DROSHA-Dependent AIM2 Inflammasome Activation Contributes to Lung Inflammation during Idiopathic Pulmonary Fibrosis


Abstract
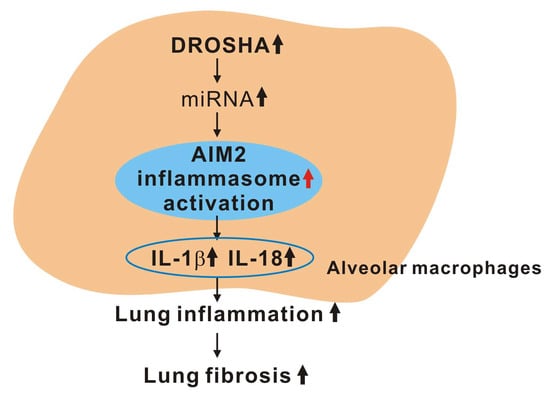
:
1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Animal Studies
2.3. Reagents and Antibodies
2.4. Cell Culture
2.5. Transduction of Drosha gRNA/Cas9 pPlasmid or Drosha Small Interfering RNA (siRNA)
2.6. Synthetic miRNAs
2.7. Immunoblot Analysis
2.8. Immunohistochemistry and Immunofluorescence Analysis
2.9. Cytokine Analysis
2.10. ASC Speck Formation Assay
2.11. Statistical Analysis
3. Results
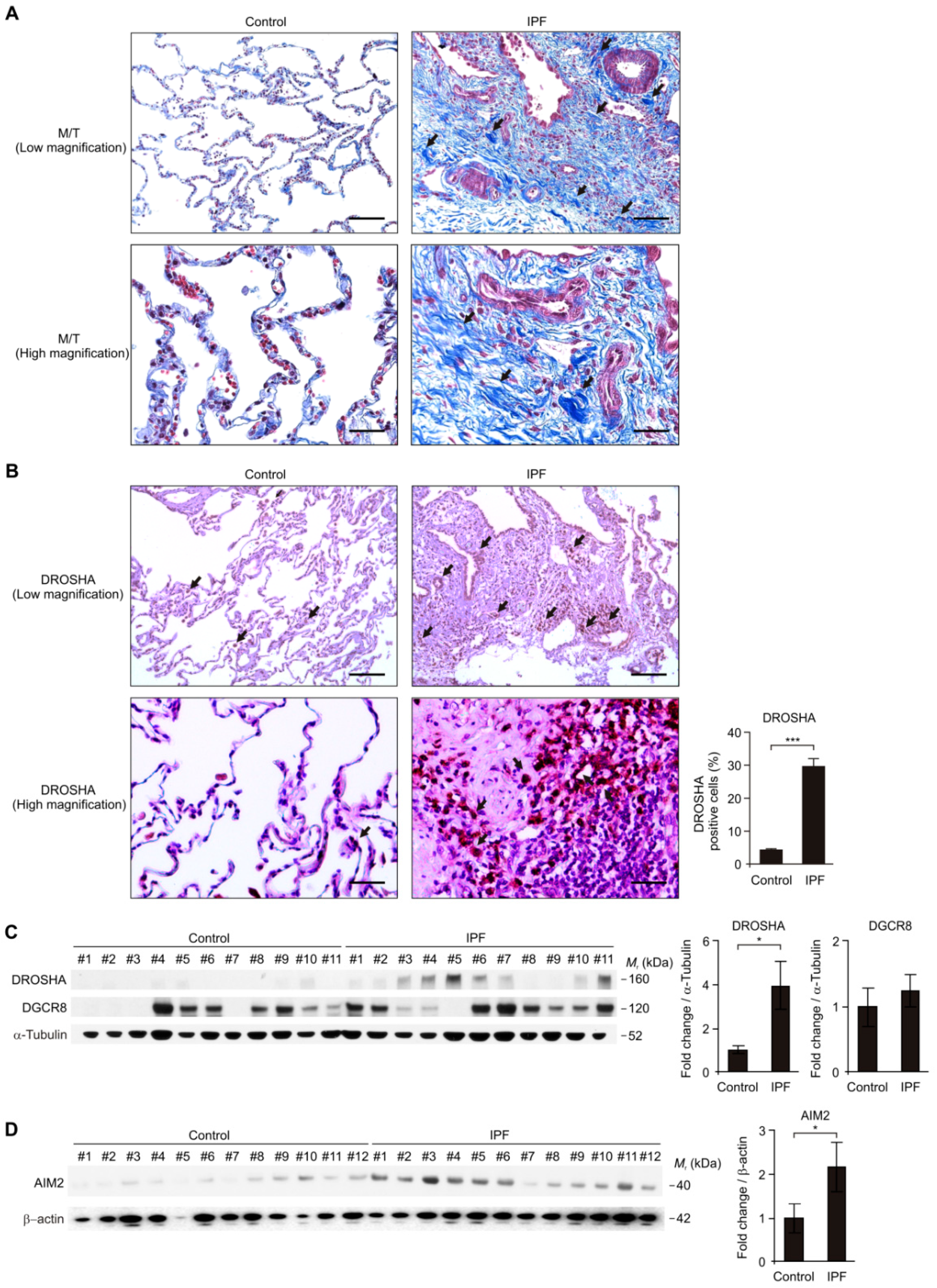
3.1. The DROSHA and AIM2 Protein Levels were Elevated in Lung Tissues of Patients with IPF
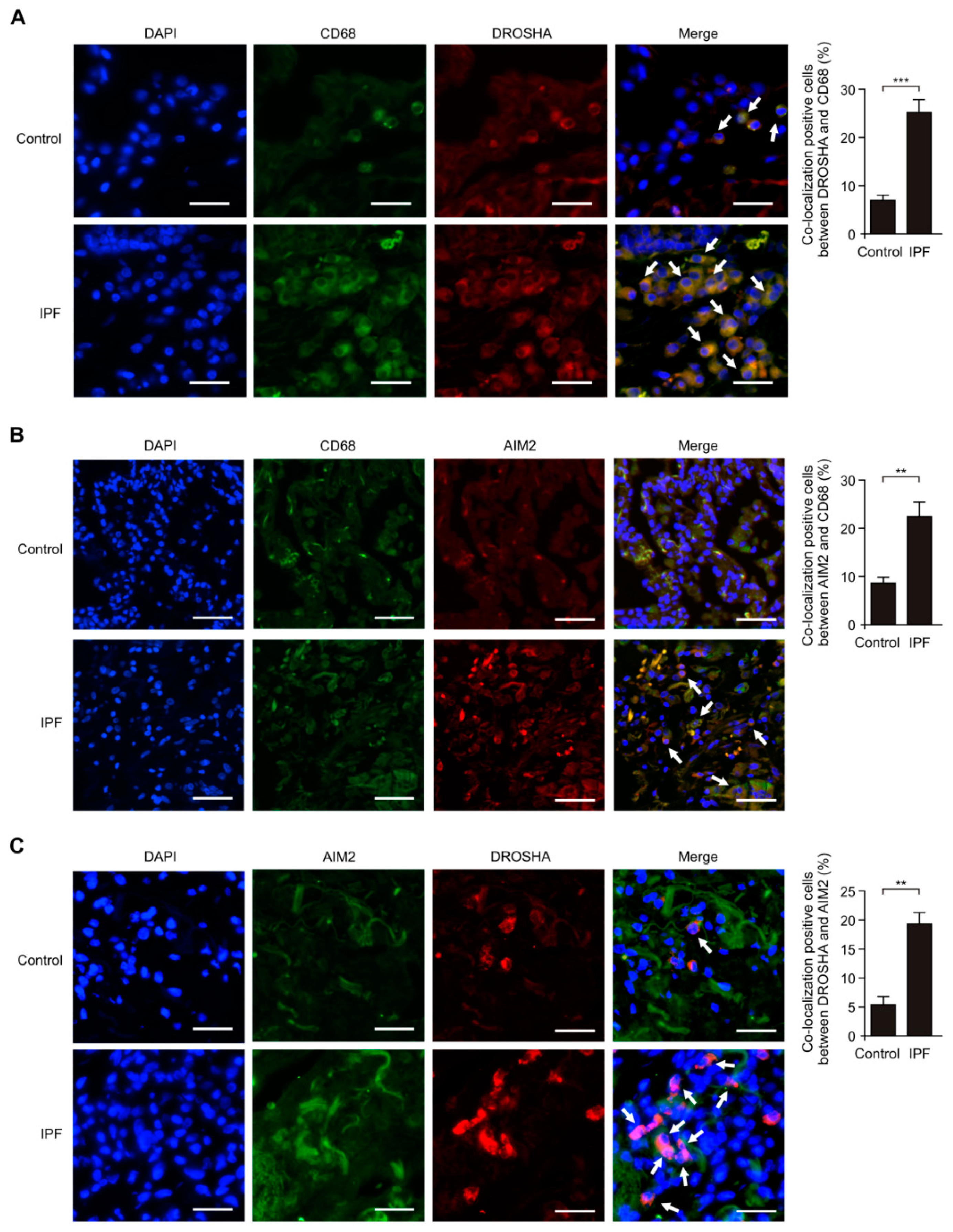
3.2. The DROSHA and AIM2 Expression Levels were Elevated in Alveolar Macrophages of Patients with IPF
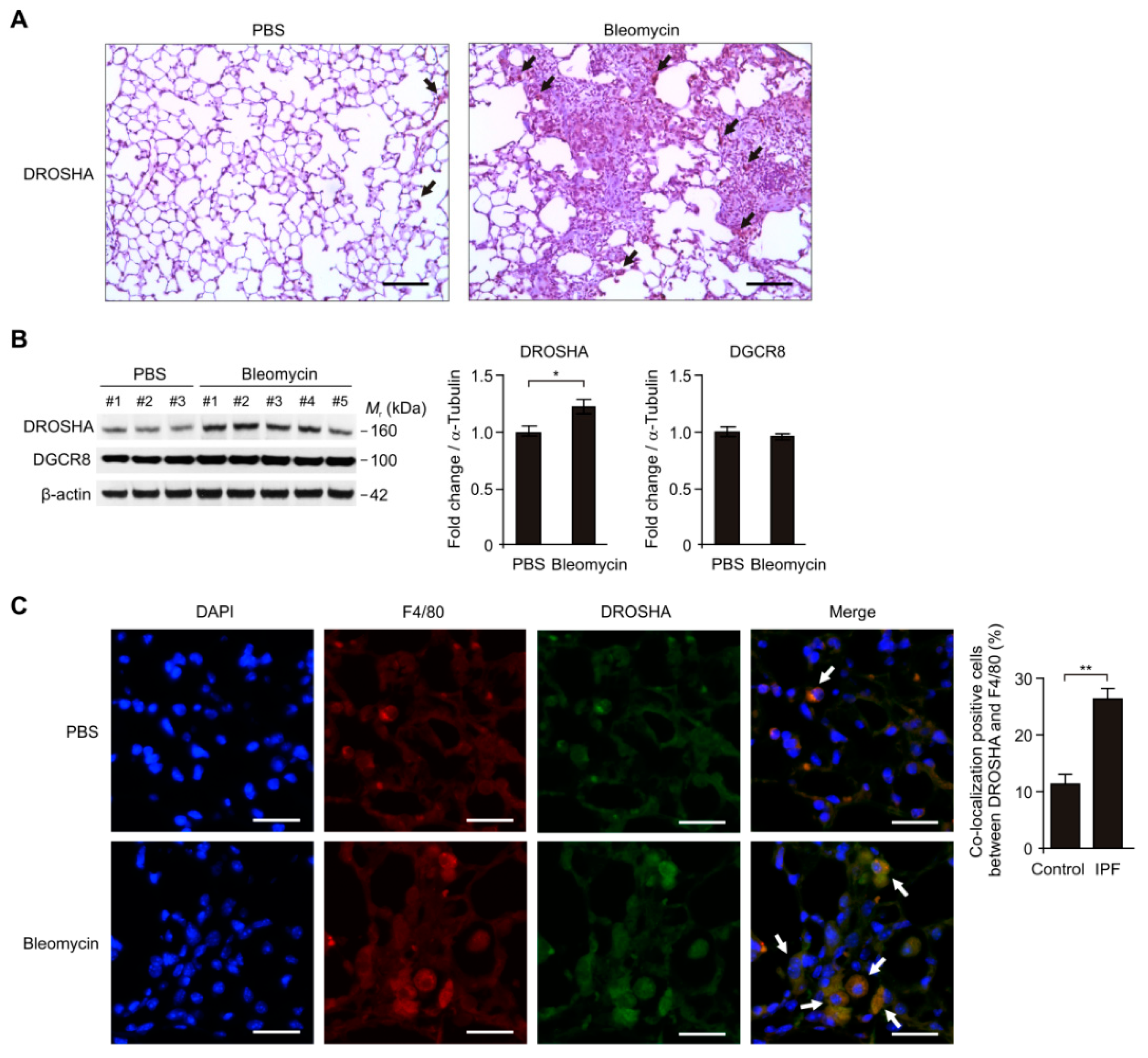
3.3. The DROSHA Protein Levels were Elevated in Alveolar Macrophages during Bleomycin-Induced Pulmonary Fibrosis
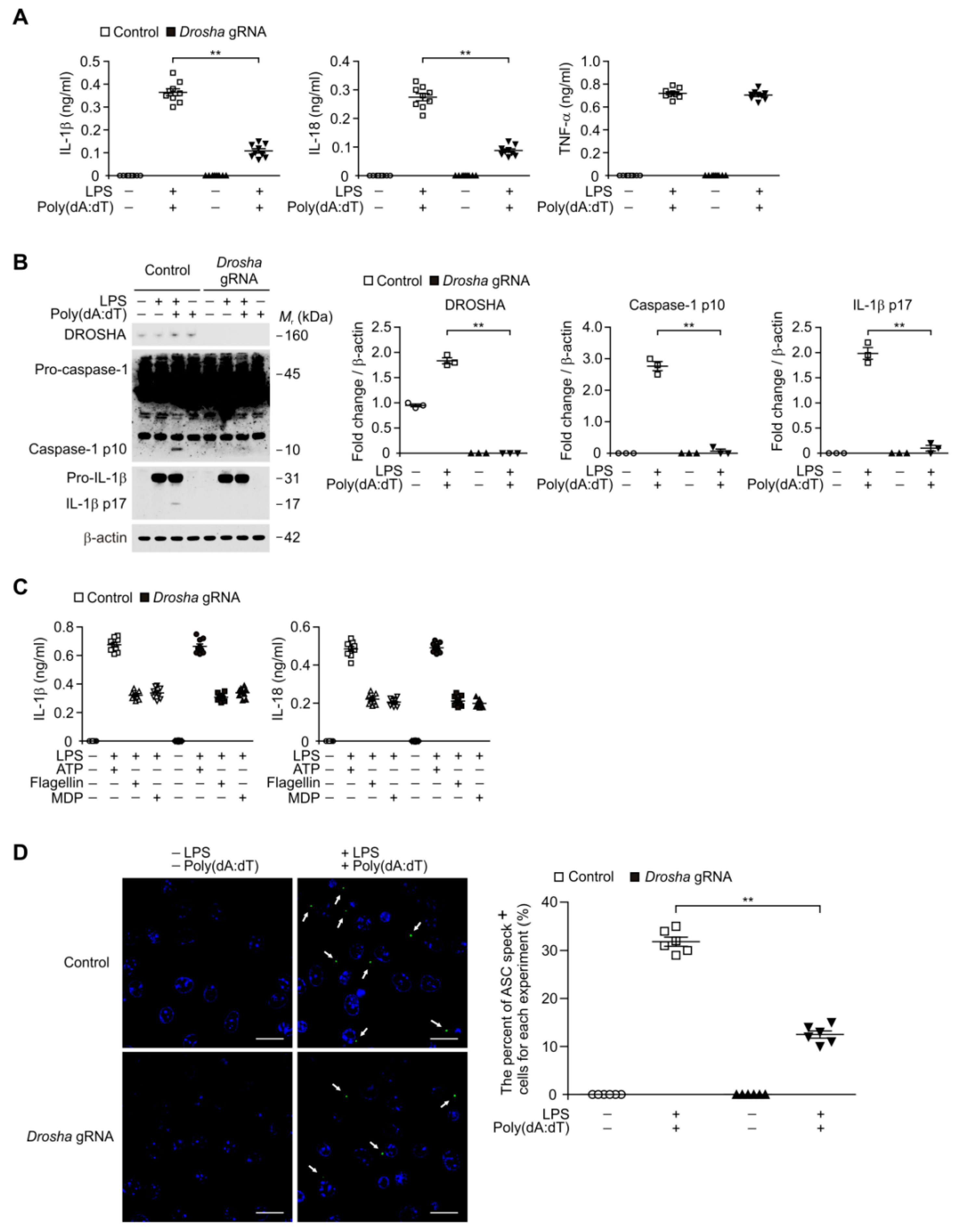
3.4. Deficiency of DROSHA Suppresses the AIM2 Inflammasome Activation in Alveolar Macrophages
