mRNA-Driven Generation of Transgene-Free Neural Stem Cells from Human Urine-Derived Cells


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
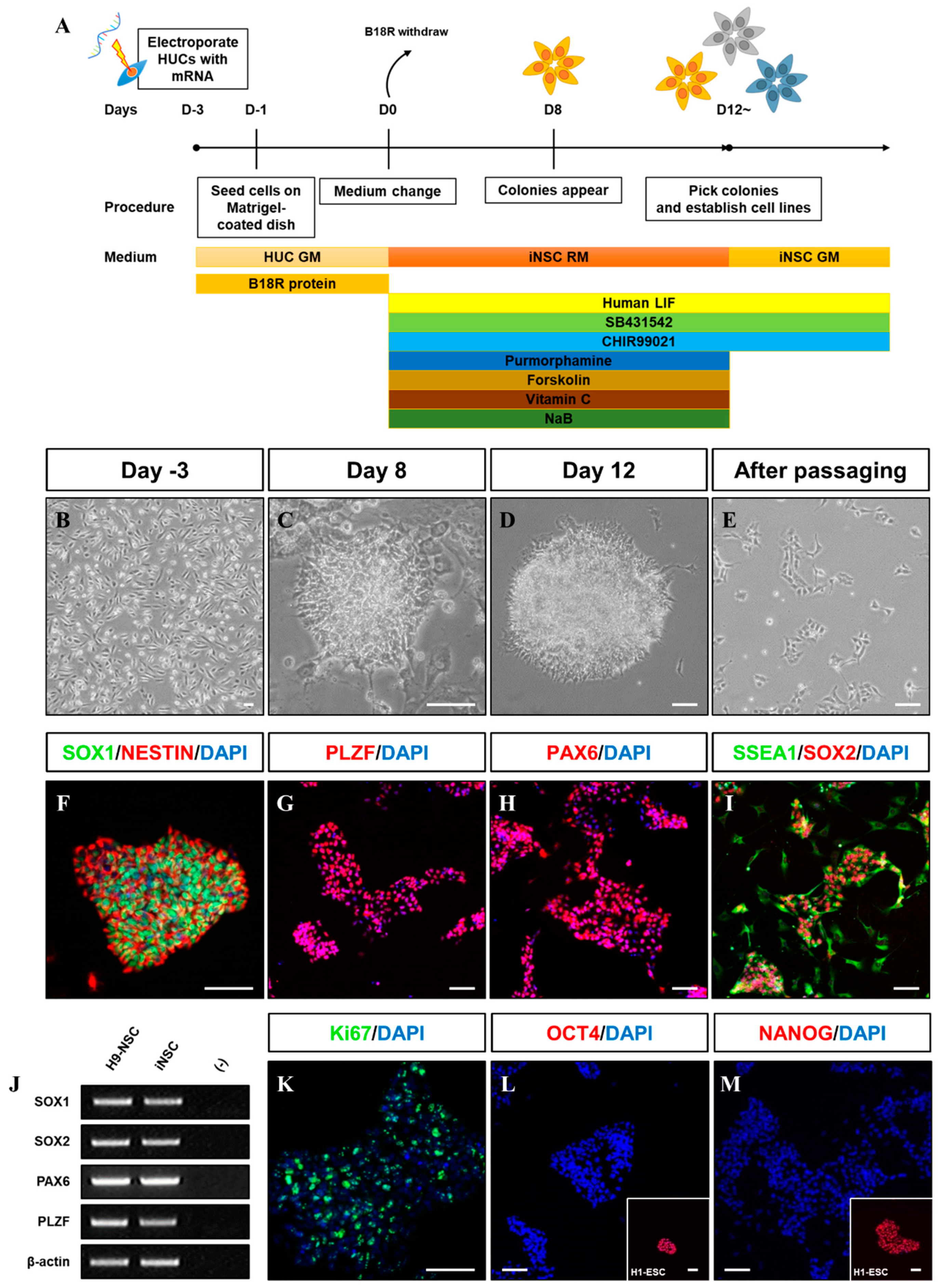
2.1. Generation of iNSCs from HUCs
2.2. In Vitro Differentiation of iNSCs
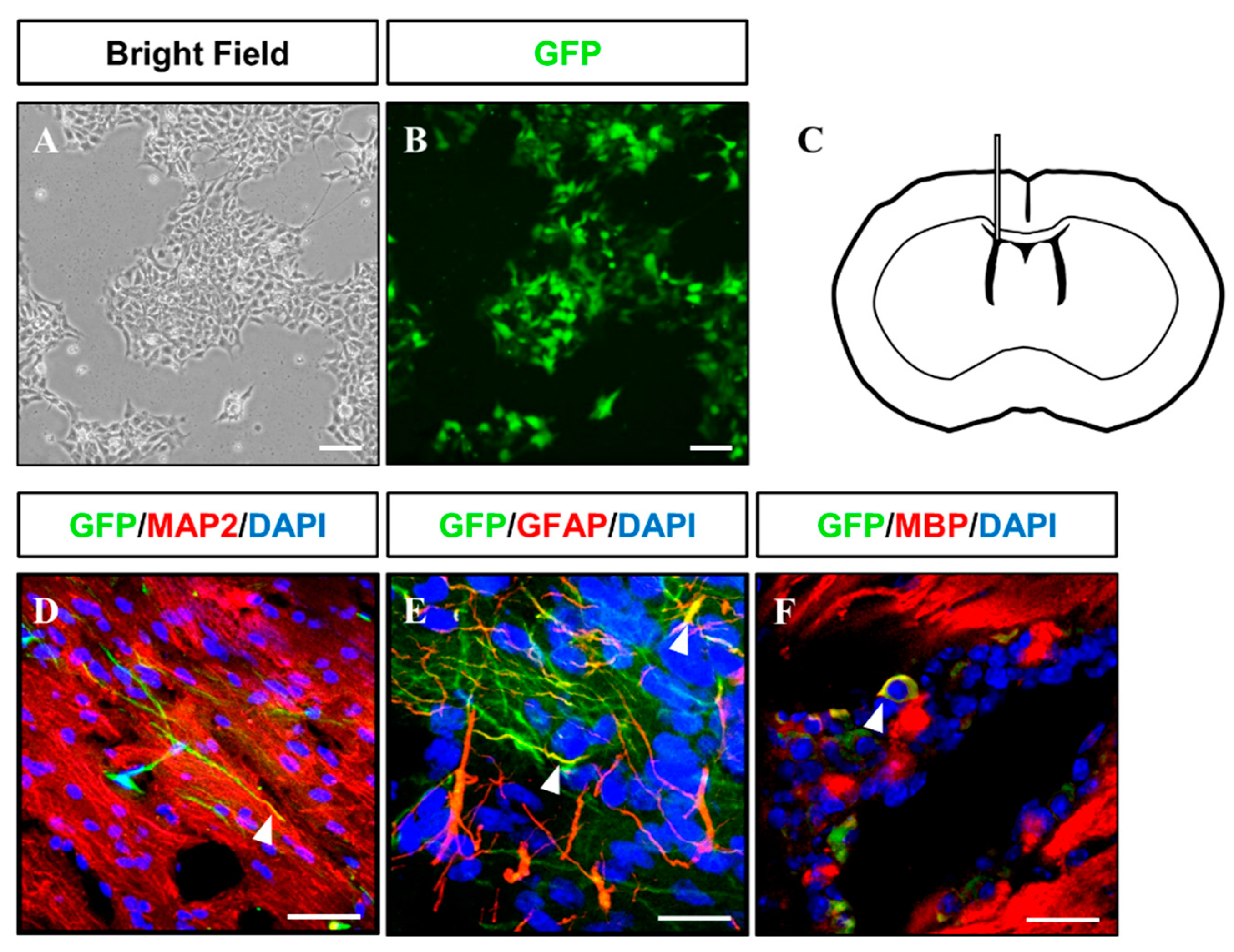
2.3. In Vivo Differentiation of iNSCs
2.4. RT-PCR and qRT-PCR
2.5. Immunocytochemistry
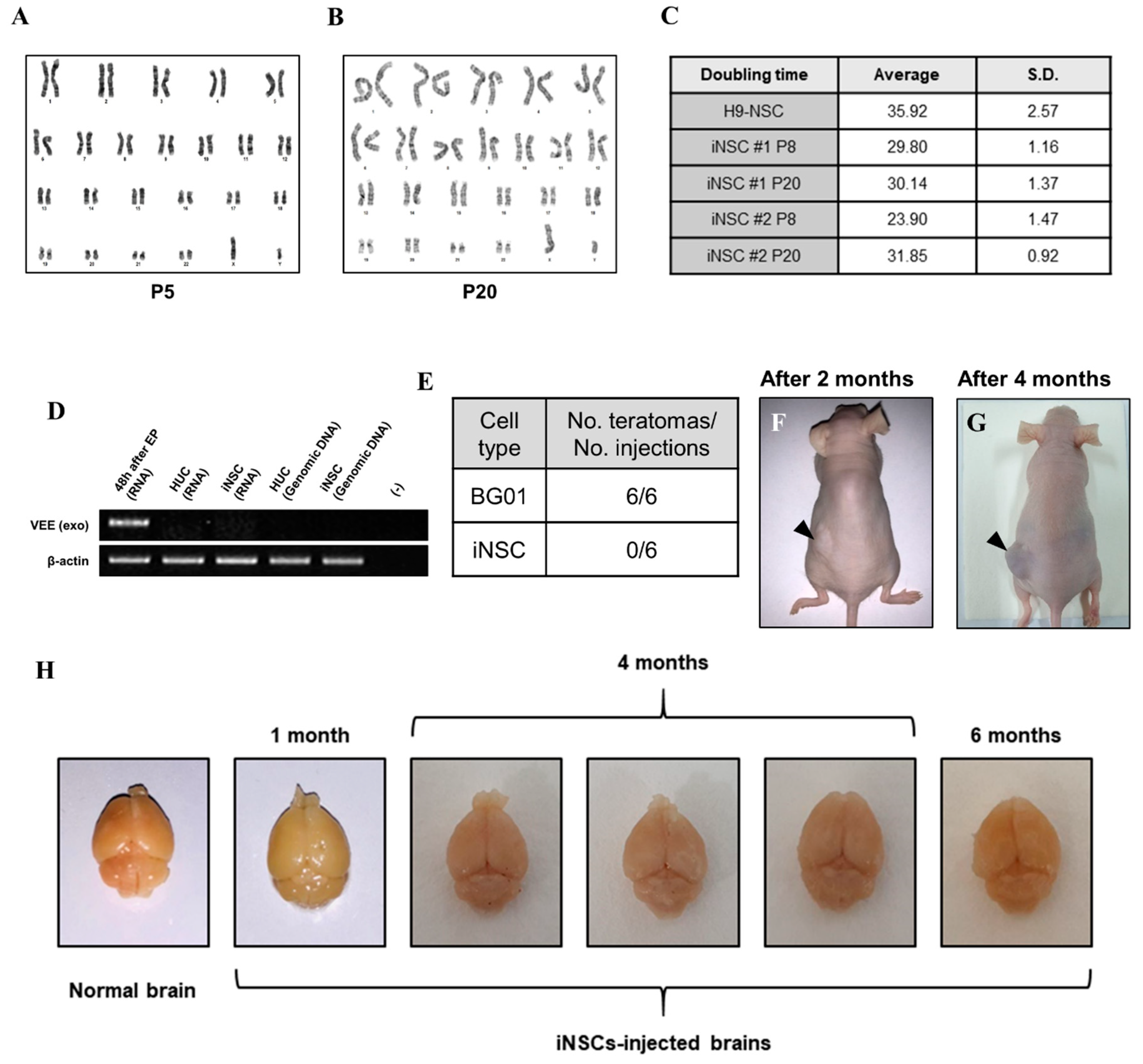
2.6. Karyotype Analysis
2.7. Teratoma Formation
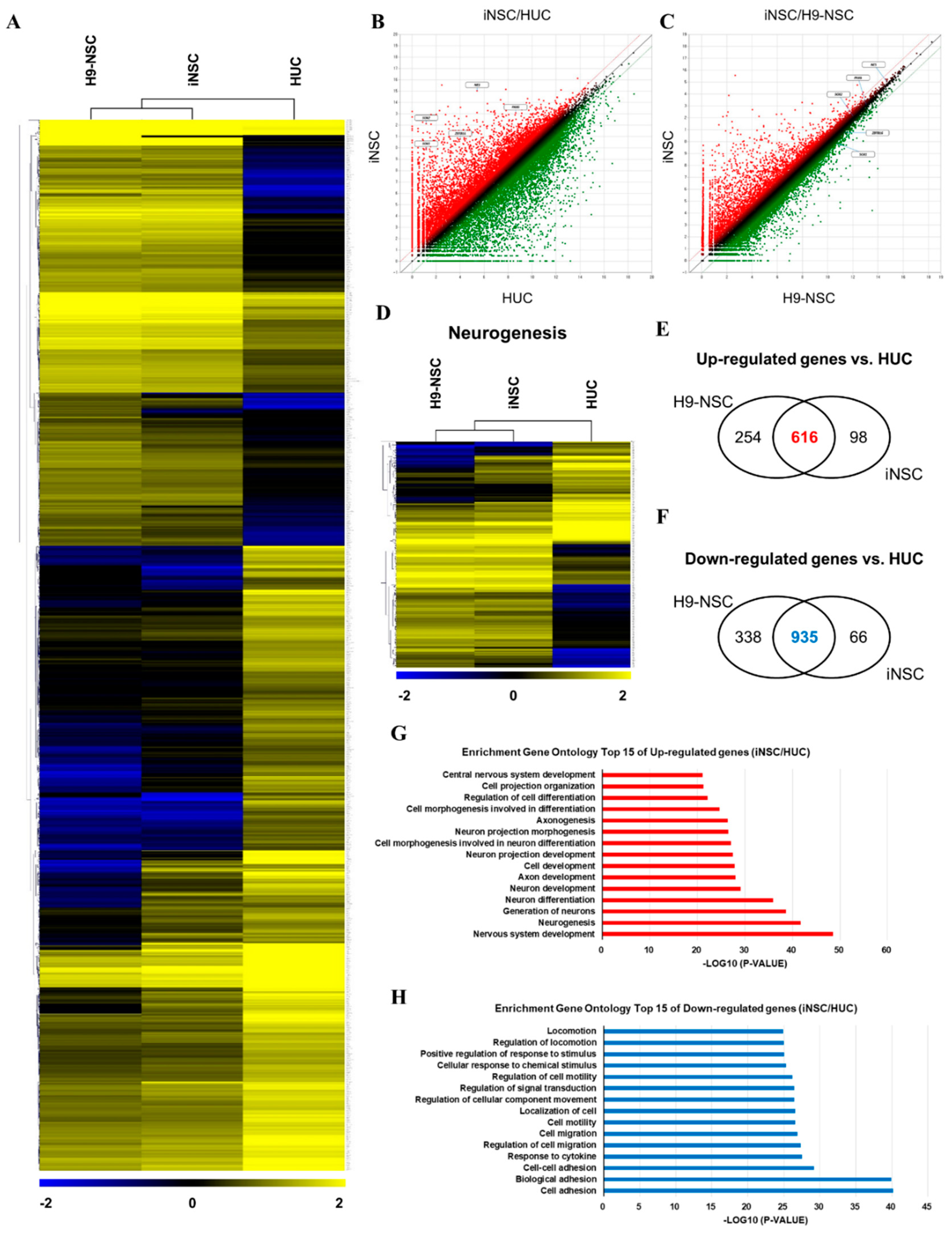
2.8. RNA-Sequencing Experiment
2.9. Whole Cell Patch Clamp Recordings
2.10. Statistical Analysis
2.11. Accession Numbers
3. Results
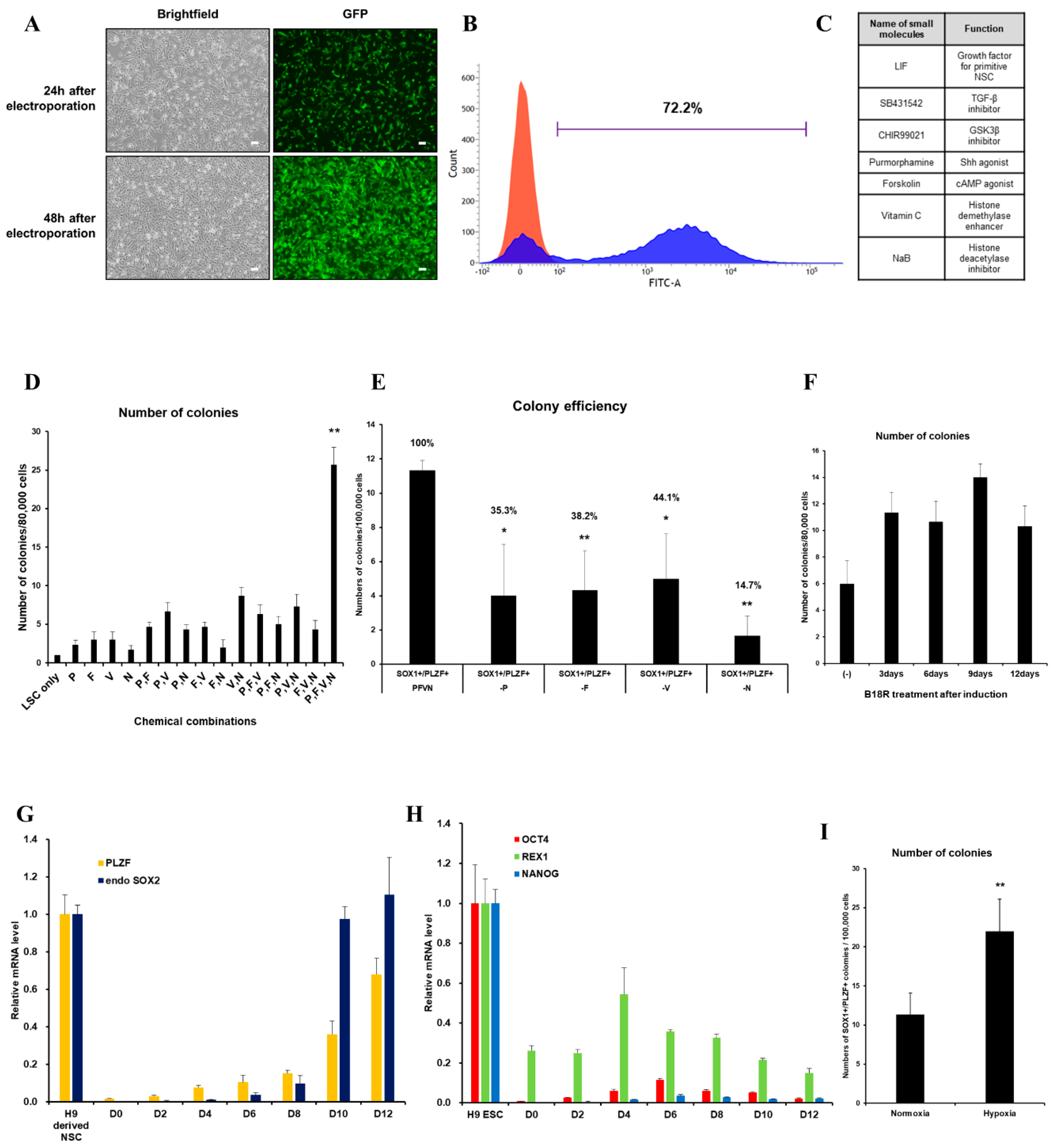
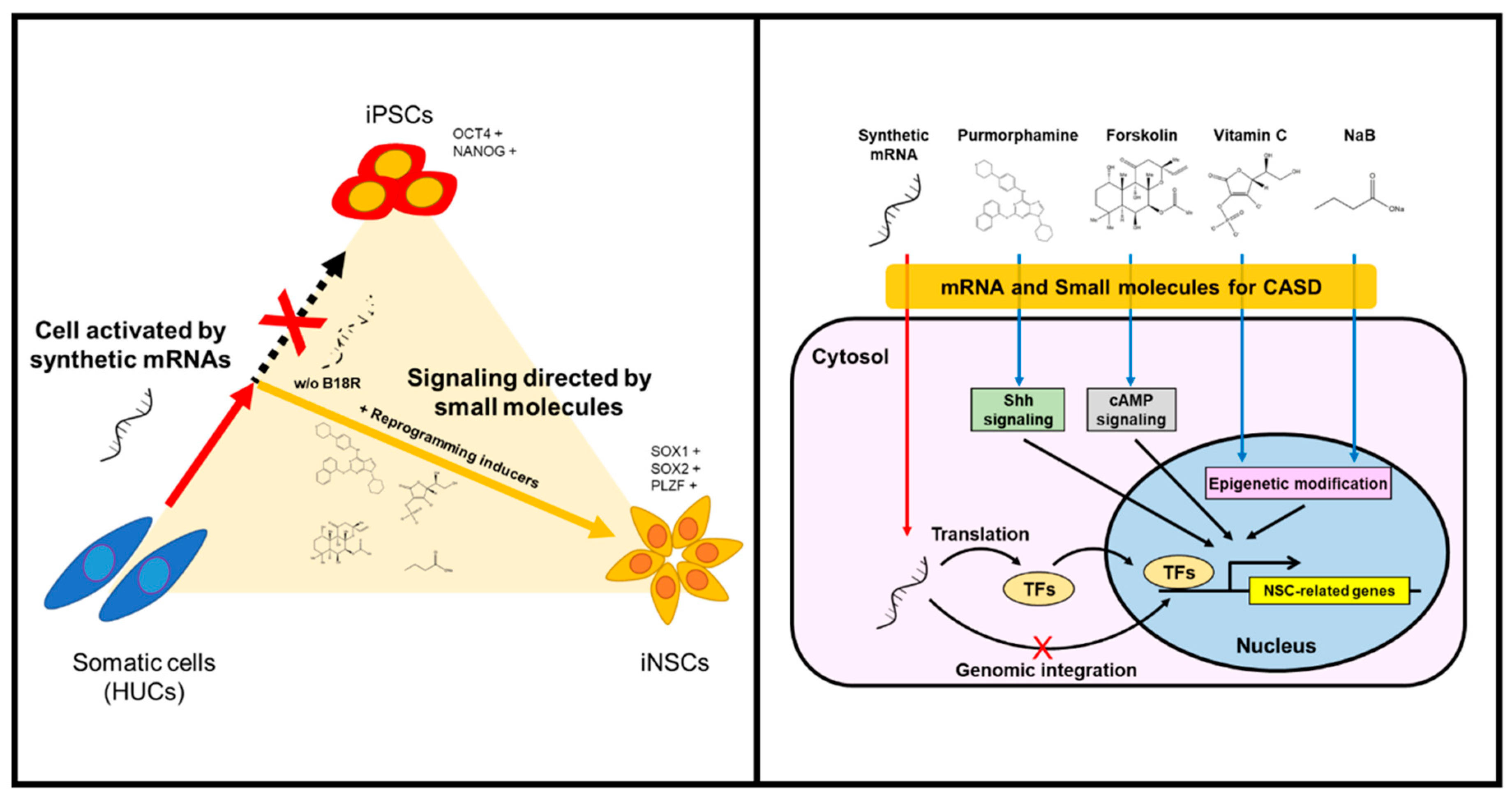
3.1. Optimized-Conditioning by Small Molecules for Generating iNSCs from HUCs
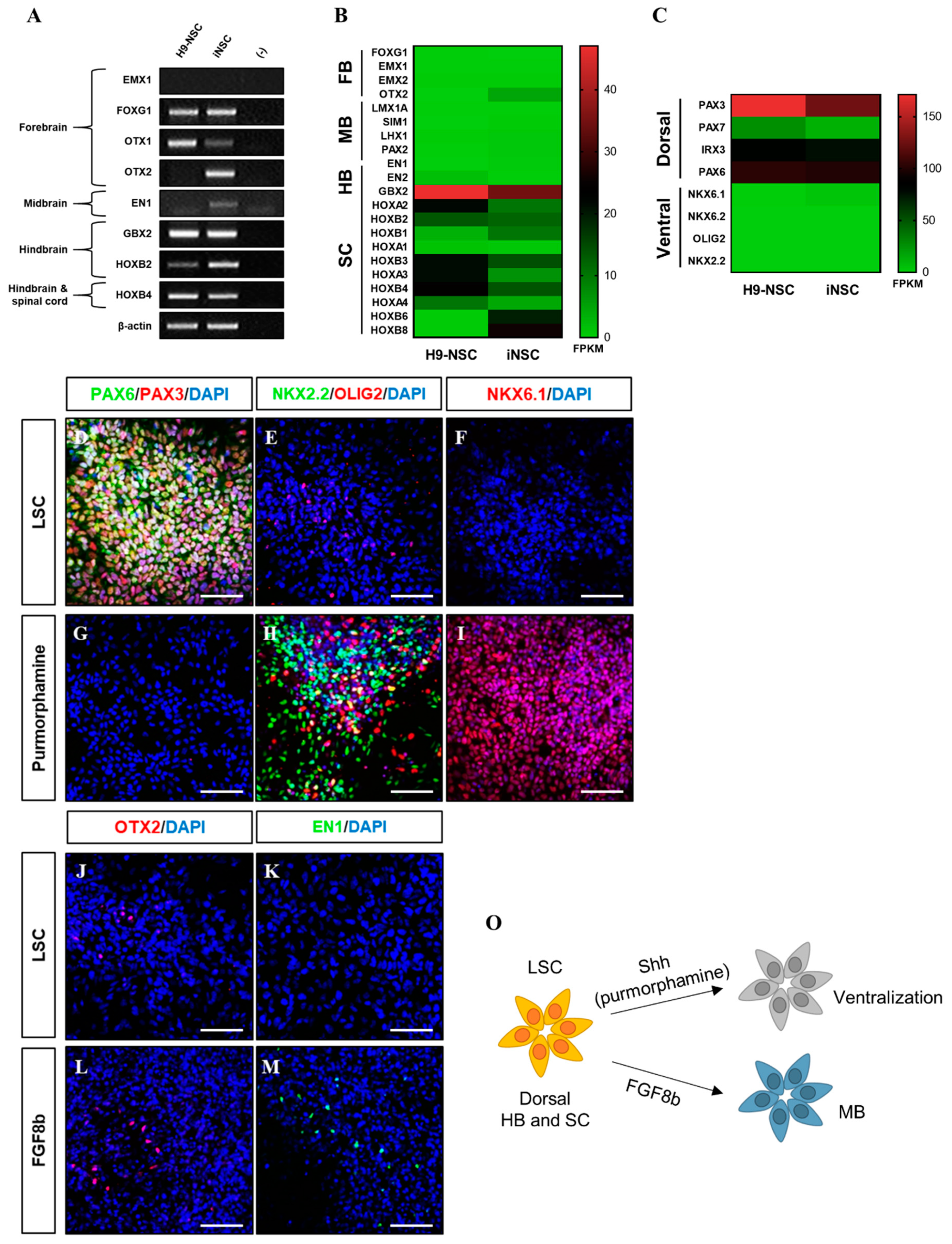
3.2. Characterization of iNSCs from HUCs by Synthetic mRNA with Small Molecules
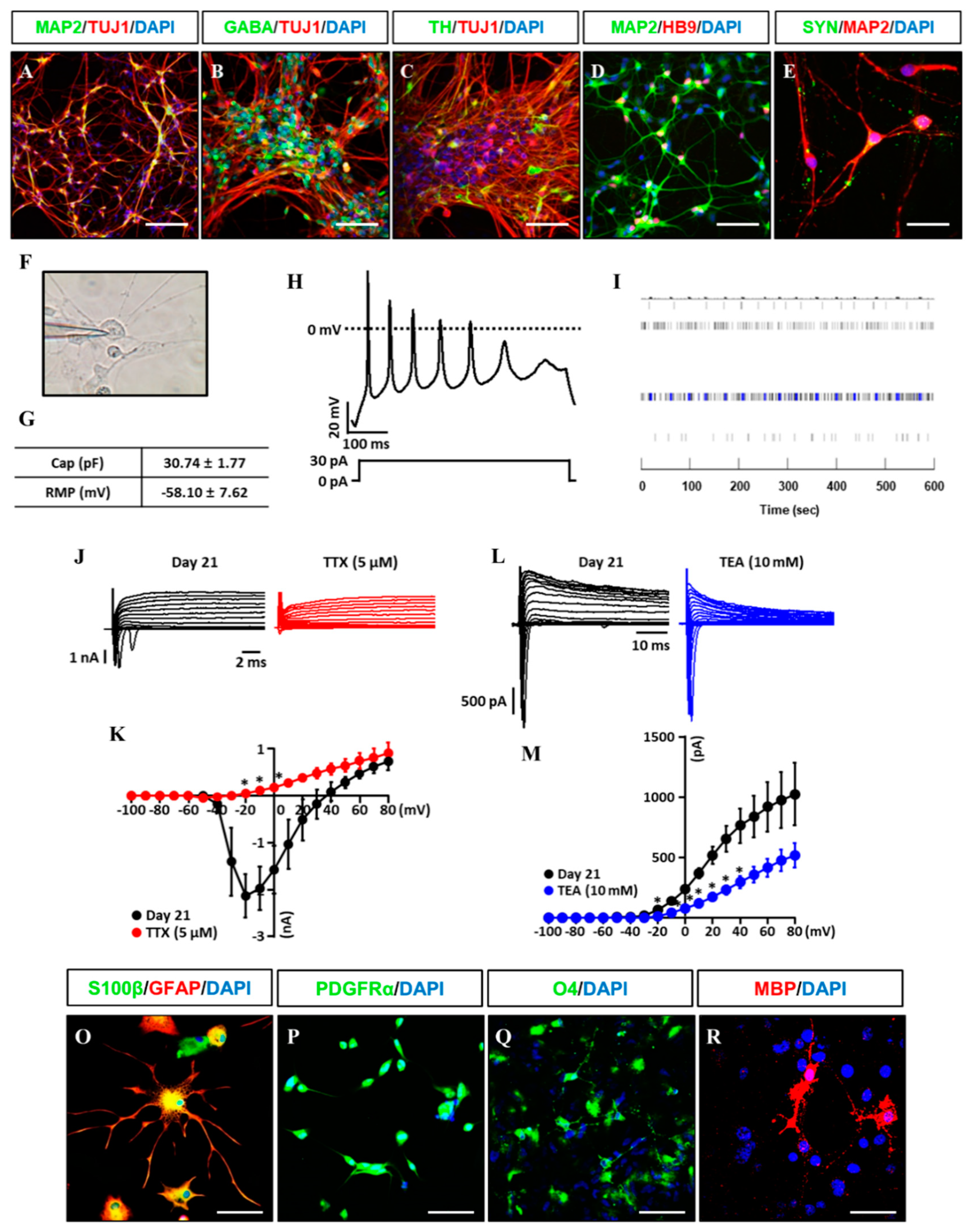
3.3. In Vitro/Vivo Differentiation Potential of iNSCs and Electrophysiological Assessment
