Prediction of Potential miRNA–Disease Associations Through a Novel Unsupervised Deep Learning Framework with Variational Autoencoder



Abstract
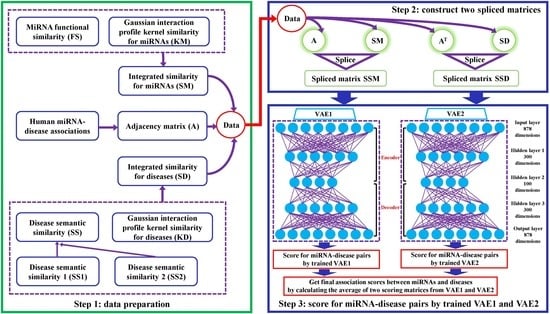
1. Introduction
2. Materials and Methods
2.1. Data Preparation
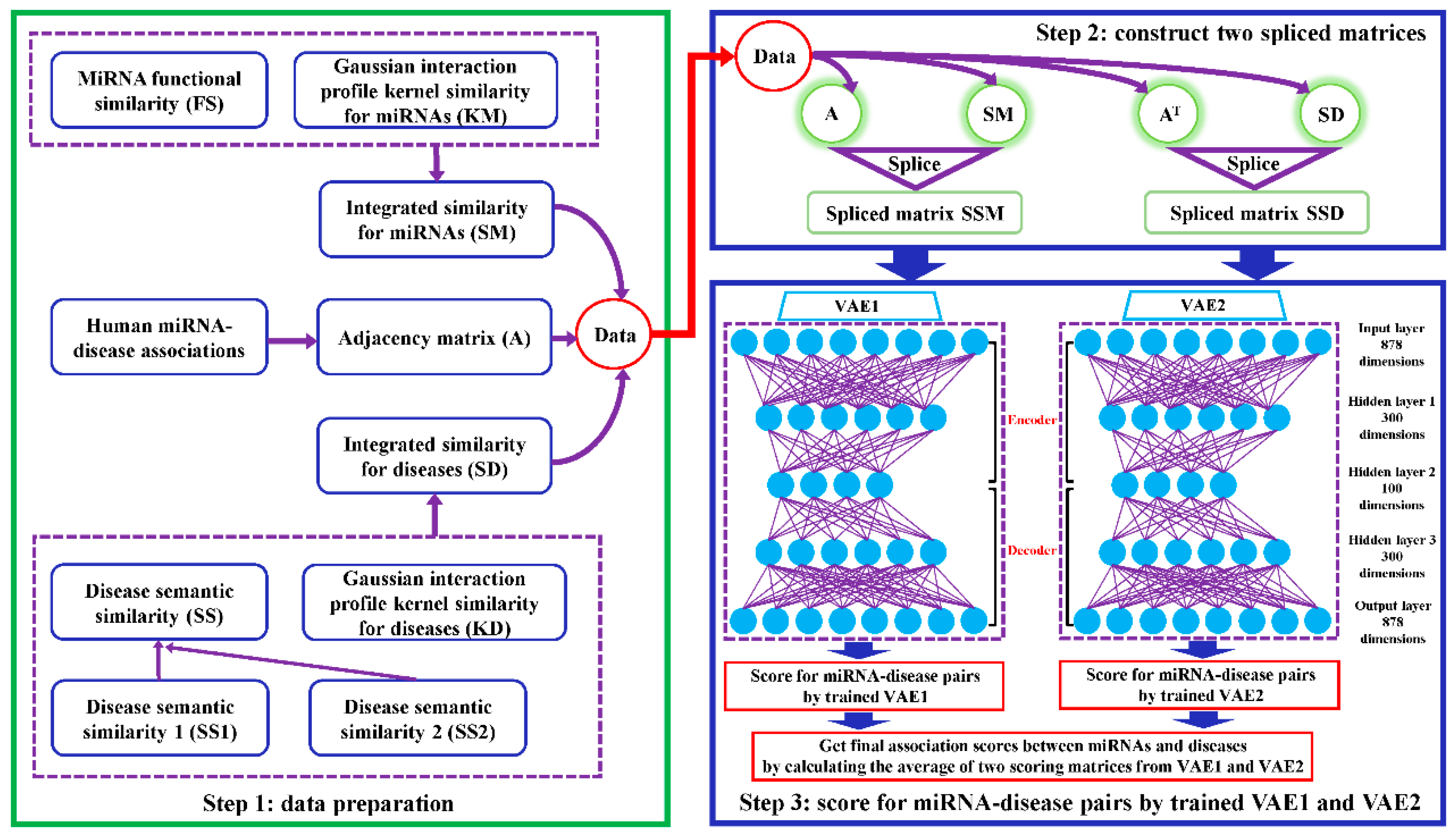
2.2. VAEMDA
3. Results
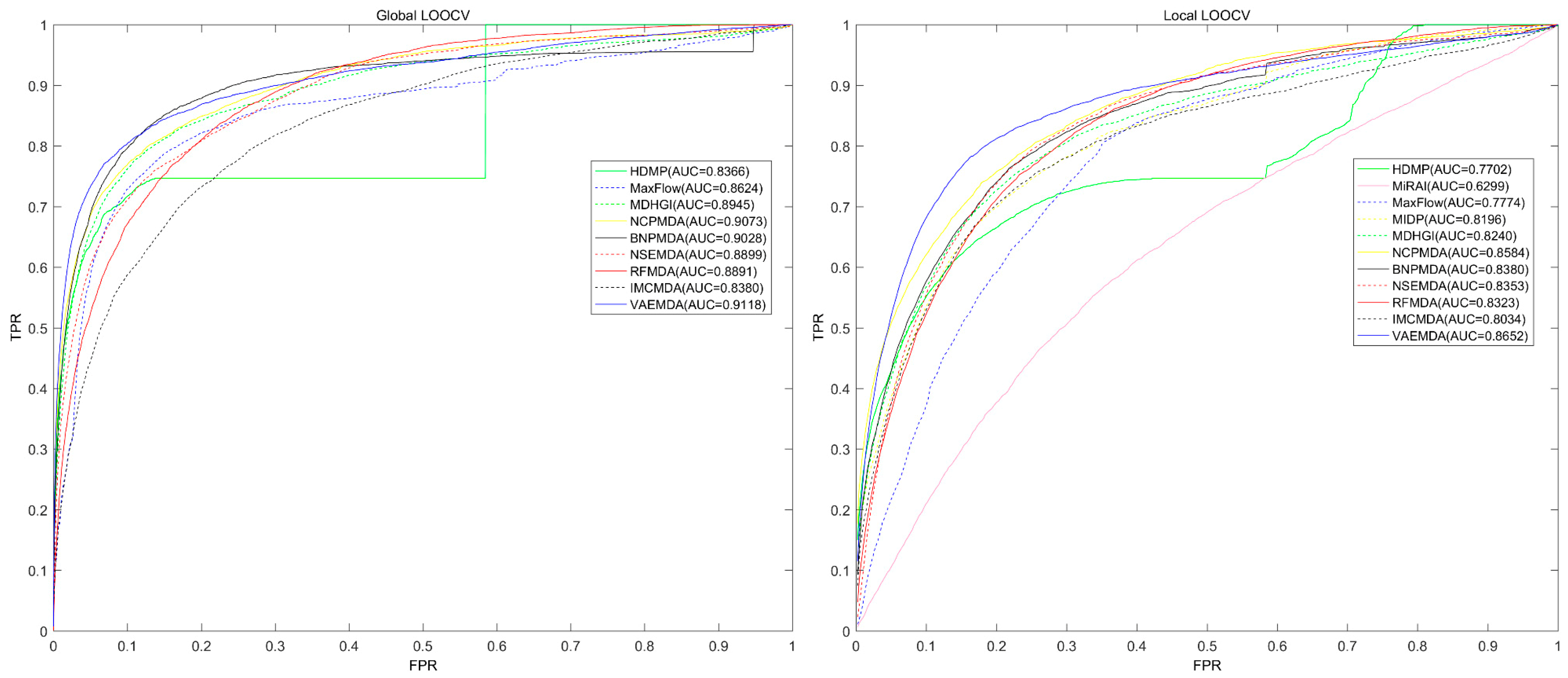
3.1. Performance Evaluation
3.2. Case Studies
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Tavazoie, S.F.; Alarcon, C.; Oskarsson, T.; Padua, D.; Wang, Q.; Bos, P.D.; Gerald, W.L.; Massague, J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 2008, 451, 147–152. [Google Scholar] [CrossRef]
- Ma, L.; Teruya-Feldstein, J.; Weinberg, R.A. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 2007, 449, 682–688. [Google Scholar] [CrossRef]
- Wahlquist, C.; Jeong, D.; Rojas-Munoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.; Doevendans, P.A.; et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef]
- Rostamian Delavar, M.; Baghi, M.; Safaeinejad, Z.; Kiani-Esfahani, A.; Ghaedi, K.; Nasr-Esfahani, M.H. Differential expression of miR-34a, miR-141, and miR-9 in MPP+-treated differentiated PC12 cells as a model of Parkinson’s disease. Gene 2018, 662, 54–65. [Google Scholar] [CrossRef]
- Amin, N.D.; Bai, G.; Klug, J.R.; Bonanomi, D.; Pankratz, M.T.; Gifford, W.D.; Hinckley, C.A.; Sternfeld, M.J.; Driscoll, S.P.; Dominguez, B.; et al. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 2015, 350, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.F.; Xu, Y.W.; Li, J.; Niu, H.L.; Ma, W.X.; Xu, J.; Zhou, P.R.; Liu, X.; Ye, D.L.; Liu, X.R.; et al. Mir20a/106a-WTX axis regulates RhoGDIa/CDC42 signaling and colon cancer progression. Nat. Commun. 2019, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Chen, L. A Highly Predictive Model for Diagnosis of Colorectal Neoplasms Using Plasma MicroRNA: Improving Specificity and Sensitivity. Ann. Surg. 2018, 267, e57–e58. [Google Scholar] [CrossRef]
- Creevey, L.; Ryan, J.; Harvey, H.; Bray, I.M.; Meehan, M.; Khan, A.R.; Stallings, R.L. MicroRNA-497 increases apoptosis in MYCN amplified neuroblastoma cells by targeting the key cell cycle regulator WEE1. Mol. Cancer 2013, 12, 23. [Google Scholar] [CrossRef]
- Xia, J.; Li, L.; Li, T.; Fang, Z.; Zhang, K.; Zhou, J.; Peng, H.; Zhang, W. Detecting and characterizing microRNAs of diverse genomic origins via miRvial. Nucleic Acids Res. 2017, 45, e176. [Google Scholar] [CrossRef][Green Version]
