Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies


Abstract
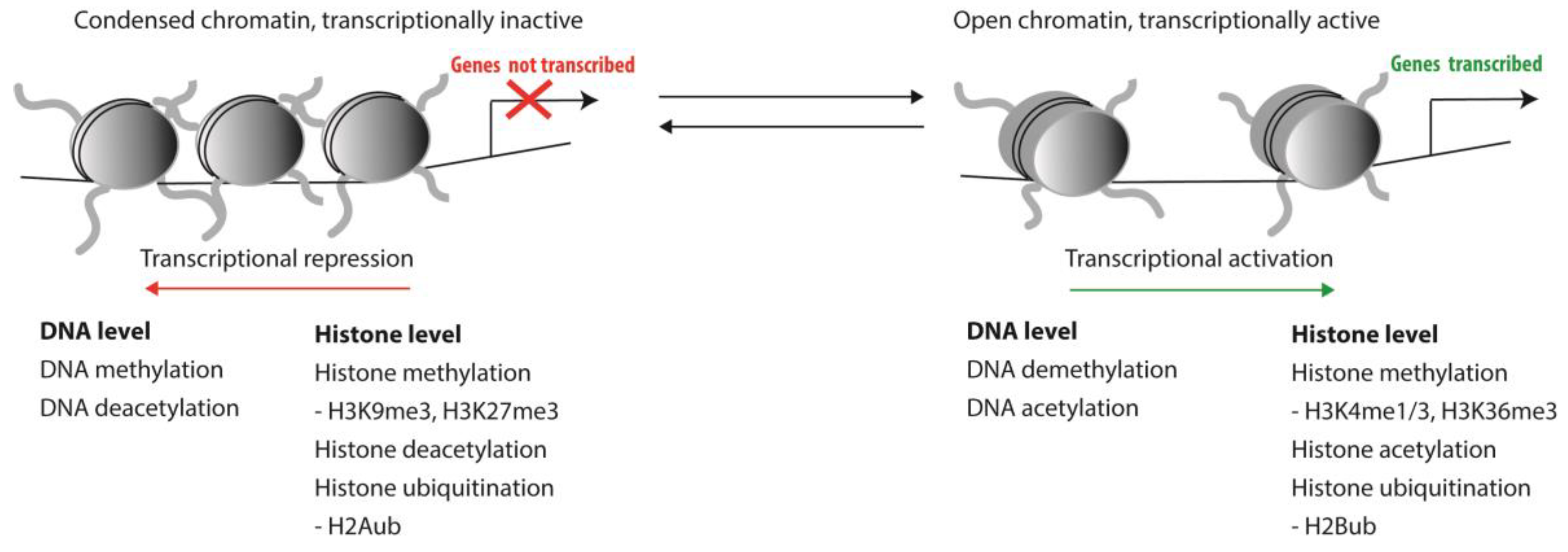
1. Epigenetic Regulation of HSCs
2. Epigenetic Changes during Normal Aging of Murine HSCs
3. Epigenetic Changes during Normal Aging of Human HSCs
4. Epigenetic Changes in Age-Related Haematopoietic Malignancies
4.1. Aberrant DNA Methylation
4.2. Aberrant Histone Modifications
5. Epigenome-Targeted Therapies
5.1. DNA Methylation Inhibitors (DNMTi)
5.2. Histone Deacetylase Inhibitors (HDACi) and Histone Acetyltransferase (HATi)
5.3. Histone Methyltransferase Inhibitors (HMTi) and Histone Demethylase Inhibitors (HDMi)
5.4. Inhibiting Epigenetic Readers
6. Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vedi, A.; Santoro, A.; Dunant, C.F.; Dick, J.E.; Laurenti, E. Molecular Landscapes of Human Hematopoietic Stem Cells in Health and Leukemia. Ann. N. Y. Acad. Sci. 2015, 1370, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Li, X.; Liang, D.; Li, T.; Zhu, P.; Guo, H.; Wu, X.; Wen, L.; Gu, T.-P.; Hu, B. Active and Passive Demethylation of Male and Female Pronuclear DNA in the Mammalian Zygote. Cell Stem Cell 2014, 15, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Oberdoerffer, P. Replication Stress: A Lifetime of Epigenetic Change. Genes 2015, 6, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA Methyltransferase 1 Is Essential for and Uniquely Regulates Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2009, 5, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, J.J.; Sinha, A.U.; Zhu, N.; Li, M.; Armstrong, S.A.; Orkin, S.H. Haploinsufficiency of Dnmt1 Impairs Leukemia Stem Cell Function through Derepression of Bivalent Chromatin Domains. Genes Dev. 2012, 26, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Bröske, A.-M.; Vockentanz, L.; Kharazi, S.; Huska, M.R.; Mancini, E.; Scheller, M.; Kuhl, C.; Enns, A.; Prinz, M.; Jaenisch, R.; et al. DNA Methylation Protects Hematopoietic Stem Cell Multipotency from Myeloerythroid Restriction. Nat. Genet. 2009, 41, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, Y.; Ema, H.; Okano, M.; Li, E.; Nakauchi, H. De Novo DNA Methyltransferase Is Essential for Self-Renewal, but Not for Differentiation, in Hematopoietic Stem Cells. J. Exp. Med. 2007, 204, 715–722. [Google Scholar] [CrossRef]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.; Xia, Z.; et al. Dnmt3a and Dnmt3b Have Overlapping and Distinct Functions in Hematopoietic Stem Cells. Cell Stem Cell 2014, 15, 350–364. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a Is Essential for Hematopoietic Stem Cell Differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef]
- Trowbridge, J.J.; Orkin, S.H. Dnmt3a Silences Hematopoietic Stem Cell Self-Renewal. Nat. Genet. 2011, 44, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Dawlaty, M.M.; Ndiaye-Lobry, D.; Yap, Y.S.; Bakogianni, S.; Yu, Y.; Bhattacharyya, S.; Shaknovich, R.; Geng, H.; Lobry, C.; et al. Tet1 Is a Tumor Suppressor of Hematopoietic Malignancy. Nat. Immunol. 2015, 16, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Guillamot, M.; Cimmino, L.; Aifantis, I. The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer 2016, 2, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Thompson, E.C.; Hastie, R.; Tsangaratou, A.; Rajewsky, K.; Koralov, S.B.; Rao, A. Ten-Eleven-Translocation 2 (TET2) Negatively Regulates Homeostasis and Differentiation of Hematopoietic Stem Cells in Mice. PNAS 2011, 108, 14566–14571. [Google Scholar] [CrossRef] [PubMed]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef]