3.4. Biosafety Evaluations of iNSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Takagi, Y. History of neural stem cell research and its clinical application. Neurol. Med. Chir. 2016, 56, 110–124. [Google Scholar] [CrossRef]
- Flax, J.D.; Aurora, S.; Yang, C.; Simonin, C.; Wills, A.M.; Billinghurst, L.L.; Jendoubi, M.; Sidman, R.L.; Wolfe, J.H.; Kim, S.U.; et al. Engraftable human neural stem cells respond to developmental cues, replace neurons, and express foreign genes. Nat. Biotechnol. 1998, 16, 1033–1039. [Google Scholar] [CrossRef]
- Gage, F.H.; Temple, S. Neural stem cells: Generating and regenerating the brain. Neuron 2013, 80, 588–601. [Google Scholar] [CrossRef]
- Nunes, M.C.; Roy, N.S.; Keyoung, H.M.; Goodman, R.R.; McKhann, G., 2nd; Jiang, L.; Kang, J.; Nedergaard, M.; Goldman, S.A. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 2003, 9, 439–447. [Google Scholar] [CrossRef]
- Studer, L.; Tabar, V.; McKay, R.D. Transplantation of expanded mesencephalic precursors leads to recovery in parkinsonian rats. Nat. Neurosci. 1998, 1, 290–295. [Google Scholar] [CrossRef]
- Casarosa, S.; Bozzi, Y.; Conti, L. Neural stem cells: Ready for therapeutic applications? Mol. Cell. Ther. 2014, 2, 31. [Google Scholar] [CrossRef]
