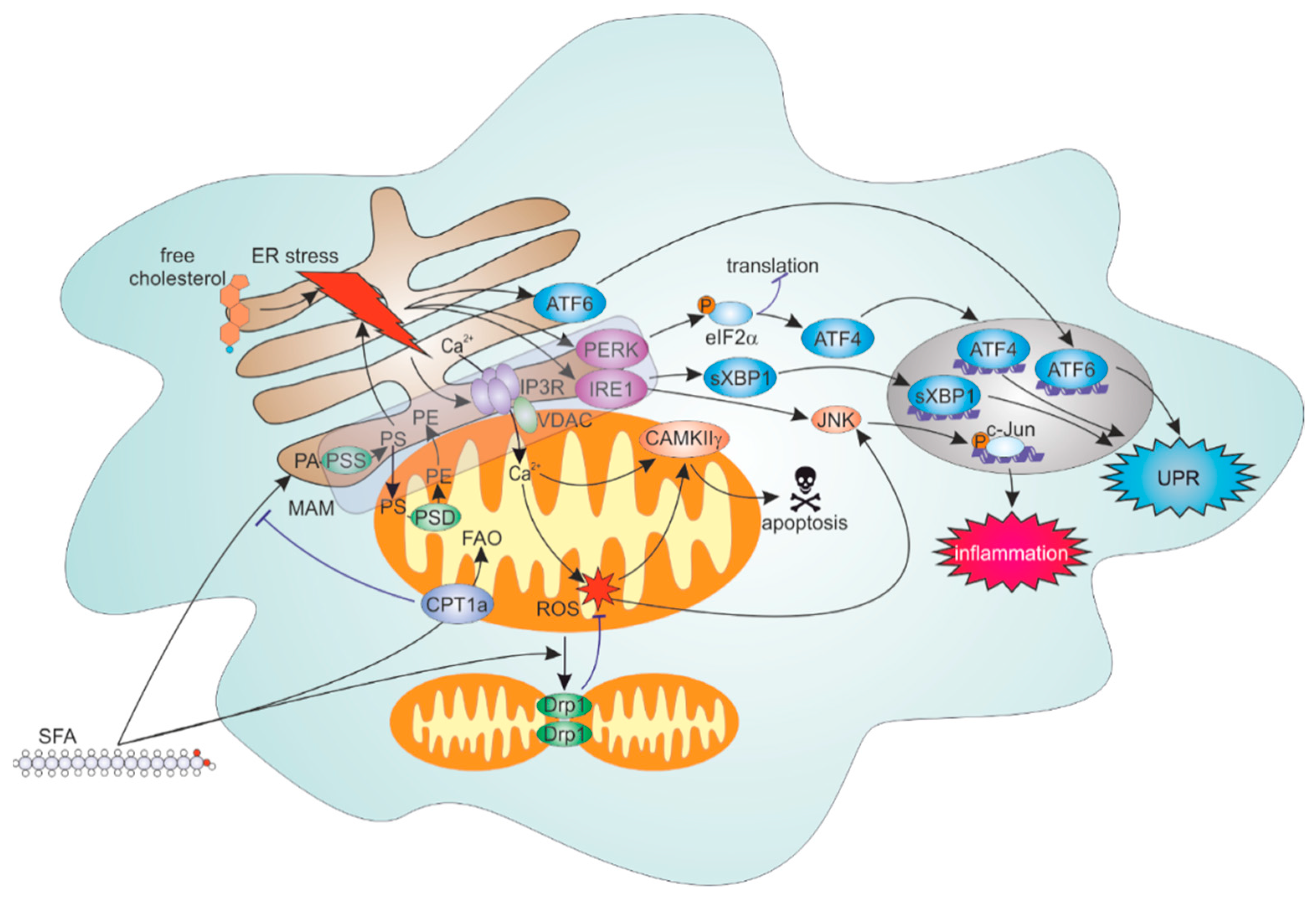
4.2. ER Stress Response to Lipid Overload
4.3. Roles of FAO and Mitochondrial Dynamics in Inflammatory Responses to SFA
4.4. ER-Mitochondrial Contacts Modulate Lipid Overload-Induced ER and Inflammation
5. ER-Mitochondria Contacts in Efferocytosis
6. ER-Mitochondria in Inflammasome
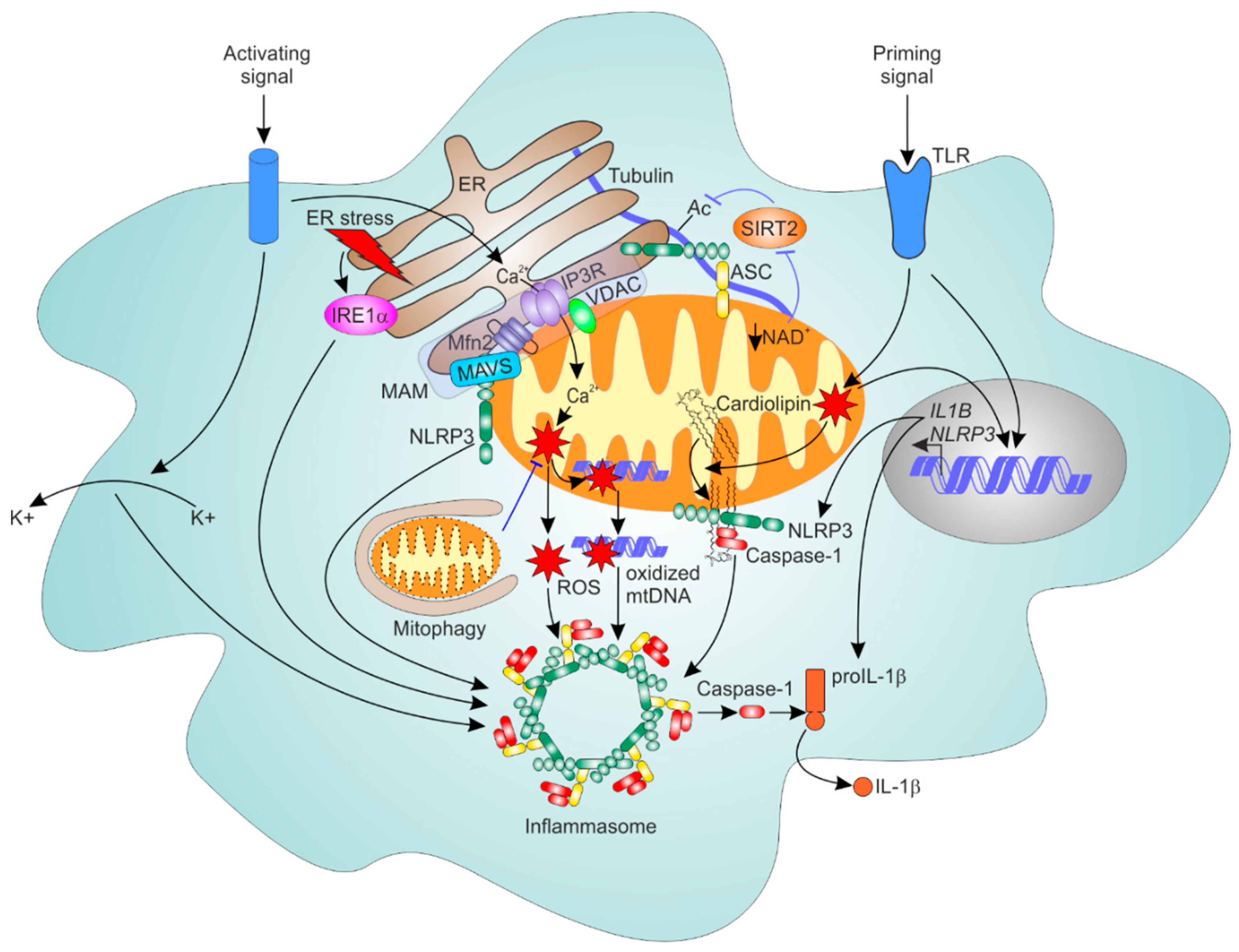
6.1. General Mechanism of NLRP3 Inflammasome Activation
6.2. Mitochondria as Platforms for NLRP3 Inflammasome Assembly
6.3. ER-Mitochondrial Contacts Regulate NLRP3 Inflammasome
6.4. Mitophagy Dampens NLRP3 Inflammasome Activation
7. ER-Mitochondria Contacts and Antiviral Responses
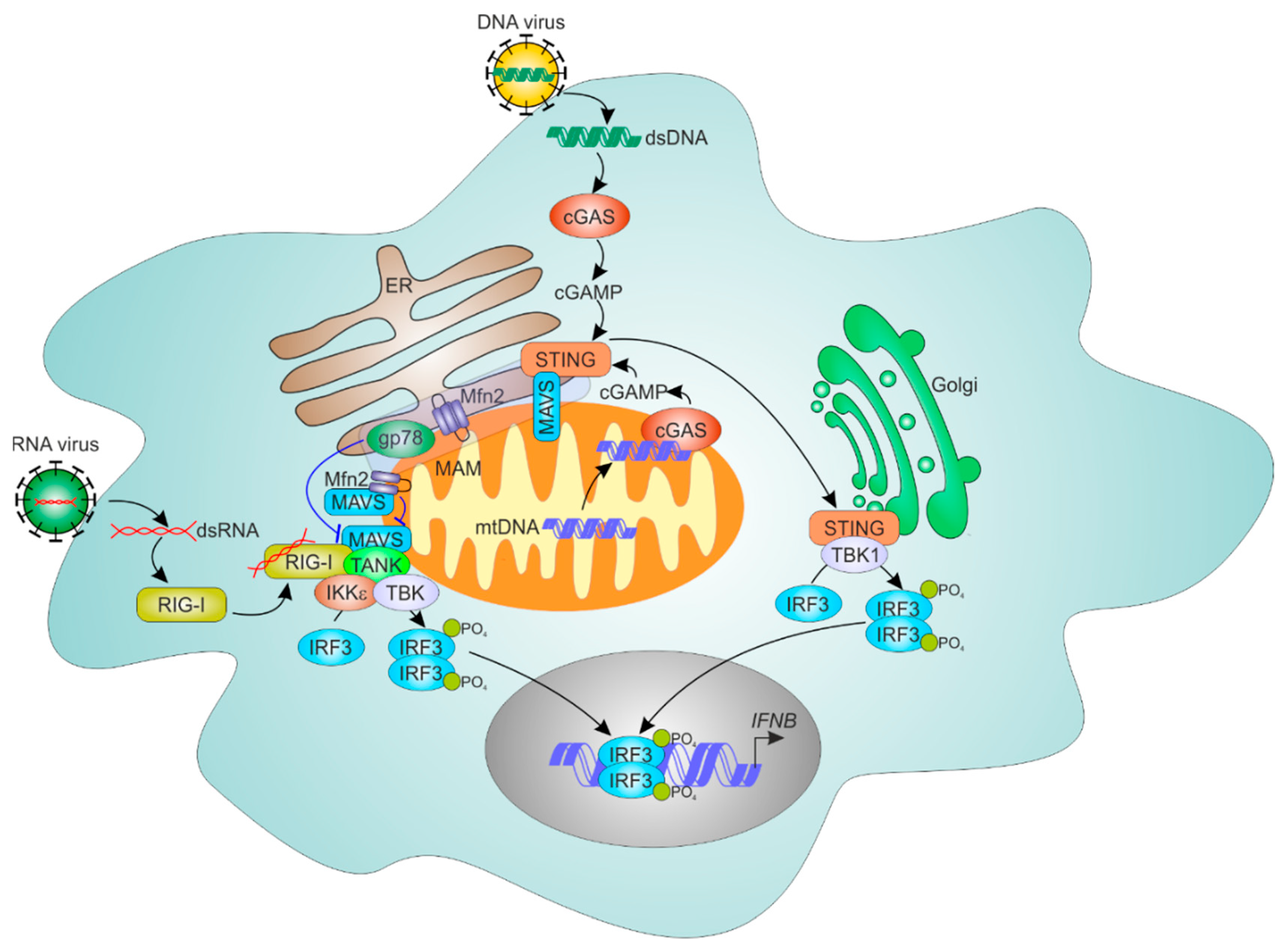
7.1. Mechanisms of Cellular RNA Sensing
7.2. ER and Mitochondria Control MAVS-Dependent RNA Sensing Mechanisms
7.3. ER-Mitochondrial Regulation of Cytosolic DNA Sensing
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Gatta, A.T.; Levine, T.P. Piecing together the patchwork of contact sites. Trends Cell Biol. 2017, 27, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [PubMed]
- Rusiñol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [PubMed]
- Lackner, L.L. The expanding and unexpected functions of mitochondria contact sites. Trends Cell Biol. 2019, 29, 580–590. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic reticulum-mitochondrial contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Kwon, S.-K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [PubMed]
- Schrepfer, E.; Scorrano, L. Mitofusins, from Mitochondria to metabolism. Mol. Cell 2016, 61, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; de Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Csordás, G.; Thomas, A.P.; Hajnóczky, G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999, 18, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Brill, A.L.; Lemos, F.O.; Jiang, J.Y.; Falcone, J.L.; Kimmerling, E.P.; Cai, Y.; Dong, K.; Kaplan, D.L.; Wallace, D.P.; et al. Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 1991, 266, 89–97. [Google Scholar] [PubMed]
- Shiao, Y.J.; Lupo, G.; Vance, J.E. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J. Biol. Chem. 1995, 270, 11190–11198. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Chao, J.T.; Tavassoli, S.; Wong, A.K.; Choudhary, V.; Young, B.P.; Loewen, C.J.; Prinz, W.A. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014, 12, e1001969. [Google Scholar] [CrossRef] [PubMed]