- Quivoron, C.; Couronné, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.-H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-verschueren, C.; Christos, P.J.; Schifano, E.; Booth, J.; Van Putten, W.; Campagne, F.; Mazumdar, M.; et al. DNA Methylation Signatures Identify Biologically Distinct Subtypes in Acute Myeloid Leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Görlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 Promotes Leukemogenesis in Vivo and Can Be Specifically Targeted in Human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brüstle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) Mutation Increases Murine Haematopoietic Progenitors and Alters Epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, I.; Herrera-Merchan, A.; Ligos, J.M.; Carramolino, L.; Nuñez, J.; Martinez, F.; Dominguez, O.; Torres, M.; Gonzalez, S. Ezh1 Is Required for Hematopoietic Stem Cell Maintenance and Prevents Senescence-like Cell Cycle Arrest. Cell Stem Cell 2012, 11, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Kamminga, L.M.; Bystrykh, L.V.; de Boer, A.; Houwer, S.; Douma, J.; Weersing, E.; Dontje, B.; de Haan, G. The Polycomb Group Gene Ezh2 Prevents Hematopoietic Stem Cell Exhaustion. Blood 2006, 107, 2170–2179. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Merchan, A.; Arranz, L.; Ligos, J.M.; De Molina, A.; Dominguez, O.; Gonzalez, S. Ectopic Expression of the Histone Methyltransferase Ezh2 in Haematopoietic Stem Cells Causes Myeloproliferative Disease. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki-Kashio, M.; Wendt, G.R.; Iwama, A. Tumor Suppressor Function of the Polycomb Group Genes. Cell Cycle 2012, 11, 2043–2044. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nguyen, A.T.; He, J.; Taranova, O.; Zhang, Y. Essential Role of DOT1L in Maintaining Normal Adult Hematopoiesis. Cell Res. 2011, 21, 1370–1373. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, Y.; Ortega, M.M.; Copeland, J.N.; Zhang, M.; Jacob, J.B.; Fields, T.A.; Vivian, J.L.; Fields, P.E. Early Mammalian Erythropoiesis Requires the Dot1L Methyltransferase. Blood 2010, 116, 4483–4491. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.Y.; Granowicz, E.M.; Maillard, I.; Thomas, D.; Hess, J.L. Requirement for Dot1l in Murine Postnatal Hematopoiesis and Leukemogenesis by MLL Translocation. Blood 2011, 117, 4759–4768. [Google Scholar] [CrossRef]
- Gallipoli, P.; Giotopoulos, G.; Huntly, B.J.P. Epigenetic Regulators as Promising Therapeutic Targets in Acute Myeloid Leukemia. Ther. Adv. Hematol. 2015, 6, 103–119. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lawton, L.N.; Rozovskaia, T.; Frampton, G.M.; Levine, S.S.; Volkert, T.L.; Croce, C.M.; Nakamura, T.; Canaani, E.; Young, R.A. Aberrant Chromatin at Genes Encoding Stem Cell Regulators in Human Mixed-Lineage Leukemia. Genes Dev. 2008, 22, 3403–3408. [Google Scholar] [CrossRef]
- Greenblatt, S.M.; Nimer, S.D. Chromatin Modifiers and the Promise of Epigenetic Therapy in Acute Leukemia. Leukemia 2014, 28, 1396–1406. [Google Scholar] [CrossRef]