3.5. Deficiency of DROSHA Suppresses the AIM2 Inflammasome Activation in Macrophages
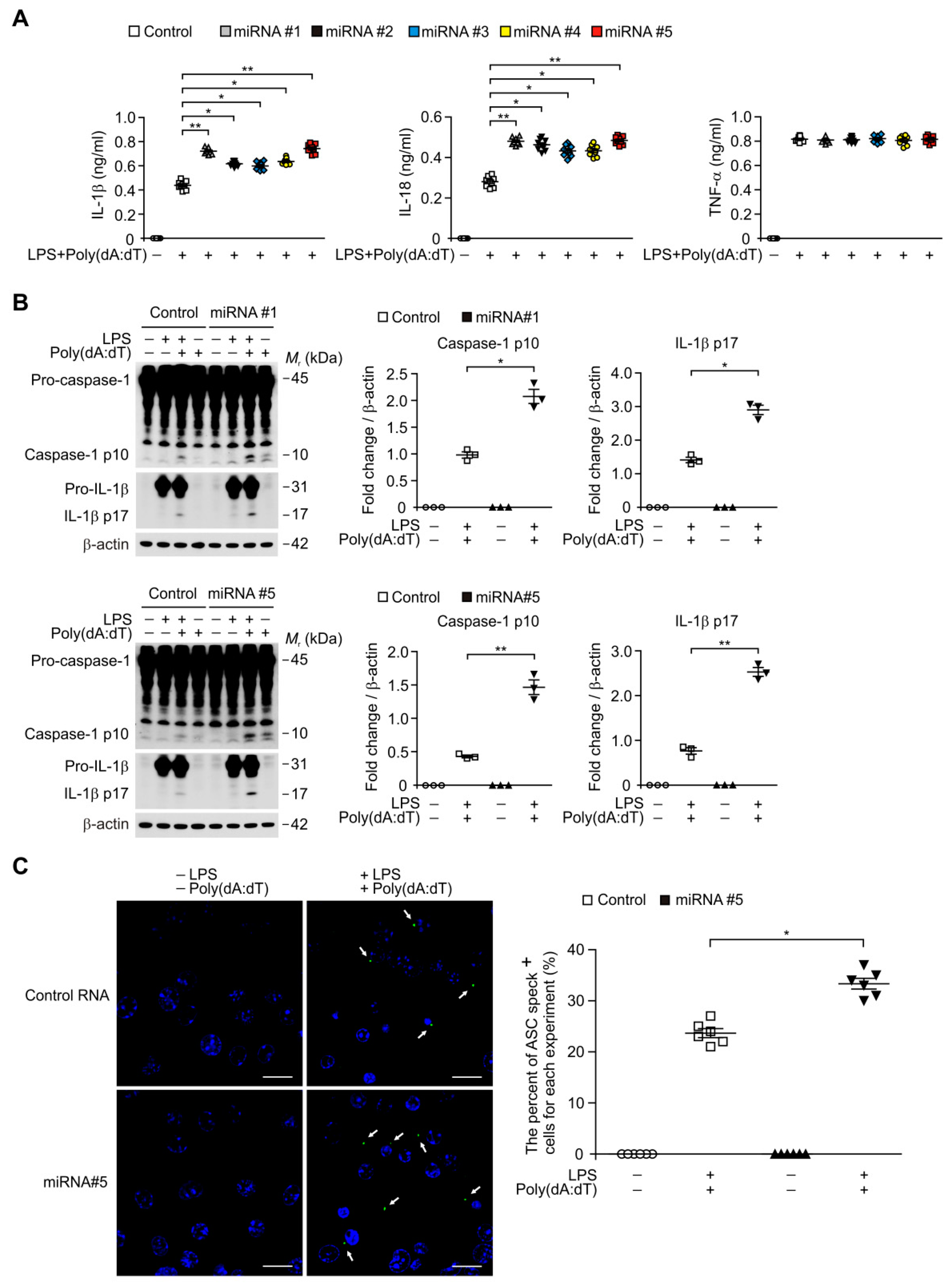
3.6. Transduction of miRNA Promotes the ASC Speck Formation for AIM2 Inflammasome Activation
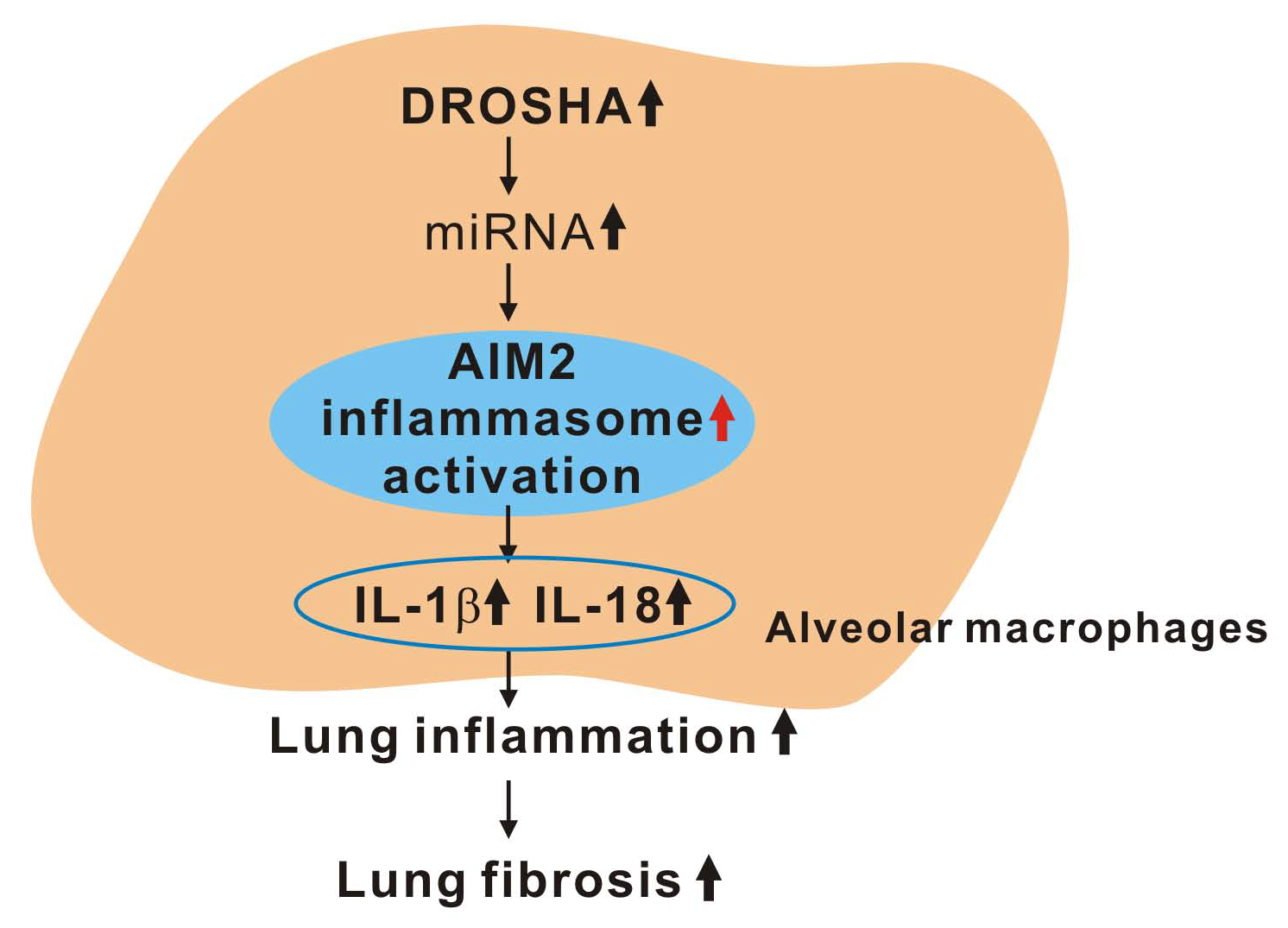
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 379, 797–798. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, K.B.; Samet, J.M.; Stidley, C.A.; Colby, T.V.; Waldron, J.A. Cigarette smoking: A risk factor for idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1997, 155, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Bringardner, B.D.; Baran, C.P.; Eubank, T.D.; Marsh, C.B. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2008, 10, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Proell, M.; Gerlic, M.; Mace, P.D.; Reed, J.C.; Riedl, S.J. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 2013, 449, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef]
- Morrone, S.R.; Matyszewski, M.; Yu, X.; Delannoy, M.; Egelman, E.H.; Sohn, J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 2015, 6, 7827. [Google Scholar] [CrossRef]