- Jain, M.; Armstrong, R.J.; Tyers, P.; Barker, R.A.; Rosser, A.E. Gabaergic immunoreactivity is predominant in neurons derived from expanded human neural precursor cells in vitro. Exp. Neurol. 2003, 182, 113–123. [Google Scholar] [CrossRef]
- Yamanaka, S. Induced pluripotent stem cells: Past, present, and future. Cell Stem Cell 2012, 10, 678–684. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Ostermeier, A.; Pang, Z.P.; Kokubu, Y.; Sudhof, T.C.; Wernig, M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 2010, 463, 1035–1041. [Google Scholar] [CrossRef]
- Lujan, E.; Chanda, S.; Ahlenius, H.; Sudhof, T.C.; Wernig, M. Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2527–2532. [Google Scholar] [CrossRef]
- Han, D.W.; Tapia, N.; Hermann, A.; Hemmer, K.; Hoing, S.; Arauzo-Bravo, M.J.; Zaehres, H.; Wu, G.; Frank, S.; Moritz, S.; et al. Direct reprogramming of fibroblasts into neural stem cells by defined factors. Cell Stem Cell 2012, 10, 465–472. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Huang, P.; He, Z.; Ji, S.; Sun, H.; Xiang, D.; Liu, C.; Hu, Y.; Wang, X.; Hui, L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011, 475, 386–389. [Google Scholar] [CrossRef]