- Smith, L.L.; Yeung, J.; Zeisig, B.B.; Popov, N.; Huijbers, I.; Barnes, J.; Wilson, A.J.; Taskesen, E.; Delwel, R.; Gil, J.; et al. Functional Crosstalk between Bmi1 and MLL/Hoxa9 Axis in Establishment of Normal Hematopoietic and Leukemic Stem Cells. Cell Stem Cell 2011, 8, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Barrett, N.A.; Malouf, C.; Kapeni, C.; Bacon, W.A.; Giotopoulos, G.; Jacobsen, S.E.W.; Huntly, B.J.; Ottersbach, K. Mll-AF4 Confers Enhanced Self-Renewal and Lymphoid Potential during a Restricted Window in Development. Cell Rep. 2016, 16, 1039–1054. [Google Scholar] [CrossRef] [PubMed]
- Koide, S.; Oshima, M.; Takubo, K.; Yamazaki, S.; Nitta, E.; Saraya, A.; Aoyama, K.; Kato, Y.; Miyagi, S.; Nakajima-Takagi, Y.; et al. Setdb1 Maintains Hematopoietic Stem and Progenitor Cells by Restricting the Ectopic Activation of Non-Hematopoietic Genes. Blood 2016, 128, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Chittka, A.; Nitarska, J.; Grazini, U.; Richardson, W.D. Transcription Factor Positive Regulatory Domain 4 (PRDM4) Recruits Protein Arginine Methyltransferase 5 (PRMT5) to Mediate Histone Arginine Methylation and Control Neural Stem Cell Proliferation and Differentiation. J. Biol. Chem. 2012, 287, 42995–43006. [Google Scholar] [CrossRef] [PubMed]
- Stopa, N.; Krebs, J.E.; Shechter, D. The PRMT5 Arginine Methyltransferase: Many Roles in Development, Cancer and Beyond. Cell. Mol. Life Sci. 2015, 72, 2041–2059. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.-W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 Is Essential for Early Mouse Development and Acts in the Cytoplasm to Maintain ES Cell Pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cheng, G.; Hamard, P.-J.; Greenblatt, S.; Wang, L.; Man, N.; Perna, F.; Xu, H.; Tadi, M.; Luciani, L.; et al. Arginine Methyltransferase PRMT5 Is Essential for Sustaining Normal Adult Hematopoiesis. J. Clin. Investig. 2015, 125, 3532–3544. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.-I.; Hannah, R.L.; Dawson, M.A.; Pridans, C.; Foster, D.; Joshi, A.; Gottgens, B.; Van Deursen, J.M.; Huntly, B.J.P. The Transcriptional Coactivator Cbp Regulates Self-Renewal and Differentiation in Adult Hematopoietic Stem Cells. Mol. Cell. Biol. 2011, 31, 5046–5060. [Google Scholar] [CrossRef]
- Katsumoto, T.; Yoshida, N.; Kitabayashi, I. Roles of the Histone Acetyltransferase Monocytic Leukemia Zinc Finger Protein in Normal and Malignant Hematopoiesis. Cancer Sci. 2008, 99, 1523–1527. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Adli, M.; LaFave, L.M.; Gao, J.; Hricik, T.; Shih, A.H.; Pandey, S.; Patel, J.P.; Chung, Y.R.; Koche, R.; et al. ASXL1 Mutations Promote Myeloid Transformation through Loss of PRC2-Mediated Gene Repression. Cancer Cell 2012, 22, 180–193. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; He, Y.; Pan, F.; Chen, S.; Rhodes, S.; Nguyen, L.; Yuan, J.; Jiang, L.; Yang, X.; et al. Loss of Asxl1 Leads to Myelodysplastic Syndrome–like Disease in Mice. Blood 2014, 123, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Uni, M.; Masamoto, Y.; Sato, T.; Kamikubo, Y.; Arai, S.; Kurokawa, M. Modeling ASXL1 Mutation Revealed Impaired Hematopoiesis Caused by Derepression of P16Ink4a through Aberrant PRC1-Mediated Histone Modi Fi Cation. Leukemia 2018, 33, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Van Den Boom, V.; Rozenveld-geugien, M.; Bonardi, F.; Malanga, D.; Gosliga, V.; Heijink, A.M.; Viglietto, G.; Morrone, G.; Fusetti, F.; Schuringa, J.J.; et al. Nonredundant and Locus-Specific Gene Repression Functions of PRC1 Paralog Family Members in Human Hematopoietic Stem/Progenitor Cells. Blood 2013, 121, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Core, N.; Bel, S.; Gaunt, S.J.; Aurrand-Lions, M.; Pearce, J.; Fisher, A.; Djabali, M. Altered Cellular Proliferation and Mesoderm Patterning in Polycomb-M33-Deficient Mice. Development 1997, 124, 721–729. [Google Scholar] [PubMed]