- Hornung, V.; Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 2010, 10, 123–130. [Google Scholar] [CrossRef]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Lee, S.; Park, M.A.; Siempos, I.I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J. Clin. Investig. 2015, 125, 665–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Inui, M.; Martello, G.; Piccolo, S. MicroRNA control of signal transduction. Nat. Rev. Mol. Cell Biol. 2010, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahamtan, A.; Teymoori-Rad, M.; Nakstad, B.; Salimi, V. Anti-inflammatory microRNAs and their potential for inflammatory diseases treatment. Front. Immunol. 2018, 9, 1377. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Rao, D.S.; Baltimore, D. microRNA regulation of inflammatory responses. Annu. Rev. Immunol. 2012, 30, 295–312. [Google Scholar] [CrossRef]
- Fan, L.; Yu, X.; Huang, Z.; Zheng, S.; Zhou, Y.; Lv, H.; Zeng, Y.; Xu, J.F.; Zhu, X.; Yi, X. Analysis of microarray-identified genes and microRNAs associated with idiopathic pulmonary fibrosis. Mediat. Inflamm. 2017, 2017, 1804240. [Google Scholar] [CrossRef]
- Pandit, K.V.; Corcoran, D.; Yousef, H.; Yarlagadda, M.; Tzouvelekis, A.; Gibson, K.F.; Konishi, K.; Yousem, S.A.; Singh, M.; Handley, D.; et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 220–229. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Dakhlallah, D.; Batte, K.; Wang, Y.; Cantemir-Stone, C.Z.; Yan, P.; Nuovo, G.; Mikhail, A.; Hitchcock, C.L.; Wright, V.P.; Nana-Sinkam, S.P.; et al. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 187, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Kurowska-Stolarska, M.; Hasoo, M.K.; Welsh, D.J.; Stewart, L.; McIntyre, D.; Morton, B.E.; Johnstone, S.; Miller, A.M.; Asquith, D.L.; Millar, N.L.; et al. The role of microRNA-155/liver X receptor pathway in experimental and idiopathic pulmonary fibrosis. J. Allergy Clin. Immunol. 2017, 139, 1946–1956. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.; Rajaguru, P.; Sudhakar Gandhi, P.S. MicroRNAs as potential targets for progressive pulmonary fibrosis. Front. Pharmacol. 