The Autophagy-Cilia Axis: An Intricate Relationship


Abstract
1. Introduction
2. The Autophagy Machinery Localizes at Cilia
3. Autophagy Controls Ciliogenesis and Cilia Length
4. Cilia Control Autophagy
5. Hedgehog Signaling Is a Regulator of Autophagy
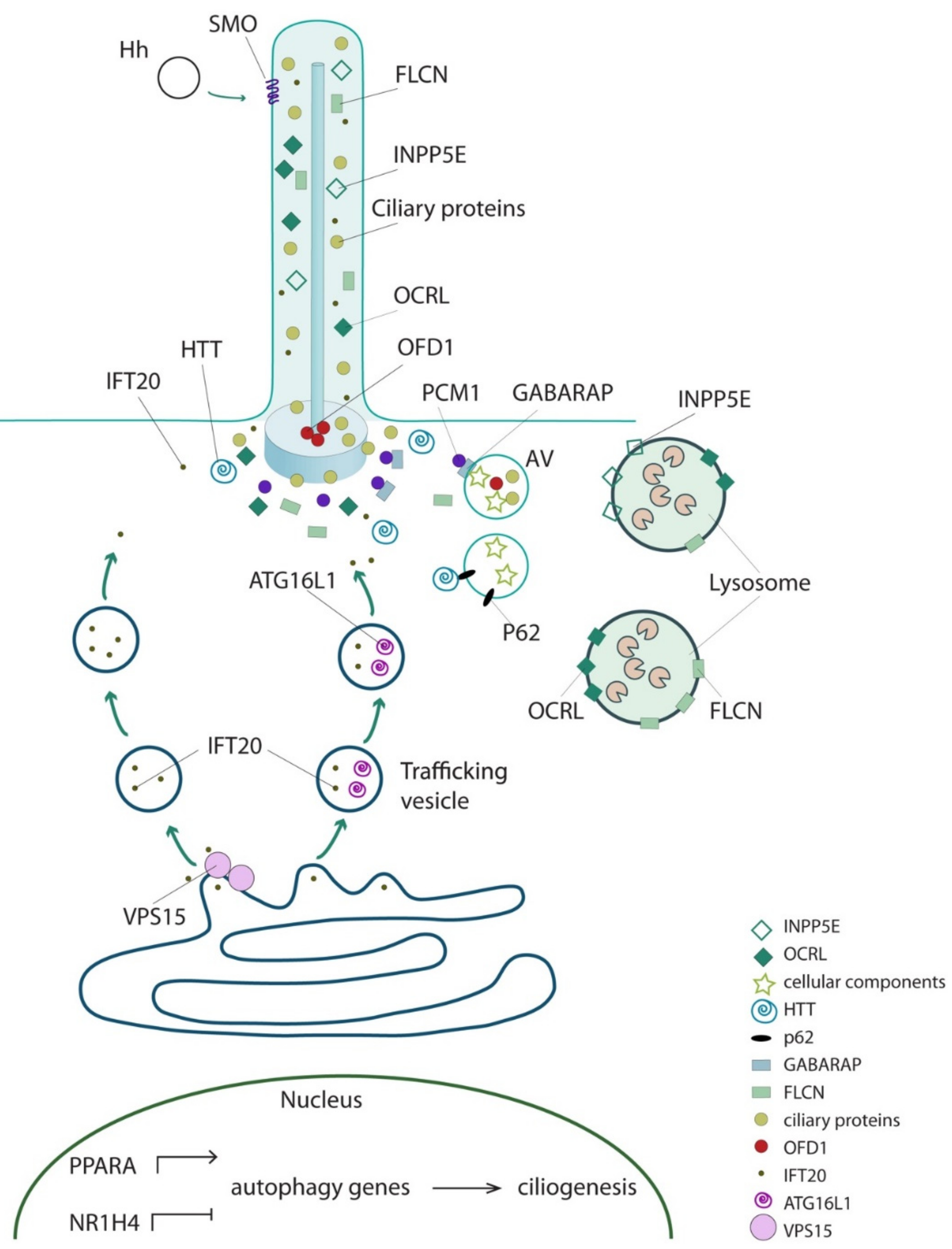
6. The Direct Functional Interplay between Ciliary and Core Autophagic Proteins
7. Autophagy-Cilia Crosstalk in Disease
7.1. Focal Cortical Dyslamination
7.2. Chronic Obstructive Pulmonary Disease
7.3. Thyroid Hurthle Cell Tumor
7.4. Renal Cystic Disease
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Zhong, Q. Cilia in autophagy and cancer. Cilia 2015, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yi, S.; Kang, Y.E.; Chang, J.Y.; Kim, J.T.; Sul, H.J.; Kim, J.O.; Kim, J.M.; Kim, J.; Porcelli, A.M.; et al. Defective ciliogenesis in thyroid hurthle cell tumors is associated with increased autophagy. Oncotarget 2016, 7, 79117–79130. [Google Scholar] [CrossRef] [PubMed]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Marshall, W.F. Intraflagellar Transport and Ciliary Dynamics. Cold Spring Harb. Perspect. Biol. 2017, 9, a021998. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.M.; Christou-Savina, S.; Xiong, Y.; Moede, T.; Moruzzi, N.; Karlsson-Edlund, P.; Leibiger, B.; Leibiger, I.B.; Ostenson, C.G.; Beales, P.L.; et al. Ciliary dysfunction impairs beta-cell insulin secretion and promotes development of type 2 diabetes in rodents. Nat. Commun. 2014, 5, 5308. [Google Scholar] [CrossRef] [PubMed]
- Insinna, C.; Besharse, J.C. Intraflagellar transport and the sensory outer segment of vertebrate photoreceptors. Dev. Dyn. 2008, 237, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Kuhara, A.; Okumura, M.; Kimata, T.; Tanizawa, Y.; Takano, R.; Kimura, K.D.; Inada, H.; Matsumoto, K.; Mori, I. Temperature sensing by an olfactory neuron in a circuit controlling behavior of C. elegans. Science 2008, 320, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.T.; Voss, J.W.; Teilmann, S.C.; Lambert, I.H. High expression of the taurine transporter TauT in primary cilia of NIH3T3 fibroblasts. Cell Biol. Int. 2005, 29, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Moorman, S.J.; Shorr, A.Z. The primary cilium as a gravitational force transducer and a regulator of transcriptional noise. Dev. Dyn. 2008, 237, 1955–1959. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Petrova, R.; Joyner, A.L. Roles for Hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Pampliega, O.; Cuervo, A.M. Autophagy and primary cilia: Dual interplay. Curr. Opin. Cell Biol. 2016, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Lohr, F.; Wakatsuki, S.; Dotsch, V.; Dikic, I.; et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef]