- Klauke, K.; Radulović, V.; Broekhuis, M.; Weersing, E.; Zwart, E.; Olthof, S.; Ritsema, M.; Bruggeman, S.; Wu, X.; Helin, K.; et al. Polycomb Cbx Family Members Mediate the Balance between Haematopoietic Stem Cell Self-Renewal and Differentiation. Nat. Cell Biol. 2013, 15, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.L.; Gil, J.; Hernando, E.; Teruya-Feldstein, J.; Narita, M.; Martínez, D.; Visakorpi, T.; Mu, D.; Cordon-Cardo, C.; Peters, G.; et al. Role of the Chromobox Protein CBX7 in Lymphomagenesis. PNAS 2007, 104, 5389–5394. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Buisman, S.C.; Weersing, E.; Dethmers-Ausema, A.; Zwart, E.; Schepers, H.; Dekker, M.R.; Lazare, S.S.; Hammerl, F.; Skokova, Y.; et al. CBX7 Induces Self-Renewal of Human Normal and Malignant Hematopoietic Stem and Progenitor Cells by Canonical and Non-Canonical Interactions. Cell Rep. 2019, 26, 1906–1918. [Google Scholar] [CrossRef]
- Calés, C.; Román-Trufero, M.; Pavón, L.; Serrano, I.; Melgar, T.; Endoh, M.; Pérez, C.; Koseki, H.; Vidal, M. Inactivation of the Polycomb Group Protein Ring1B Unveils an Antiproliferative Role in Hematopoietic Cell Expansion and Cooperation with Tumorigenesis Associated with Ink4a Deletion. Mol. Cell. Biol. 2008, 28, 1018–1028. [Google Scholar] [CrossRef]
- Park, I.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 Is Required for Maintenance of Adult Self-Renewing Haematopoietic Stem Cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Lessard, J.; Sauvageau, G. Bmi-1 Determines the Proliferative Capacity of Normal and Leukaemic Stem Cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef]
- Rizo, A.; Olthof, S.; Han, L.; Vellenga, E.; De Haan, G.; Schuringa, J.J. Repression of BMI1 in Normal and Leukemic Human CD34+ Cells Impairs Self-Renewal and Induces Apoptosis Repression of BMI1 in Normal and Leukemic Human CD34+ Cells Impairs Self-Renewal and Induces Apoptosis. Hematology 2009, 114, 1498–1504. [Google Scholar]
- Schuringa, J.J.; Vellenga, E. Role of the Polycomb Group Gene BMI1 in Normal and Leukemic Hematopoietic Stem and Progenitor Cells. Curr. Opin. Hematol. 2010, 17, 294–299. [Google Scholar] [CrossRef]
- Wada, T.; Koyama, D.; Kikuchi, J.; Honda, H.; Furukawa, Y. Overexpression of the Shortest Isoform of Histone Demethylase LSD1 Primes Hematopoietic Stem Cells for Malignant Transformation. Blood 2015, 125, 3731–3747. [Google Scholar] [CrossRef]
- Kerenyi, M.A.; Shao, Z.; Hsu, Y.-J.; Guo, G.; Luc, S.; O’Brien, K.; Fujiwara, Y.; Peng, C.; Nguyen, M.; Orkin, S.H. Histone Demethylase Lsd1 Represses Hematopoietic Stem and Progenitor Cell Signatures during Blood Cell Maturation. Elife 2013, 2. [Google Scholar] [CrossRef]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-Specific Demethylase 1 Restricts Hematopoietic Progenitor Proliferation and Is Essential for Terminal Differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef]
- Stewart, M.H.; Albert, M.; Sroczynska, P.; Cruickshank, V.A.; Guo, Y.; Rossi, D.J.; Helin, K.; Enver, T. The Histone Demethylase Jarid1b Is Required for Hematopoietic Stem Cell Self-Renewal in Mice. Blood 2015, 125, 2075–2078. [Google Scholar] [CrossRef]