- Romsos, E.L.; Vallone, P.M. Rapid PCR of STR markers: Applications to human identification. Forensic Sci. Int. Genet. 2015, 18, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Rupprom, K.; Chavalitshewinkoon-Petmitr, P.; Diraphat, P.; Kittigul, L. Evaluation of real-time RT-PCR assays for detection and quantification of norovirus genogroups I and II. Virol. Sin. 2017, 32, 139–146. [Google Scholar] [CrossRef]
- Zhang, X.; Ping, X.; Zhuang, H. Author Correction: Ultrasensitive Nano-rt-iPCR for Determination of Polybrominated Diphenyl Ethers in Natural Samples. Sci. Rep. 2018, 8, 9666. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Favaloro, E.J. Laboratory Testing for Activated Protein C Resistance (APCR). Methods Mol. Biol. 2017, 1646, 137–143. [Google Scholar]
- Fan, X.; Kurgan, L. Comprehensive overview and assessment of computational prediction of microRNA targets in animals. Brief. Bioinform. 2015, 16, 780–794. [Google Scholar] [CrossRef]
- Chen, X.; Xie, D.; Zhao, Q.; You, Z.H. MicroRNAs and complex diseases: From experimental results to computational models. Brief. Bioinform. 2019, 20, 515–539. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.X.; Yan, G.Y. RWRMDA: Predicting novel human microRNA-disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Xu, J.; Zhang, G.; Xu, L.; Li, C.; Wang, L.; Zhao, Z.; Jiang, W.; Guo, Z.; Li, X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Biol. 2013, 7, 101. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, Y.; Li, J.; Li, X.; Zhong, Y.; Zhang, Z.; Ding, J. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics 2015, 31, 1805–1815. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z.; et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar] [CrossRef]
- Mork, S.; Pletscher-Frankild, S.; Palleja Caro, A.; Gorodkin, J.; Jensen, L.J. Protein-driven inference of miRNA-disease associations. Bioinformatics 2014, 30, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.H.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef]
- Pasquier, C.; Gardes, J. Prediction of miRNA-disease associations with a vector space model. Sci. Rep. 2016, 6, 27036. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Liao, B.; Li, X.; Li, K. Network Consistency Projection for Human miRNA-Disease Associations Inference. Sci. Rep. 2016, 6, 36054. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Chen, X.; Lu, L. Large-scale prediction of microRNA-disease associations by combinatorial prioritization algorithm. Sci. Rep. 2017, 7, 43792. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xie, D.; Wang, L.; Zhao, Q.; You, Z.H.; Liu, H. BNPMDA: Bipartite Network Projection for MiRNA-Disease Association prediction. Bioinformatics 2018, 34, 3178–3186. [Google Scholar] [CrossRef]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018, 14, e1006418. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, G.Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2014, 4, 5501. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.N.; Li, J.Q. Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.F.; Yan, G.Y. RKNNMDA: Ranking-based KNN for MiRNA-Disease Association prediction. RNA Biol. 2017, 14, 952–962. [Google Scholar] [CrossRef]
- Chen, X.; Wang, C.C.; Yin, J.; You, Z.H. Novel Human miRNA-Disease Association Inference Based on Random Forest. Mol. Ther. Nucleic Acids 2018, 13, 568–579. [Google Scholar] [CrossRef]
- Wang, C.C.; Chen, X.; Yin, J.; Qu, J. An integrated framework for the identification of potential miRNA-disease association based on novel negative samples extraction strategy. RNA Biol. 2019, 16, 257–269. [Google Scholar] [CrossRef]
- Rampasek, L.; Hidru, D.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Dr.VAE: Improving drug response prediction via modeling of drug perturbation effects. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Rashid, S.; Shah, S.; Bar-Joseph, Z.; Pandya, R. Dhaka: Variational Autoencoder for Unmasking Tumor Heterogeneity from Single Cell Genomic Data. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Tezcan, K.C.; Baumgartner, C.F.; Luechinger, R.; Pruessmann, K.P.; Konukoglu, E. MR Image Reconstruction Using Deep Density Priors. IEEE Trans. Med. Imaging 2019, 38, 1633–1642. [Google Scholar] [CrossRef]
- Xu, W.; Tan, Y. Semisupervised Text Classification by Variational Autoencoder. IEEE Trans. Neural Netw. Learn. Syst. 2019, 1–14. [Google Scholar] [CrossRef]
- Titus, A.J.; Wilkins, O.M.; Bobak, C.A.; Christensen, B.C. An unsupervised deep learning framework with variational autoencoders for genome-wide DNA methylation analysis and biologic feature extraction applied to breast cancer. bioRxiv 2018, 433763. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014, 42, D1070–D1074. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, S.; Yang, Z.; Wang, Y. Tracking the Time Varying Neural Tuning via Adam on Point Process Observations. In Proceedings of the 40th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Honolulu, HI, USA, 18–21 July 2018; pp. 195–198. [Google Scholar]
- Wang, P.; Ge, R.; Xiao, X.; Cai, Y.; Wang, G.; Zhou, F. Rectified-Linear-Unit-Based Deep Learning for Biomedical Multi-label Data. Interdiscip. Sci. Comput. Life Sci. 2017, 9, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Wongsuphasawat, K.; Smilkov, D.; Wexler, J.; Wilson, J.; Mane, D.; Fritz, D.; Krishnan, D.; Viegas, F.B.; Wattenberg, M. Visualizing Dataflow Graphs of Deep Learning Models in TensorFlow. IEEE Trans. Vis. Comput. Graph. 2018, 24, 1–12. [Google Scholar] [CrossRef]