- Munch, C.; Dikic, I. Publisher Correction: Hitchhiking on selective autophagy. Nat. Cell Biol. 2018, 20, 990. [Google Scholar] [CrossRef] [PubMed]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Diaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional interaction between autophagy and ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Orhon, I.; Dupont, N.; Pampliega, O.; Cuervo, A.M.; Codogno, P. Autophagy and regulation of cilia function and assembly. Cell Death Differ. 2015, 22, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. The plasma membrane brings autophagosomes to life. Nat. Cell Biol. 2010, 12, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell’s antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef]
- Hershko, A.; Tomkins, G.M. Studies on the degradation of tyrosine aminotransferase in hepatoma cells in culture. Influence of the composition of the medium and adenosine triphosphate dependence. J. Biol. Chem. 1971, 246, 710–714. [Google Scholar]
- Struchtrup, A.; Wiegering, A.; Stork, B.; Ruther, U.; Gerhardt, C. The ciliary protein RPGRIP1L governs autophagy independently of its proteasome-regulating function at the ciliary base in mouse embryonic fibroblasts. Autophagy 2018, 14, 567–583. [Google Scholar] [CrossRef]
- Lam, H.C.; Cloonan, S.M.; Bhashyam, A.R.; Haspel, J.A.; Singh, A.; Sathirapongsasuti, J.F.; Cervo, M.; Yao, H.; Chung, A.L.; Mizumura, K.; et al. Histone deacetylase 6-mediated selective autophagy regulates COPD-associated cilia dysfunction. J. Clin. Investig. 2013, 123, 5212–5230. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Follit, J.A.; Tuft, R.A.; Fogarty, K.E.; Pazour, G.J. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol. Biol. Cell 2006, 17, 3781–3792. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Dikic, I. The Three Musketeers of Autophagy: Phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 2011, 21, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Cebotaru, V.; Cebotaru, L.; Kim, H.; Chiaravalli, M.; Boletta, A.; Qian, F.; Guggino, W.B. Polycystin-1 negatively regulates Polycystin-2 expression via the aggresome/autophagosome pathway. J. Biol. Chem. 2014, 289, 6404–6414. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Lee, J.N.; Son, M.; Lim, J.Y.; Dutta, R.K.; Maharjan, Y.; Kwak, S.; Oh, G.T.; Byun, K.; Choe, S.K.; et al. Ciliogenesis is reciprocally regulated by PPARA and NR1H4/FXR through controlling autophagy in vitro and in vivo. Autophagy 2018, 14, 1011–1027. [Google Scholar] [CrossRef]
- Wang, S.; Livingston, M.J.; Su, Y.; Dong, Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 2015, 11, 607–616. [Google Scholar] [CrossRef]
- Kim, E.S.; Shin, J.H.; Park, S.J.; Jo, Y.K.; Kim, J.S.; Kang, I.H.; Nam, J.B.; Chung, D.Y.; Cho, Y.; Lee, E.H.; et al. Inhibition of autophagy suppresses sertraline-mediated primary ciliogenesis in retinal pigment epithelium cells. PLoS ONE 2015, 10, e0118190. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Huang, W. Thioridazine promotes primary ciliogenesis in lung cancer cells through enhancing cell autophagy. Int. J. Clin. Exp. Med. 2017, 10, 13960–13969. [Google Scholar]
- Massa, F.; Tammaro, R.; Prado, M.A.; Cesana, M.; Lee, B.H.; Finley, D.; Franco, B.; Morleo, M. The deubiquitinating enzyme USP14 controls ciliogenesis and hedgehog signalling. Hum. Mol. Genet. 2018, 28, 764–777. [Google Scholar] [CrossRef]