- Cellot, S.; Hope, K.J.; Chagraoui, J.; Sauvageau, M.; Deneault, É.; MacRae, T.; Mayotte, N.; Wilhelm, B.T.; Landry, J.R.; Ting, S.B. RNAi Screen Identifies Jarid1b as a Major Regulator of Mouse HSC Activity. Blood 2013, 122, 1545–1555. [Google Scholar] [CrossRef]
- Van der Meulen, J.; Speleman, F.; Van Vlierberghe, P. The H3K27me3 Demethylase UTX in Normal Development and Disease. Epigenetics 2014, 9, 658–668. [Google Scholar] [CrossRef]
- Thieme, S.; Gyárfás, T.; Richter, C.; Özhan, G.; Fu, J.; Alexopoulou, D.; Muders, M.H.; Michalk, I.; Jakob, C.; Dahl, A.; et al. The Histone Demethylase UTX Regulates Stem Cell Migration and Hematopoiesis. Blood 2013, 121, 2462–2473. [Google Scholar] [CrossRef]
- Nakata, Y.; Yamasaki, N.; Ueda, T.; Ikeda, K.; Nagamachi, A.; Inaba, T.; Honda, H. JMJD3 Plays Essential Roles in the Maintenance of Hematopoietic Stem Cells and Leukemic Stem Cells through the Regulation of P16INK4a. Blood 2016, 128, 2653. [Google Scholar]
- Nijnik, A.; Clare, S.; Hale, C.; Raisen, C.; McIntyre, R.E.; Yusa, K.; Everitt, A.R.; Mottram, L.; Podrini, C.; Lucas, M.; et al. The Critical Role of Histone H2A-Deubiquitinase Mysm1 in Hematopoiesis and Lymphocyte Differentiation. Blood 2012, 119, 1370–1379. [Google Scholar] [CrossRef]
- Huo, Y.; Li, B.-Y.; Lin, Z.-F.; Wang, W.; Jiang, X.-X.; Chen, X.; Xi, W.-J.; Yang, A.-G.; Chen, S.-Y.; Wang, T. MYSM1 Is Essential for Maintaining Hematopoietic Stem Cell (HSC) Quiescence and Survival. Med. Sci. Monit. 2018, 24, 2541–2549. [Google Scholar] [CrossRef]
- Wang, T.; Nandakumar, V.; Jiang, X.-X.; Jones, L.; Yang, A.-G.; Huang, X.F.; Chen, S.-Y. The Control of Hematopoietic Stem Cell Maintenance, Self-Renewal, and Differentiation by Mysm1-Mediated Epigenetic Regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef]
- Wang, H.; Diao, D.; Shi, Z.; Zhu, X.; Gao, Y.; Gao, S.; Liu, X.; Wu, Y.; Rudolph, K.L.; Liu, G.; et al. SIRT6 Controls Hematopoietic Stem Cell Homeostasis through Epigenetic Regulation of Wnt Signaling. Cell Stem Cell 2016, 18, 495–507. [Google Scholar] [CrossRef]
- Singh, S.K.; Williams, C.A.; Klarmann, K.; Burkett, S.S.; Keller, J.R.; Oberdoerffer, P. Sirt1 Ablation Promotes Stress-Induced Loss of Epigenetic and Genomic Hematopoietic Stem and Progenitor Cell Maintenance. J. Exp. Med. 2013, 210, 987–1001. [Google Scholar] [CrossRef]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 Reverses Aging-Associated Degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef]
- Mohrin, M.; Shin, J.; Liu, Y.; Brown, K.; Luo, H.; Xi, Y.; Haynes, C.M.; Chen, D. A Mitochondrial UPR-Mediated Metabolic Checkpoint Regulates Hematopoietic Stem Cell Aging. Science 2015, 347, 1374–1377. [Google Scholar] [CrossRef]
- Luo, H.; Mu, W.-C.; Karki, R.; Chiang, H.-H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.-D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging Hematopoietic Stem Cells Decline in Function and Exhibit Epigenetic Dysregulation. PLoS Biol. 2007, 5, 1750–1762. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell Intrinsic Alterations Underlie Hematopoietic Stem Cell Aging. PNAS 2005, 102, 9194–9199. [Google Scholar] [CrossRef]
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D. Functionally Distinct Hematopoietic Stem Cells Modulate Hematopoietic Lineage Potential during Aging by a Mechanism of Clonal Expansion. PNAS 2010, 107. [Google Scholar] [CrossRef]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; De Haan, G. Clonal Analysis Reveals Multiple Functional Defects of Aged Murine Hematopoietic Stem Cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef]