2015, 6, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Liang, J.; Guo, R.; Liu, N.; Noble, P.W.; Jiang, D. Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation. Physiol. Genom. 2011, 43, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahid, M.A.; Satoh, M.; Chan, E.K. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol. Immunol. 2011, 8, 388–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butz, H.; Racz, K.; Hunyady, L.; Patocs, A. Crosstalk between TGF-beta signaling and the microRNA machinery. Trends Pharmacol. Sci. 2012, 33, 382–393. [Google Scholar] [CrossRef]
- Bi, Y.; Liu, G.; Yang, R. MicroRNAs: Novel regulators during the immune response. J. Cell Physiol. 2009, 218, 467–472. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Seluanov, A.; Vaidya, A.; Gorbunova, V. Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. 2010, 5, 2033. [Google Scholar] [CrossRef]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabón, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.W.; Wu, J.; Zhang, Z.; Datta, P.; Ibrahimi, I.; Taniguchi, S.; Sagara, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Cryopyrin and pyrin activate caspase-1, but not NF-B, via ASC oligomerization. Cell Death Differ. 2006, 13, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, H.; Zissel, G.; Goldmann, T.; Tschernig, T.; Vollmer, E.; Pabst, R.; Müller-Quernheim, J. Alveolar macrophages are the main source for tumour necrosis factor-alpha in patients with sarcoidosis. Eur. Respir. J. 2003, 21, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Mutlu, G.M.; Budinger, G.R.; Perlman, H. Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am. J. Respir. Cell Mol. Biol. 2013, 49, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Paugh, S.W.; Coss, D.R.; Bao, J.; Laudermilk, L.T.; Grace, C.R.; Ferreira, A.M.; Waddell, M.B.; Ridout, G.; Naeve, D.; Leuze, M.; et al. MicroRNAs form triplexes with double stranded DNA at sequence-specific binding sites; a eukaryotic mechanism via which microRNAs could directly alter gene expression. PLoS Comput. Biol. 2016, 12, e1004744. [Google