- Wiegering, A.; Ruther, U.; Gerhardt, C. The Role of Primary Cilia in the Crosstalk between the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 241. [Google Scholar] [CrossRef]
- Orhon, I.; Dupont, N.; Codogno, P. Primary cilium and autophagy: The avengers of cell-size regulation. Autophagy 2016, 12, 2258–2259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orhon, I.; Dupont, N.; Zaidan, M.; Boitez, V.; Burtin, M.; Schmitt, A.; Capiod, T.; Viau, A.; Beau, I.; Kuehn, E.W.; et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat. Cell Biol. 2016, 18, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Wang, Y.; Lalli, M.A.; Guzman, E.; Godshalk, S.E.; Zhou, H.; Kosik, K.S. Primary Cilium-Autophagy-Nrf2 (PAN) Axis Activation Commits Human Embryonic Stem Cells to a Neuroectoderm Fate. Cell 2016, 165, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Stoetzel, C.; Bar, S.; De Craene, J.O.; Scheidecker, S.; Etard, C.; Chicher, J.; Reck, J.R.; Perrault, I.; Geoffroy, V.; Chennen, K.; et al. A mutation in VPS15 (PIK3R4) causes a ciliopathy and affects IFT20 release from the cis-Golgi. Nat. Commun. 2016, 7, 13586. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Dhand, R.; Vanhaesebroeck, B.; MacDougall, L.K.; Stein, R.; Zvelebil, M.J.; Domin, J.; Panaretou, C.; Waterfield, M.D. A human phosphatidylinositol 3-kinase complex related to the yeast Vps34p-Vps15p protein sorting system. EMBO J. 1995, 14, 3339–3348. [Google Scholar] [CrossRef]
- Lindmo, K.; Brech, A.; Finley, K.D.; Gaumer, S.; Contamine, D.; Rusten, T.E.; Stenmark, H. The PI 3-kinase regulator Vps15 is required for autophagic clearance of protein aggregates. Autophagy 2008, 4, 500–506. [Google Scholar] [CrossRef]
- Hasegawa, J.; Iwamoto, R.; Otomo, T.; Nezu, A.; Hamasaki, M.; Yoshimori, T. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J. 2016, 35, 1853–1867. [Google Scholar] [CrossRef]
- Joachim, J.; Razi, M.; Judith, D.; Wirth, M.; Calamita, E.; Encheva, V.; Dynlacht, B.D.; Snijders, A.P.; O’Reilly, N.; Jefferies, H.B.J.; et al. Centriolar Satellites Control GABARAP Ubiquitination and GABARAP-Mediated Autophagy. Curr. Biol. 2017, 27, 2123–2136. [Google Scholar] [CrossRef]
- De Leo, M.G.; Staiano, L.; Vicinanza, M.; Luciani, A.; Carissimo, A.; Mutarelli, M.; Di Campli, A.; Polishchuk, E.; Di Tullio, G.; Morra, V.; et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 2016, 18, 839–850. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Seifan, S.; Claessens, T.; Behrends, C.; Kamps, M.A.; Rozycka, E.; Kemp, A.J.; Nookala, R.K.; Blenis, J.; Coull, B.J.; et al. FLCN, a novel autophagy component, interacts with GABARAP and is regulated by ULK1 phosphorylation. Autophagy 2014, 10, 1749–1760. [Google Scholar] [CrossRef]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Petralia, R.S.; Schwartz, C.M.; Wang, Y.X.; Kawamoto, E.M.; Mattson, M.P.; Yao, P.J. Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2013, 2, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Li, Y.; Singh, P.; Cao, L.; Xu, L.J.; Li, D.; Wang, Y.; Xie, Z.; Gui, Y.; et al. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1319–H1331. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Menzies, F.M.; Chang, Y.Y.; Simecek, N.; Neufeld, T.P.; Rubinsztein, D.C. The Hedgehog signalling pathway regulates autophagy. Nat. Commun. 2012, 3, 1200. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.J.; Chang, C.H.; Ibrahim, R.B.; Lin, I.H.; Wang, C.H.; Wang, W.J.; Tsai, J.W. Gli2 modulates cell cycle re-entry through autophagy-mediated regulation on the length of primary cilia. J. Cell Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, B.; Zhao, Q. Hedgehog signaling regulates osteoblast differentiation in zebrafish larvae through modulation of autophagy. Biol. Open 2019, 8, bio040840. [Google Scholar] [CrossRef]
- Tang, X.; Deng, L.; Chen, Q.; Wang, Y.; Xu, R.; Shi, C.; Shao, J.; Hu, G.; Gao, M.; Rao, H.; et al. Inhibition of Hedgehog signaling pathway impedes cancer cell proliferation by promotion of autophagy. Eur. J. Cell Biol. 2015, 94, 223–233. [Google Scholar] [CrossRef]
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013, 58, 995–1010. [Google Scholar] [CrossRef]
- Xu, Y.; An, Y.; Wang, X.; Zha, W.; Li, X. Inhibition of the Hedgehog pathway induces autophagy in pancreatic ductal adenocarcinoma cells. Oncol. Rep. 2014, 31, 707–712. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Deleyrolle, L.P.; Nakamura, N.S.; Parker, A.K.; Martuscello, R.T.; Reynolds, B.A.; Sarkisian, M.R. PCM1 Depletion Inhibits Glioblastoma Cell Ciliogenesis and Increases Cell Death and Sensitivity to Temozolomide. Transl. Oncol. 2016, 9, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Keryer, G.; Pineda, J.R.; Liot, G.; Kim, J.; Dietrich, P.; Benstaali, C.; Smith, K.; Cordelieres, F.P.; Spassky, N.; Ferrante, R.J.; et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Investig. 2011, 121, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Bielas, S.L.; Silhavy, J.L.; Brancati, F.; Kisseleva, M.V.; Al-Gazali, L.; Sztriha, L.; Bayoumi, R.A.; Zaki, M.S.; Abdel-Aleem, A.; Rosti, R.O.; et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009, 41, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Chavez, M.; Ena, S.; Van Sande, J.; de Kerchove d’Exaerde, A.; Schurmans, S.; Schiffmann, S.N. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Dev. Cell 2015, 34, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Phua, S.C.; Roberson, E.C.; Garcia, G., 3rd; Abedin, M.; Schurmans, S.; Inoue, T.; Reiter, J.F. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev. Cell 2015, 34, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Luo, N.; West, C.C.; Murga-Zamalloa, C.A.; Sun, L.; Anderson, R.M.; Wells, C.D.; Weinreb, R.N.; Travers, J.B.; Khanna, H.; Sun, Y. OCRL localizes to the primary cilium: A new role for cilia in Lowe syndrome. Hum. Mol. Genet. 2012, 21, 3333–3344. [Google Scholar] [CrossRef] [PubMed]
- Conduit, S.E.; Dyson, J.M.; Mitchell, C.A. Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. FEBS Lett. 2012, 586, 2846–2857. [Google Scholar] [CrossRef]