- Sudo, B.K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-Associated Characteristics of Murine Hematopoietic Stem Cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef]
- Säwén, P.; Lang, S.; Mandal, P.; Rossi, D.J.; Soneji, S.; Bryder, D. Mitotic History Reveals Distinct Stem Cell Populations and Their Contributions to Hematopoiesis. Cell Rep. 2016, 14, 2809–2818. [Google Scholar] [CrossRef]
- Yamamoto, R.; Wilkinson, A.C.; Ooehara, J.; Nakauchi, Y.; Pritchard, J.K.; Yamamoto, R.; Wilkinson, A.C.; Ooehara, J.; Lan, X.; Lai, C.; et al. Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment Resource Large-Scale Clonal Analysis Resolves Aging of the Mouse Hematopoietic Stem Cell Compartment. Cell Stem Cell 2018, 22, 600–607. [Google Scholar] [CrossRef]
- Cho, R.H.; Sieburg, H.B.; Muller-Sieburg, C.E. A New Mechanism for the Aging of Hematopoietic Stem Cells: Aging Changes the Clonal Composition of the Stem Cell Compartment but Not Individual Stem Cells. Blood 2008, 111, 5553–5562. [Google Scholar] [CrossRef]
- Bernitz, J.M.; Kim, H.S.; MacArthur, B.; Sieburg, H.; Moore, K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell 2016, 167, 1296–1309. [Google Scholar] [CrossRef]
- Quéré, R.; Saint-paul, L.; Carmignac, V.; Martin, R.Z.; Chrétien, M. Tif1 γ Regulates the TGF- β 1 Receptor and Promotes Physiological Aging of Hematopoietic Stem Cells. PNAS 2014, 111, 10592–10597. [Google Scholar]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging That Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Norddahl, G.L.; Sten, G.; Ugale, A.; Frisk, M.A.M.; Mattsson, R.; Deierborg, T.; Sigvardsson, M.; Bryder, D. An Epigenetic Component of Hematopoietic Stem Cell Aging Amenable to Reprogramming into a Young State. Blood 2013, 121, 4257–4264. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic Nerve Degeneration in Bone Marrow Drives Aging of the Hematopoietic Stem Cell Niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef]
- Beerman, I.; Rossi, D.J. Epigenetic Regulation of Hematopoietic Stem Cell Aging. Exp. Cell Res. 2014, 329, 192–199. [Google Scholar] [CrossRef]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Article Proliferation-Dependent Alterations of the DNA Methylation Landscape Underlie Hematopoietic Stem Cell Aging. Stem Cell 2013, 12, 413–425. [Google Scholar]
- Bersenev, A.; Rozenova, K.; Balcerek, J.; Jiang, J.; Wu, C.; Tong, W. Lnk Deficiency Partially Mitigates Hematopoietic Stem Cell Aging. Aging Cell 2012, 11, 949–959. [Google Scholar] [CrossRef]
- Flach, J.; Bakker, S.T.; Mohrin, M.; Conroy, P.C.; Pietras, E.M.; Reynaud, D.; Alvarez, S.; Diolaiti, M.E.; Ugarte, F.; Camilla, E.; et al. Replication Stress Is a Potent Driver of Functional Decline in Ageing Haematopoietic Stem Cells. Nature 2014, 512, 198–202. [Google Scholar] [CrossRef]
- Kirschner, K.; Chandra, T.; Kiselev, V.; Hemberg, M.; Reik, W.; Green, A.R.; Kirschner, K.; Chandra, T.; Kiselev, V.; Cruz, D.F.; et al. Proliferation Drives Aging-Related Functional Decline in a Subpopulation of the Hematopoietic Stem Cell Compartment Report Proliferation Drives Aging-Related Functional Decline in a Subpopulation of the Hematopoietic Stem Cell Compartment. Cell Rep. 2017, 19, 1503–1511. [Google Scholar] [CrossRef]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-Cell RNA-Seq Reveals Changes in Cell Cycle and Differentiation Programs upon Aging of Hematopoietic Stem Cells. Genome Res. 2015, 25, 1860–1872. [Google Scholar] [CrossRef]