- Lu, J.; Liu, H.; Huang, C.T.; Chen, H.; Du, Z.; Liu, Y.; Sherafat, M.A.; Zhang, S.C. Generation of integration-free and region-specific neural progenitors from primate fibroblasts. Cell Rep. 2013, 3, 1580–1591. [Google Scholar] [CrossRef]
- Okita, K.; Yamanaka, S. Induced pluripotent stem cells: Opportunities and challenges. Philos. Trans. R. Soc. B Biol. Sci. 2011, 366, 2198–2207. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mrna. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef]
- Yoshioka, N.; Gros, E.; Li, H.R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.D.; Yu, B.D.; et al. Efficient generation of human ipscs by a synthetic self-replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Y.; Ding, S. A chemical approach to stem-cell biology and regenerative medicine. Nature 2008, 453, 338–344. [Google Scholar] [CrossRef]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Kang, P.J.; Moon, J.H.; Yoon, B.S.; Hyeon, S.; Jun, E.K.; Park, G.; Yun, W.; Park, J.; Park, M.; Kim, A.; et al. Reprogramming of mouse somatic cells into pluripotent stem-like cells using a combination of small molecules. Biomaterials 2014, 35, 7336–7345. [Google Scholar] [CrossRef]
- Park, G.; Yoon, B.S.; Kim, Y.S.; Choi, S.C.; Moon, J.H.; Kwon, S.; Hwang, J.; Yun, W.; Kim, J.H.; Park, C.Y.; et al. Conversion of mouse fibroblasts into cardiomyocyte-like cells using small molecule treatments. Biomaterials 2015, 54, 201–212. [Google Scholar] [CrossRef]
- Zhang, M.; Lin, Y.H.; Sun, Y.J.; Zhu, S.; Zheng, J.; Liu, K.; Cao, N.; Li, K.; Huang, Y.; Ding, S. Pharmacological reprogramming of fibroblasts into neural stem cells by signaling-directed transcriptional activation. Cell Stem Cell 2016, 18, 653–667. [Google Scholar] [CrossRef]
- Zheng, J.; Choi, K.A.; Kang, P.J.; Hyeon, S.; Kwon, S.; Moon, J.H.; Hwang, I.; Kim, Y.I.; Kim, Y.S.; Yoon, B.S.; et al. A combination of small molecules directly reprograms mouse fibroblasts into neural stem cells. Biochem. Biophys. Res. Commun. 2016, 476, 42–48. [Google Scholar] [CrossRef]
- Li, W.; Sun, W.; Zhang, Y.; Wei, W.; Ambasudhan, R.; Xia, P.; Talantova, M.; Lin, T.; Kim, J.; Wang, X.; et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. USA 2011, 108, 8299–8304. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martinez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in es cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Esteban, M.A.; Wang, T.; Qin, B.; Yang, J.; Qin, D.; Cai, J.; Li, W.; Weng, Z.; Chen, J.; Ni, S.; et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 2010, 6, 71–79. [Google Scholar] [CrossRef]
- Kim, H.J.; Leeds, P.; Chuang, D.M. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J. Neurochem. 2009, 110, 1226–1240. [Google Scholar] [CrossRef]
- Mali, P.; Chou, B.K.; Yen, J.; Ye, Z.; Zou, J.; Dowey, S.; Brodsky, R.A.; Ohm, J.E.; Yu, W.; Baylin, S.B.; et al. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells 2010, 28, 713–720. [Google Scholar] [CrossRef]
- Merz, K.; Herold, S.; Lie, D.C. CREB in adult neurogenesis—Master and partner in the development of adult-born neurons? Eur. J. Neurosci. 2011, 33, 1078–1086. [Google Scholar] [CrossRef]
- Nakagawa, S.; Kim, J.E.; Lee, R.; Malberg, J.E.; Chen, J.; Steffen, C.; Zhang, Y.J.; Nestler, E.J.; Duman, R.S. Regulation of neurogenesis in adult mouse hippocampus by camp and the camp response element-binding protein. J. Neurosci. 2002, 22, 3673–3682. [Google Scholar] [CrossRef]