- Galmes, R.; Houcine, A.; van Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016, 17, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; de Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed]
- Giordano, F. Non-vesicular lipid trafficking at the endoplasmic reticulum-mitochondria interface. Biochem. Soc. Trans. 2018, 46, 437–452. [Google Scholar] [CrossRef]
- Prudent, J.; McBride, H.M. The mitochondria-endoplasmic reticulum contact sites: A signalling platform for cell death. Curr. Opin. Cell Biol. 2017, 47, 52–63. [Google Scholar] [CrossRef]
- Prudent, J.; Zunino, R.; Sugiura, A.; Mattie, S.; Shore, G.C.; McBride, H.M. MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Mol. Cell 2015, 59, 941–955. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage immunometabolism: Where are we (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and function in different stages of atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Namgaladze, D.; Brüne, B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim. Biophys. Acta 2016, 1861, 1796–1807. [Google Scholar] [CrossRef]
- Norris, P.C.; Dennis, E.A. A lipidomic perspective on inflammatory macrophage eicosanoid signaling. Adv. Biol. Regul. 2014, 54, 99–110. [Google Scholar] [CrossRef]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: Implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [CrossRef] [PubMed]
- Håversen, L.; Danielsson, K.N.; Fogelstrand, L.; Wiklund, O. Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis 2009, 202, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and oleic acid: The yin and yang of fatty acids in type 2 diabetes mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Naugler, W.; Galimi, F.; Lee, M.-S.; Karin, M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA 2006, 103, 16454–16459. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef]
- Namgaladze, D.; Lips, S.; Leiker, T.J.; Murphy, R.C.; Ekroos, K.; Ferreiros, N.; Geisslinger, G.; Brüne, B. Inhibition of macrophage fatty acid β-oxidation exacerbates palmitate-induced inflammatory and endoplasmic reticulum stress responses. Diabetologia 2014, 57, 1067–1077. [Google Scholar] [CrossRef]
- Gonzalez-Hurtado, E.; Lee, J.; Choi, J.; Selen Alpergin, E.S.; Collins, S.L.; Horton, M.R.; Wolfgang, M.J. Loss of macrophage fatty acid oxidation does not potentiate systemic metabolic dysfunction. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E381–E393. [Google Scholar] [CrossRef] [PubMed]