- Noda, S.; Ichikawa, H.; Miyoshi, H. Hematopoietic Stem Cell Aging Is Associated with Functional Decline and Delayed Cell Cycle Progression. Biochem. Biophys. Res. Commun. 2009, 383, 210–215. [Google Scholar] [CrossRef]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating Mitochondrial DNA Mutations Drive Premature Hematopoietic Aging Phenotypes Distinct from Physiological Stem Cell Aging. Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef]
- Kramer, A.; Challen, G.A. The Epigenetic Basis of Hematopoietic Stem Cell Aging Functional and Molecular Manifestations of HSC Aging. Semin. Hematol. 2017, 54, 19–24. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Bryder, D. The Slippery Slope of Hematopoietic Stem Cell Aging. Exp. Hematol. 2017, 56, 1–6. [Google Scholar] [CrossRef]
- Pang, W.W.; Schrier, S.L.; Weissman, I.L. Age-Associated Changes in Human Hematopoietic Stem Cells. Semin. Hematol. 2017, 54, 39–42. [Google Scholar] [CrossRef]
- Huidobro, C.; Fernandez, A.F.; Fraga, M.F. Aging Epigenetics: Causes and Consequences. Mol. Aspects Med. 2013, 34, 765–781. [Google Scholar] [CrossRef]
- Zane, L.; Sharma, V.; Misteli, T. Common Features of Chromatin in Aging and Cancer: Cause or Coincidence? Trends Cell Biol. 2014, 24, 686–694. [Google Scholar] [CrossRef]
- Fuke, C.; Shimabukuro, M.; Petronis, A.; Sugimoto, J.; Oda, T.; Miura, K.; Miyazaki, T.; Ogura, C.; Okazaki, Y.; Jinno, Y. Age Related Changes in 5-Methylcytosine Content in Human Peripheral Leukocytes and Placentas: An HPLC-Based Study. Ann. Hum. Genet. 2004, 68, 196–204. [Google Scholar] [CrossRef]
- Wagner, W. Epigenetic Aging Clocks in Mice and Men. Genome Biol. 2017, 18, 17–19. [Google Scholar] [CrossRef]
- Horvath, S. Erratum to DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2015, 16. [Google Scholar] [CrossRef]
- Søraas, A.; Matsuyama, M.; de Lima, M.; Wald, D.; Buechner, J.; Gedde-Dahl, T.; Søraas, C.L.; Chen, B.; Ferrucci, L.; Dahl, J.A.; et al. Epigenetic Age Is a Cell-Intrinsic Property in Transplanted Human Hematopoietic Cells. Aging Cell 2019. [Google Scholar] [CrossRef]
- Weidner, C.I.; Ziegler, P.; Hahn, M.; Brümmendorf, T.H.; Ho, A.D.; Dreger, P.; Wagner, W. Epigenetic Aging upon Allogeneic Transplantation: The Hematopoietic Niche Does Not Affect Age-Associated DNA Methylation. Leukemia 2015, 29, 985–988. [Google Scholar] [CrossRef]
- Stölzel, F.; Brosch, M.; Horvath, S.; Kramer, M.; Thiede, C.; von Bonin, M.; Ammerpohl, O.; Middeke, M.; Schetelig, J.; Ehninger, G.; et al. Dynamics of Epigenetic Age Following Hematopoietic Stem Cell Transplantation. Haematologica 2017, 102, e321–e323. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Shlush, L.I. Age-Related Clonal Hematopoiesis. Blood 2018, 131, 496–504. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-Related Mutations Associated with Clonal Hematopoietic Expansion and Malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Steensma, D.P. Clinical Consequences of Clonal Hematopoiesis of Indeterminate Potential. Blood 2018, 2, 264–269. [Google Scholar]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357. [Google Scholar] [CrossRef]