Scholar] [CrossRef] [PubMed]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.; Dentler, J.; Luttmann, W.; Friedrich, K.; Müller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Postlethwaite, A.E.; Raghow, R.; Stricklin, G.P.; Poppleton, H.; Seyer, J.M.; Kang, A.H. Modulation of fibroblast functions by interleukin 1: Increased steady-state accumulation of type I procollagen messenger RNAs and stimulation of other functions but not chemotaxis by human recombinant interleukin 1α and-β. J. Cell Biol. 1988, 106, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Investig. 2001, 107, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Redente, E.F.; Jacobsen, K.M.; Solomon, J.J.; Lara, A.R.; Faubel, S.; Keith, R.C.; Henson, P.M.; Downey, G.P.; Riches, D.W. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L510–L518. [Google Scholar] [CrossRef]
- Mehta, A.; Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | IPF | |
|---|---|---|
| Subjects | 12 | 12 |
| Age years | 46.5+/−11.4 | 65+/−9.2 |
| Male/female | 11/1 | 11/1 |
| Smoking status yes/no | 4/8 | 6/6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, S.J.; Hong, K.S.; Jeong, J.H.; Lee, M.; Choi, A.M.K.; Stout-Delgado, H.W.; Moon, J.-S. DROSHA-Dependent AIM2 Inflammasome Activation Contributes to Lung Inflammation during Idiopathic Pulmonary Fibrosis. Cells 2019, 8, 938. https://doi.org/10.3390/cells8080938
Cho SJ, Hong KS, Jeong JH, Lee M, Choi AMK, Stout-Delgado HW, Moon J-S. DROSHA-Dependent AIM2 Inflammasome Activation Contributes to Lung Inflammation during Idiopathic Pulmonary Fibrosis. Cells. 2019; 8(8):938. https://doi.org/10.3390/cells8080938
Chicago/Turabian StyleCho, Soo Jung, Kyoung Sook Hong, Ji Hun Jeong, Mihye Lee, Augustine M. K. Choi, Heather W. Stout-Delgado, and Jong-Seok Moon. 2019. "DROSHA-Dependent AIM2 Inflammasome Activation Contributes to Lung Inflammation during Idiopathic Pulmonary Fibrosis" Cells 8, no. 8: 938. https://doi.org/10.3390/cells8080938
APA StyleCho, S. J., Hong, K. S., Jeong, J. H., Lee, M., Choi, A. M. K., Stout-Delgado, H. W., & Moon, J.-S. (2019). DROSHA-Dependent AIM2 Inflammasome Activation Contributes to Lung Inflammation during Idiopathic Pulmonary Fibrosis. Cells, 8(8), 938. https://doi.org/10.3390/cells8080938