- Zezina, E.; Snodgrass, R.G.; Schreiber, Y.; Zukunft, S.; Schürmann, C.; Heringdorf, D.M.Z.; Geisslinger, G.; Fleming, I.; Brandes, R.P.; Brüne, B.; et al. Mitochondrial fragmentation in human macrophages attenuates palmitate-induced inflammatory responses. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, R.G.; Boß, M.; Zezina, E.; Weigert, A.; Dehne, N.; Fleming, I.; Brüne, B.; Namgaladze, D. Hypoxia potentiates palmitate-induced pro-inflammatory activation of primary human macrophages. J. Biol. Chem. 2016, 291, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Holzer, R.G.; Park, E.-J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Standen, C.L.; Morel, C.; Jung, D.Y.; Kim, J.K.; Swat, W.; Flavell, R.A.; Davis, R.J. A Protein scaffold coordinates SRC-mediated JNK Activation in response to metabolic stress. Cell Rep. 2017, 20, 2775–2783. [Google Scholar] [CrossRef] [PubMed]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER-mitochondria contact sites: Role in metabolic diseases. J. Mol. Endocrinol. 2017, 58, R87–R106. [Google Scholar] [CrossRef]
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.-P. Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, e76941. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Pers, B.M.; Parlakgül, G.; Güney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Alvarez, M.I.; Sebastián, D.; Vives, S.; Ivanova, S.; Bartoccioni, P.; Kakimoto, P.; Plana, N.; Veiga, S.R.; Hernández, V.; Vasconcelos, N.; et al. Deficient Endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease. Cell 2019, 177, 881–895.e17. [Google Scholar] [CrossRef]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Yao, P.M.; Li, Y.; Devlin, C.M.; Zhang, D.; Harding, H.P.; Sweeney, M.; Rong, J.X.; Kuriakose, G.; Fisher, E.A.; et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 2003, 5, 781–792. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef]
- Timmins, J.M.; Ozcan, L.; Seimon, T.A.; Li, G.; Malagelada, C.; Backs, J.; Backs, T.; Bassel-Duby, R.; Olson, E.N.; Anderson, M.E.; et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Invest. 2009, 119, 2925–2941. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) Integrity contributes to muscle insulin resistance in mice and humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef]