- Chen, X.; Huang, L.; Xie, D.; Zhao, Q. EGBMMDA: Extreme Gradient Boosting Machine for MiRNA-Disease Association prediction. Cell Death Dis. 2018, 9, 3. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Li, B.; Xu, W.W.; Han, L.; Chan, K.T.; Tsao, S.W.; Lee, N.P.Y.; Law, S.; Xu, L.Y.; Li, E.M.; Chan, K.W.; et al. MicroRNA-377 suppresses initiation and progression of esophageal cancer by inhibiting CD133 and VEGF. Oncogene 2017, 36, 3986–4000. [Google Scholar] [CrossRef]
- Koumangoye, R.B.; Andl, T.; Taubenslag, K.J.; Zilberman, S.T.; Taylor, C.J.; Loomans, H.A.; Andl, C.D. SOX4 interacts with EZH2 and HDAC3 to suppress microRNA-31 in invasive esophageal cancer cells. Mol. Cancer 2015, 14, 24. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Ng, C.K.Y.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, X.; Liu, Z.; Zhou, Z.; Wang, Y.; Tu, J.; Li, L.; Bao, H.; Yang, L.; Tu, K. MicroRNA-1296 inhibits metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma by targeting SRPK1-mediated PI3K/AKT pathway. Mol. Cancer 2017, 16, 103. [Google Scholar] [CrossRef]
- Xu, L.Z.; Li, S.S.; Zhou, W.; Kang, Z.J.; Zhang, Q.X.; Kamran, M.; Xu, J.; Liang, D.P.; Wang, C.L.; Hou, Z.J.; et al. p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene 2017, 36, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xu, C.; Guan, J.; Ping, Y.; Fan, H.; Li, Y.; Zhao, H.; Li, X. Discovering Dysfunction of Multiple MicroRNAs Cooperation in Disease by a Conserved MicroRNA Co-Expression Network. PLoS ONE 2012, 7, e32201. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-195 | dbDEMC | hsa-mir-144 | dbDEMC |
| hsa-mir-221 | dbDEMC | hsa-mir-30d | dbDEMC |
| hsa-mir-146b | dbDEMC | hsa-mir-7 | dbDEMC |
| hsa-mir-125b | dbDEMC | hsa-mir-337 | unconfirmed |
| hsa-mir-200b | dbDEMC | hsa-mir-107 | dbDEMC; miR2Disease |
| hsa-mir-9 | dbDEMC | hsa-mir-30c | dbDEMC |
| hsa-mir-29b | dbDEMC | hsa-mir-378a | unconfirmed |
| hsa-mir-24 | dbDEMC | hsa-mir-513a | unconfirmed |
| hsa-mir-106b | dbDEMC | hsa-mir-16 | dbDEMC |
| hsa-mir-30a | dbDEMC | hsa-mir-204 | 26722467 |
| hsa-mir-429 | dbDEMC | hsa-mir-367 | dbDEMC |
| hsa-mir-206 | dbDEMC | hsa-mir-422a | dbDEMC |
| hsa-mir-182 | dbDEMC | hsa-let-7g | dbDEMC |
| hsa-mir-103a | unconfirmed | hsa-mir-127 | dbDEMC |
| hsa-let-7e | dbDEMC | hsa-mir-142 | dbDEMC |
| hsa-mir-27b | dbDEMC | hsa-mir-198 | dbDEMC |