- Shin, D.M.; Ahn, J.I.; Lee, K.H.; Lee, Y.S.; Lee, Y.S. Ascorbic acid responsive genes during neuronal differentiation of embryonic stem cells. Neuroreport 2004, 15, 1959–1963. [Google Scholar] [CrossRef]
- Cheng, L.; Hu, W.; Qiu, B.; Zhao, J.; Yu, Y.; Guan, W.; Wang, M.; Yang, W.; Pei, G. Generation of neural progenitor cells by chemical cocktails and hypoxia. Cell Res. 2014, 24, 665–679. [Google Scholar] [CrossRef]
- Rowitch, D.H. Glial specification in the vertebrate neural tube. Nat. Rev. Neurosci. 2004, 5, 409–419. [Google Scholar] [CrossRef]
- De Filippis, L.; Delia, D. Hypoxia in the regulation of neural stem cells. Cell. Mol. Life Sci. 2011, 68, 2831–2844. [Google Scholar] [CrossRef]
- Mohyeldin, A.; Garzon-Muvdi, T.; Quinones-Hinojosa, A. Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor glis1. Nature 2011, 474, 225–229. [Google Scholar] [CrossRef]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef]
- Sommer, C.A.; Christodoulou, C.; Gianotti-Sommer, A.; Shen, S.S.; Sailaja, B.S.; Hezroni, H.; Spira, A.; Meshorer, E.; Kotton, D.N.; Mostoslavsky, G. Residual expression of reprogramming factors affects the transcriptional program and epigenetic signatures of induced pluripotent stem cells. PLoS ONE 2012, 7, e51711. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, P.J.; Son, D.; Ko, T.H.; Hong, W.; Yun, W.; Jang, J.; Choi, J.-I.; Song, G.; Lee, J.; Kim, I.Y.; et al. mRNA-Driven Generation of Transgene-Free Neural Stem Cells from Human Urine-Derived Cells. Cells 2019, 8, 1043. https://doi.org/10.3390/cells8091043
Kang PJ, Son D, Ko TH, Hong W, Yun W, Jang J, Choi J-I, Song G, Lee J, Kim IY, et al. mRNA-Driven Generation of Transgene-Free Neural Stem Cells from Human Urine-Derived Cells. Cells. 2019; 8(9):1043. https://doi.org/10.3390/cells8091043
Chicago/Turabian StyleKang, Phil Jun, Daryeon Son, Tae Hee Ko, Wonjun Hong, Wonjin Yun, Jihoon Jang, Jong-Il Choi, Gwonhwa Song, Jangbo Lee, In Yong Kim, and et al. 2019. "mRNA-Driven Generation of Transgene-Free Neural Stem Cells from Human Urine-Derived Cells" Cells 8, no. 9: 1043. https://doi.org/10.3390/cells8091043
APA StyleKang, P. J., Son, D., Ko, T. H., Hong, W., Yun, W., Jang, J., Choi, J.-I., Song, G., Lee, J., Kim, I. Y., & You, S. (2019). mRNA-Driven Generation of Transgene-Free Neural Stem Cells from Human Urine-Derived Cells. Cells, 8(9), 1043. https://doi.org/10.3390/cells8091043