- Thoudam, T.; Ha, C.-M.; Leem, J.; Chanda, D.; Park, J.-S.; Kim, H.-J.; Jeon, J.-H.; Choi, Y.-K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER-mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Morioka, S.; Maueröder, C.; Ravichandran, K.S. Living on the edge: efferocytosis at the interface of homeostasis and pathology. Immunity 2019, 50, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. 2019, 29, 443–456.e5. [Google Scholar] [CrossRef] [PubMed]
- Morioka, S.; Perry, J.S.A.; Raymond, M.H.; Medina, C.B.; Zhu, Y.; Zhao, L.; Serbulea, V.; Onengut-Gumuscu, S.; Leitinger, N.; Kucenas, S.; et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature 2018, 563, 714–718. [Google Scholar] [CrossRef]
- Wang, Y.; Subramanian, M.; Yurdagul, A.; Barbosa-Lorenzi, V.C.; Cai, B.; de Juan-Sanz, J.; Ryan, T.A.; Nomura, M.; Maxfield, F.R.; Tabas, I. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell 2017, 171, 331–345.e22. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Han, C.Z.; Elliott, M.R.; Kinchen, J.M.; Trampont, P.C.; Das, S.; Collins, S.; Lysiak, J.J.; Hoehn, K.L.; Ravichandran, K.S. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature 2011, 477, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Viaud, M.; Ivanov, S.; Vujic, N.; Duta-Mare, M.; Aira, L.-E.; Barouillet, T.; Garcia, E.; Orange, F.; Dugail, I.; Hainault, I.; et al. Lysosomal cholesterol hydrolysis couples efferocytosis to anti-inflammatory oxysterol production. Circ. Res. 2018, 122, 1369–1384. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46. [Google Scholar] [CrossRef]
- Doran, A.C.; Ozcan, L.; Cai, B.; Zheng, Z.; Fredman, G.; Rymond, C.C.; Dorweiler, B.; Sluimer, J.C.; Hsieh, J.; Kuriakose, G.; et al. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J. Clin. Invest. 2017, 127, 4075–4089. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 Inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Yabal, M.; Calleja, D.J.; Simpson, D.S.; Lawlor, K.E. Stressing out the mitochondria: Mechanistic insights into NLRP3 inflammasome activation. J. Leukoc. Biol. 2019, 105, 377–399. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Núñez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting edge: Mitochondrial assembly of the NLRP3 inflammasome complex is initiated at priming. J. Immunol. 2018, 200, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Natarajan, K.; Clatworthy, M.R.; Wang, Z.; Germain, R.N. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013, 153, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.-W.; Alnemri, E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [PubMed]