- Coon, B.G.; Hernandez, V.; Madhivanan, K.; Mukherjee, D.; Hanna, C.B.; Barinaga-Rementeria Ramirez, I.; Lowe, M.; Beales, P.L.; Aguilar, R.C. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum. Mol. Genet. 2012, 21, 1835–1847. [Google Scholar] [CrossRef]
- Montjean, R.; Aoidi, R.; Desbois, P.; Rucci, J.; Trichet, M.; Salomon, R.; Rendu, J.; Faure, J.; Lunardi, J.; Gacon, G.; et al. OCRL-mutated fibroblasts from patients with Dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Hum. Mol. Genet. 2015, 24, 994–1006. [Google Scholar] [CrossRef]
- Luijten, M.N.; Basten, S.G.; Claessens, T.; Vernooij, M.; Scott, C.L.; Janssen, R.; Easton, J.A.; Kamps, M.A.; Vreeburg, M.; Broers, J.L.; et al. Birt-Hogg-Dube syndrome is a novel ciliopathy. Hum. Mol. Genet. 2013, 22, 4383–4397. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Warren, M.B.; Toro, J.R.; Matrosova, V.; Glenn, G.; Turner, M.L.; Duray, P.; Merino, M.; Choyke, P.; Pavlovich, C.P.; et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2002, 2, 157–164. [Google Scholar] [CrossRef]
- Haywood, A.M. Relationships between binding, phagocytosis and membrane fusion of enveloped viruses. Prog. Clin. Biol. Res. 1990, 343, 117–132. [Google Scholar] [PubMed]
- Kaliszewski, M.; Knott, A.B.; Bossy-Wetzel, E. Primary cilia and autophagic dysfunction in Huntington’s disease. Cell Death Differ. 2015, 22, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Kim, W.I.; Kang, H.C.; Kim, S.H.; Park, A.H.; Park, E.K.; Cho, Y.W.; Kim, S.; Kim, H.M.; Kim, J.A.; et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat. Med. 2015, 21, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Gopalappa, R.; Kim, S.H.; Ramakrishna, S.; Lee, M.; Kim, W.I.; Kim, J.; Park, S.M.; Lee, J.; Oh, J.H.; et al. Somatic Mutations in TSC1 and TSC2 Cause Focal Cortical Dysplasia. Am. J. Hum. Genet. 2017, 100, 454–472. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.C.; Crino, P.B. Focal malformations of cortical development: New vistas for molecular pathogenesis. Neuroscience 2013, 252, 262–276. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.C.; Reiter, J.F.; Kim, D.S.; Kim, H.H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97. [Google Scholar] [CrossRef]
- Singla, V.; Romaguera-Ros, M.; Garcia-Verdugo, J.M.; Reiter, J.F. Ofd1, a human disease gene, regulates the length and distal structure of centrioles. Dev. Cell 2010, 18, 410–424. [Google Scholar] [CrossRef]
- Ferrante, M.I.; Zullo, A.; Barra, A.; Bimonte, S.; Messaddeq, N.; Studer, M.; Dolle, P.; Franco, B. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat. Genet. 2006, 38, 112–117. [Google Scholar] [CrossRef]
- Barnes, P.J. Chronic obstructive pulmonary disease. N. Engl. J. Med. 2000, 343, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Leopold, P.L.; O’Mahony, M.J.; Lian, X.J.; Tilley, A.E.; Harvey, B.G.; Crystal, R.G. Smoking is associated with shortened airway cilia. PLoS ONE 2009, 4, e8157. [Google Scholar] [CrossRef] [PubMed]
- Frasca, J.M.; Auerbach, O.; Carter, H.W.; Parks, V.R. Morphologic alterations induced by short-term cigarette smoking. Am. J. Pathol. 1983, 111, 11–20. [Google Scholar] [PubMed]