| hsa-mir-193b | dbDEMC | hsa-mir-125a | dbDEMC |
| hsa-mir-224 | dbDEMC | hsa-mir-23a | dbDEMC |
| hsa-mir-10b | dbDEMC | hsa-mir-197 | dbDEMC |
| hsa-mir-1 | dbDEMC | hsa-mir-96 | dbDEMC |
| hsa-mir-424 | dbDEMC | hsa-mir-20b | dbDEMC |
| hsa-mir-708 | 27092874 | hsa-mir-133b | dbDEMC |
| hsa-mir-32 | dbDEMC | hsa-mir-191 | dbDEMC |
| hsa-mir-17 | dbDEMC | hsa-mir-132 | dbDEMC |
| hsa-mir-222 | dbDEMC | hsa-mir-103b | unconfirmed |
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-mir-484 | HMDD v2.0 | hsa-mir-608 | HMDD v2.0 |
| hsa-mir-148a | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-218 | HMDD v2.0 |
| hsa-mir-29b | dbDEMC; HMDD v2.0 | hsa-mir-21 | miR2Disease; HMDD v2.0 |
| hsa-let-7b | miR2Disease; HMDD v2.0 | hsa-mir-490 | HMDD v2.0 |
| hsa-mir-181b | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-301a | HMDD v2.0 |
| hsa-mir-483 | HMDD v2.0 | hsa-mir-10b | HMDD v2.0 |
| hsa-mir-96 | miR2Disease;HMDD v2.0 | hsa-mir-638 | 28529597 |
| hsa-mir-34b | 28337312 | hsa-mir-221 | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-let-7e | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-326 | HMDD v2.0 |
| hsa-mir-320e | HMDD v2.0 | hsa-mir-362 | HMDD v2.0 |
| hsa-mir-1271 | HMDD v2.0 | hsa-mir-26 | HMDD v2.0 |
| hsa-mir-30c | miR2Disease; HMDD v2.0 | hsa-mir-320b | HMDD v2.0 |
| hsa-mir-26a | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-320d | HMDD v2.0 |
| hsa-mir-450b | HMDD v2.0 | hsa-mir-1202 | HMDD v2.0 |
| hsa-mir-629 | HMDD v2.0 | hsa-mir-519e | HMDD v2.0 |
| hsa-mir-409 | HMDD v2.0 | hsa-mir-187 | HMDD v2.0 |
| hsa-mir-503 | HMDD v2.0 | hsa-let-7g | miR2Disease; HMDD v2.0 |
| hsa-mir-320c | HMDD v2.0 | hsa-mir-92 | dbDEMC; HMDD v2.0 |
| hsa-mir-219 | miR2Disease; HMDD v2.0 | hsa-mir-302b | HMDD v2.0 |
| hsa-mir-181d | dbDEMC; HMDD v2.0 | hsa-mir-125a | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-mir-491 | HMDD v2.0 | hsa-let-7d | miR2Disease; HMDD v2.0 |
| hsa-let-7a | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-345 | HMDD v2.0 |
| hsa-mir-526a | HMDD v2.0 | hsa-mir-527 | HMDD v2.0 |
| hsa-mir-450a | HMDD v2.0 | hsa-mir-34c | HMDD v2.0 |
| hsa-let-7f | miR2Disease; HMDD v2.0 | hsa-let-7c | dbDEMC; miR2Disease; HMDD v2.0 |
| miRNA | Evidence | miRNA | Evidence |
|---|---|---|---|
| hsa-let-7b | dbDEMC; HMDD v2.0 | hsa-mir-126 | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-let-7g | dbDEMC; HMDD v2.0 | hsa-mir-135a | dbDEMC; HMDD v2.0 |
| hsa-mir-92b | dbDEMC | hsa-mir-128b | miR2Disease |
| hsa-mir-16 | dbDEMC; HMDD v2.0 | hsa-mir-24 | dbDEMC; HMDD v2.0 |