- Ermler, M.E.; Traylor, Z.; Patel, K.; Schattgen, S.A.; Vanaja, S.K.; Fitzgerald, K.A.; Hise, A.G. Rift Valley fever virus infection induces activation of the NLRP3 inflammasome. Virology 2014, 449, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Banerjee, S.; Franchi, L.; Loo, Y.-M.; Gale, M.; Núñez, G.; Silverman, R.H. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 2015, 17, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Ozkurede, U.; Kim, Y.-G.; Arindam, C.; Gale, M.; Silverman, R.H.; Colonna, M.; Akira, S.; et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 2014, 193, 4214–4222. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Lawlor, K.E.; Yu, E.C.-W.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA. 2012, 109, 11282–11287. [Google Scholar] [CrossRef]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Current Biology 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.-M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.-C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Gelmetti, V.; de Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef]
- Erpapazoglou, Z.; Mouton-Liger, F.; Corti, O. From dysfunctional endoplasmic reticulum-mitochondria coupling to neurodegeneration. Neurochemistry International 2017, 109, 171–183. [Google Scholar] [CrossRef]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic reticulum stress activates the inflammasome via NLRP3- and Caspase-2-driven mitochondrial damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Robblee, M.M.; Kim, C.C.; Porter Abate, J.; Valdearcos, M.; Sandlund, K.L.M.; Shenoy, M.K.; Volmer, R.; Iwawaki, T.; Koliwad, S.K. Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 2016, 14, 2611–2623. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Horner, S.M. MAVS coordination of antiviral innate immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.T.; Gale, M.; Loo, Y.-M. RIG-I and other RNA sensors in antiviral immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef] [PubMed]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.Y.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-D.; Sun, L.; Seth, R.B.; Pineda, G.; Chen, Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. USA 2005, 102, 17717–17722. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Harding, A.T.; Sweeney, C.; Miao, D.; Swan, G.; Zhou, C.; Jiang, Z.; Fitzgerald, K.A.; Hammer, G.; Bergo, M.O.; et al. Control of antiviral innate immune response by protein geranylgeranylation. Sci. Adv. 2019, 5, eaav7999. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.L.; Zhu, J.; Sarkar, S.N.; Coyne, C.B. Regulation of mitochondrial antiviral signaling (MAVS) expression and signaling by the mitochondria-associated endoplasmic reticulum membrane (MAM) protein Gp78. J. Biol. Chem. 2014, 289, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Liao, Y.; Zhou, L.; Peng, S.; Chen, H.; Yuan, Z. Amplified RLR signaling activation through an interferon-stimulated gene-endoplasmic reticulum stress-mitochondrial calcium uniporter protein loop. Sci. Rep. 2016, 6, 20158. [Google Scholar] [CrossRef]
- Yasukawa, K.; Oshiumi, H.; Takeda, M.; Ishihara, N.; Yanagi, Y.; Seya, T.; Kawabata, S.-i.; Koshiba, T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009, 2, ra47. [Google Scholar] [CrossRef]
- Castanier, C.; Garcin, D.; Vazquez, A.; Arnoult, D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010, 11, 133–138. [Google Scholar] [CrossRef]
- Onoguchi, K.; Onomoto, K.; Takamatsu, S.; Jogi, M.; Takemura, A.; Morimoto, S.; Julkunen, I.; Namiki, H.; Yoneyama, M.; Fujita, T. Virus-infection or 5’ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 2010, 6, e1001012. [Google Scholar] [CrossRef]
- Hwang, M.-S.; Boulanger, J.; Howe, J.D.; Albecka, A.; Pasche, M.; Mureşan, L.; Modis, Y. MAVS polymers smaller than 80 nm induce mitochondrial membrane remodeling and interferon signaling. FEBS J. 2019, 286, 1543–1560. [Google Scholar] [CrossRef]
- Koshiba, T.; Yasukawa, K.; Yanagi, Y.; Kawabata, S.-I. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal. 2011, 4, ra7. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Zevini, A.; Olagnier, D.; Hiscott, J. Crosstalk between cytoplasmic RIG-I and STING sensing pathways. Trends Immunol. 2017, 38, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.-Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Franz, K.M.; Neidermyer, W.J.; Tan, Y.-J.; Whelan, S.P.J.; Kagan, J.C. STING-dependent translation inhibition restricts RNA virus replication. Proc. Natl. Acad. Sci. USA 2018, 115, E2058–E2067. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Modulation | Effect on Tethering | Physiological Outcome |