- Ballenger, J.J. Experimental effect of cigarette smoke on human respiratory cilia. N. Engl. J. Med. 1960, 263, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- DeCaen, P.G.; Delling, M.; Vien, T.N.; Clapham, D.E. Direct recording and molecular identification of the calcium channel of primary cilia. Nature 2013, 504, 315–318. [Google Scholar] [CrossRef]
- Delling, M.; DeCaen, P.G.; Doerner, J.F.; Febvay, S.; Clapham, D.E. Primary cilia are specialized calcium signalling organelles. Nature 2013, 504, 311–314. [Google Scholar] [CrossRef]
- Lin, F.; Hiesberger, T.; Cordes, K.; Sinclair, A.M.; Goldstein, L.S.; Somlo, S.; Igarashi, P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2003, 100, 5286–5291. [Google Scholar] [CrossRef]
- Jonassen, J.A.; SanAgustin, J.; Baker, S.P.; Pazour, G.J. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J. Am. Soc. Nephrol. 2012, 23, 641–651. [Google Scholar] [CrossRef]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Zullo, A.; Iaconis, D.; Barra, A.; Cantone, A.; Messaddeq, N.; Capasso, G.; Dolle, P.; Igarashi, P.; Franco, B. Kidney-specific inactivation of Ofd1 leads to renal cystic disease associated with upregulation of the mTOR pathway. Hum. Mol. Genet. 2010, 19, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Pema, M.; Drusian, L.; Chiaravalli, M.; Castelli, M.; Yao, Q.; Ricciardi, S.; Somlo, S.; Qian, F.; Biffo, S.; Boletta, A. mTORC1-mediated inhibition of polycystin-1 expression drives renal cyst formation in tuberous sclerosis complex. Nat. Commun. 2016, 7, 10786. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef]
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096. [Google Scholar] [CrossRef]
- Takabatake, Y.; Kimura, T.; Takahashi, A.; Isaka, Y. Autophagy and the kidney: Health and disease. Nephrol. Dial. Transplant. 2014, 29, 1639–1647. [Google Scholar] [CrossRef]
- Belibi, F.; Zafar, I.; Ravichandran, K.; Segvic, A.B.; Jani, A.; Ljubanovic, D.G.; Edelstein, C.L. Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Ren. Physiol. 2011, 300, F1235–F1243. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Y.; Sui, Z.; Zhang, Y.; Liu, M.; Tang, H. USP14 de-ubiquitinates vimentin and miR-320a modulates USP14 and vimentin to contribute to malignancy in gastric cancer cells. Oncotarget 2017, 8, 48725–48736. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.Y.; Ma, T.L.; Hung, C.C.; Tian, Y.C.; Chen, Y.C.; Yang, C.W.; Cheng, Y.C. Metformin Inhibits Cyst Formation in a Zebrafish Model of Polycystin-2 Deficiency. Sci. Rep. 2017, 7, 7161. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Watari, M.; Saito, T.; Morishita, Y.; Ishibashi, K. Enhanced Autophagy in Polycystic Kidneys of AQP11 Null Mice. Int. J. Mol. Sci. 2016, 17, 1993. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Masyuk, T.V.; Lorenzo Pisarello, M.J.; Ding, J.F.; Loarca, L.; Huang, B.Q.; LaRusso, N.F. Cholangiocyte autophagy contributes to hepatic cystogenesis in polycystic liver disease and represents a potential therapeutic target. Hepatology 2018, 67, 1088–1108. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein | Aliases | Ciliary Localization | MIM#; Phenotype | Effect on Autophagy | Ciliated Conditions | Ref. |