| hsa-let-7i | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-191 | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-let-7e | dbDEMC; HMDD v2.0 | hsa-mir-182 | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-mir-223 | dbDEMC; HMDD v2.0 | hsa-mir-27a | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-mir-99a | dbDEMC | hsa-mir-26a | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-mir-100 | dbDEMC; HMDD v2.0 | hsa-mir-195 | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-mir-92a | HMDD v2.0 | hsa-mir-150 | dbDEMC |
| hsa-mir-196b | dbDEMC | hsa-mir-454 | 28795052 |
| hsa-mir-99b | dbDEMC | hsa-mir-183 | dbDEMC; HMDD v2.0 |
| hsa-mir-142 | 25406066 | hsa-mir-30e | unconfirmed |
| hsa-mir-203 | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-342 | dbDEMC; HMDD v2.0 |
| hsa-mir-18b | dbDEMC;HMDD v2.0 | hsa-mir-372 | dbDEMC |
| hsa-mir-181a | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-95 | dbDEMC |
| hsa-let-7c | dbDEMC;HMDD v2.0 | hsa-mir-409 | HMDD v2.0 |
| hsa-mir-335 | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-31 | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-mir-130a | dbDEMC | hsa-mir-192 | dbDEMC |
| hsa-mir-199b | dbDEMC; HMDD v2.0 | hsa-mir-96 | dbDEMC; miR2Disease; HMDD v2.0 |
| hsa-mir-29c | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-323 | unconfirmed |
| hsa-mir-23b | dbDEMC;HMDD v2.0 | hsa-mir-181d | dbDEMC; miR2Disease |
| hsa-mir-101 | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-15b | dbDEMC |
| hsa-mir-224 | dbDEMC;HMDD v2.0 | hsa-mir-32 | dbDEMC |
| hsa-mir-373 | dbDEMC; miR2Disease; HMDD v2.0 | hsa-mir-378 | 25120807 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Chen, X.; Yin, J. Prediction of Potential miRNA–Disease Associations Through a Novel Unsupervised Deep Learning Framework with Variational Autoencoder. Cells 2019, 8, 1040. https://doi.org/10.3390/cells8091040
Zhang L, Chen X, Yin J. Prediction of Potential miRNA–Disease Associations Through a Novel Unsupervised Deep Learning Framework with Variational Autoencoder. Cells. 2019; 8(9):1040. https://doi.org/10.3390/cells8091040
Chicago/Turabian StyleZhang, Li, Xing Chen, and Jun Yin. 2019. "Prediction of Potential miRNA–Disease Associations Through a Novel Unsupervised Deep Learning Framework with Variational Autoencoder" Cells 8, no. 9: 1040. https://doi.org/10.3390/cells8091040
APA StyleZhang, L., Chen, X., & Yin, J. (2019). Prediction of Potential miRNA–Disease Associations Through a Novel Unsupervised Deep Learning Framework with Variational Autoencoder. Cells, 8(9), 1040. https://doi.org/10.3390/cells8091040