|---|---|---|
| ER Stress Responses | ||
| silencing Grp75, Mfn2, inhibiting cyclophilin D | decrease | increased ER stress and reduced insulin signaling in the liver [61] |
| overexpression of ER-mitochondrial tether in the liver | increase | mitochondrial Ca2+ overload, ROS formation, reduced insulin signaling [62] |
| liver-specific Mfn2 knockout | decrease | Disturbed ER phospholipid homeostasis, ER stress, inflammation [65] |
| Antiviral Response | ||
| Mfn2 silencing | decrease | increased antiviral signaling in hepatoma and HEK 293 cells [123,127] |
| Modulation | Potential Impact of Increased Tethering | Physiological Outcome |
|---|---|---|
| ER Stress Responses | ||
| FAO | increase | attenuated ER stress and inflammation induced by SFA [50] |
| mitochondrial fragmentation induced by fatty acids | increase | attenuated mROS formation and inflammation in response to SFA [52] |
| mitochondrial CAMKIIγ translocation following ER stress | increase | mitochondrial Ca2+ overload, ΔΨm loss, apoptosis [69] |
| Efferocytosis | ||
| mitochondrial fragmentation following efferocytosis | increase | reduced mitochondrial Ca2+ uptake, enhanced efferocytosis [75] |
| LIPA- and 25-hydroxycholesterol-driven mitochondrial respiration | increase | attenuated mROS, increased mitochondrial Ca2+ and ΔΨm, attenuated NLRP3 activity [77] |
| Inflammasome Activation | ||
| ER to mitochondria Ca2+ transfer | increase | NLRP3 activation [98,99] |
| mROS generation | increase | NLRP3 activation [85] |
| cardiolipin OMM translocation | unclear | NLRP3 activation [90,91] |
| mitophagy | decrease | NLRP3 inhibition [85,103,104,105] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Namgaladze, D.; Khodzhaeva, V.; Brüne, B. ER-Mitochondria Communication in Cells of the Innate Immune System. Cells 2019, 8, 1088. https://doi.org/10.3390/cells8091088
Namgaladze D, Khodzhaeva V, Brüne B. ER-Mitochondria Communication in Cells of the Innate Immune System. Cells. 2019; 8(9):1088. https://doi.org/10.3390/cells8091088
Chicago/Turabian StyleNamgaladze, Dmitry, Vera Khodzhaeva, and Bernhard Brüne. 2019. "ER-Mitochondria Communication in Cells of the Innate Immune System" Cells 8, no. 9: 1088. https://doi.org/10.3390/cells8091088
APA StyleNamgaladze, D., Khodzhaeva, V., & Brüne, B. (2019). ER-Mitochondria Communication in Cells of the Innate Immune System. Cells, 8(9), 1088. https://doi.org/10.3390/cells8091088