|---|---|---|---|---|---|---|
| IFT20 | - | Axoneme | No disease | Interacts with ATG16L and promotes its shuttling from Golgi to cilia; autophagic activity decreased in IFT20-depleted (MEFs and human neuroectodermal) cells | YES | [22,43] |
| IFT88 | D13S1056E, DAF19, TG737, TTC10, hTg737 | Axoneme | No disease | Autophagic activity decreased in IFT88-depleted (KECs, HK2, human neuroectodermal) cells | YES | [22,36,42,43] |
| KIF3A | FLA10, KLP-20 | Basal body | No disease | Autophagic activity decreased in KIF3A-silenced human neuroectodermal cells and in murine renal tubular epithelial cells | YES | [42,43] |
| VPS15 | PIK3R4 P150 | Axoneme and basal body | cilia phenotype (retinitis pigmentosa, limb abnormalities, renal cysts) | Encodes for VPS34 regulatory subunits. Is involved in autophagosomes formation. Promotes formation and/or release of IFT20 positive vesicles from cis-Golgi to cilia | YES | [44,45,46] |
| RPGRIP1L | CORS3, FTM, JBTS7, MKS5, NPHP8, PPP1R134 | Ciliary transition zone | 611560: Joubert syndrome 7 (JBTS7); 611561: Meckel syndrome 5 (MKS5); 216360: COACH syndrome | Autophagic activity decreased in Rpgrip1l deficient MEFs | NO | [29] |
| INPP5E | CORS1, CPD4, JBTS1, MORMS, PPI5PIV, pharbin | Axoneme | 610156: mental retardation, truncal obesity, retinal dystrophy, and micropenis syndrome (MORMS); 213300: Joubert syndrome 1 (JBTS1) | Localizes to lysosomes and is required for autophagosome/lysosome fusion | NO | [47] |
| PCM1 | PTC4, RET/PCM-1 | Centriolar satellite | No disease | Interacts with GABARAP and controls its localization and degradation at centriolar satellites thus influencing GABARAP-autophagosome formation | NO | [48] |
| OCRL | INPP5F, LOCR, NPHL2-1, OCRL-1 | Axoneme and basal body | 300555: Dent disease-2; 309000: Lowe oculocerebrorenal syndrome | Recruited to lysosomes and required for autophagosome-lysosome fusion | NO | [49] |
| FLCN | Folliculin BHD FLCL | Axoneme and basal body | 135150: Birt-Hogg-Dube syndrome (BHD); 144700: nonpapillary renal carcinoma | Interacts with GABARAP and ULK1 kinase, playing a positive role in autophagy. Involved in signaling amino acid levels to mTOR kinase at lysosomes | NO | [50,51] |
| HTT | Huntingtin HD Protein | Basal body | 143100: Huntington disease | Interacts with p62 and ULK1 kinase; required for selective autophagy | NO | [52] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morleo, M.; Franco, B. The Autophagy-Cilia Axis: An Intricate Relationship. Cells 2019, 8, 905. https://doi.org/10.3390/cells8080905
Morleo M, Franco B. The Autophagy-Cilia Axis: An Intricate Relationship. Cells. 2019; 8(8):905. https://doi.org/10.3390/cells8080905
Chicago/Turabian StyleMorleo, Manuela, and Brunella Franco. 2019. "The Autophagy-Cilia Axis: An Intricate Relationship" Cells 8, no. 8: 905. https://doi.org/10.3390/cells8080905
APA StyleMorleo, M., & Franco, B. (2019). The Autophagy-Cilia Axis: An Intricate Relationship. Cells, 8(8), 905. https://doi.org/10.3390/cells8080905