- Cancer Genome Atlas. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A Mutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Schlenk, R.F.; Paschka, P.; Stölzle, A.; Späth, D.; Kuendgen, A.; von Lilienfeld-Toal, M.; Brugger, W.; Derigs, H.G.; Kremers, S.; et al. Clinical Impact of DNMT3A Mutations in Younger Adult Patients with Acute Myeloid Leukemia: Results of the AML Study Group (AMLSG). Blood 2013, 121, 4769–4777. [Google Scholar] [CrossRef]
- Gale, R.E.; Lamb, K.; Allen, C.; El-Sharkawi, D.; Stowe, C.; Jenkinson, S.; Tinsley, S.; Dickson, G.; Burnett, A.K.; Hills, R.K.; et al. Simpson’s Paradox and the Impact of Different DNMT3A Mutations on Outcome in Younger Adults With Acute Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 2072–2083. [Google Scholar] [CrossRef]
- Brunetti, L.; Gundry, M.C.; Goodell, M.A. DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef]
- Spencer, D.H.; Al-Khalil, B.; Russler-Germain, D.; Lamprecht, T.; Havey, N.; Fulton, R.S.; O’Laughlin, M.; Fronick, C.; Wilson, R.K.; Ley, T.J. Whole-Genome Bisulfite Sequencing of Primary AML Cells with the DNMT3A R882H Mutation Identifies Regions of Focal Hypomethylation That Are Associated with Open Chromatin. Blood 2014, 124, 608. [Google Scholar]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinawi, M.; et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 2017, 168, 801–816. [Google Scholar] [CrossRef]
- Yang, L.; Rodriguez, B.; Mayle, A.; Park, H.J.; Lin, X.; Luo, M.; Jeong, M.; Curry, C.V.; Kim, S.-B.; Ruau, D.; et al. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer Cell 2016, 29, 922–934. [Google Scholar] [CrossRef]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 Mutations Predict Response to Hypomethylating Agents in Myelodysplastic Syndrome Patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V. Della; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Kosmider, O.; Gelsi-Boyer, V.; Ciudad, M.; Racoeur, C.; Jooste, V.; Vey, N.; Quesnel, B.; Fenaux, P.; Bastie, J.-N.; Beyne-Rauzy, O.; et al. TET2 Gene Mutation Is a Frequent and Adverse Event in Chronic Myelomonocytic Leukemia. Haematologica 2009, 94, 1676–1681. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, J.-D.; Yang, D.; Wang, Y.; Wen, X.; Guo, H.; Yang, L.; Lian, X.; Lin, J. TET2 Expression Is a Potential Prognostic and Predictive Biomarker in Cytogenetically Normal Acute Myeloid Leukemia. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Fong, C.Y.; Morison, J.; Dawson, M.A. Epigenetics in the Hematologic Malignancies. Hematologica 2014, 99, 1079–1095. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49. [Google Scholar] [CrossRef]
- Ungerstedt, J.S. Epigenetic Modifiers in Myeloid Malignancies: The Role of Histone Deacetylase Inhibitors. Int. J. Mol. Sci. 2018, 19, 3091. [Google Scholar] [CrossRef]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression Profile of Class I Histone Deacetylases in Human Cancer Tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef]
- Yang, H.; Maddipoti, S.; Quesada, A.; Bohannan, Z.; Cabrero Calvo, M.; Colla, S.; Wei, Y.; Estecio, M.; Wierda, W.; Bueso-Ramos, C.; et al. Analysis of Class I and II Histone Deacetylase Gene Expression in Human Leukemia. Leuk. Lymphoma 2015, 56, 3426–3433. [Google Scholar] [CrossRef]
- Castelli, G.; Pelosi, E.; Testa, U. Targeting Histone Methyltransferase and Demethylase in Acute Myeloid Leukemia Therapy. Onco. Targets. Ther. 2018, 11, 131–155. [Google Scholar] [CrossRef]
- Nagase, R.; Inoue, D.; Pastore, A.; Fujino, T.; Hou, H.A.; Yamasaki, N.; Goyama, S.; Saika, M. Expression of Mutant Asxl1 Perturbs Hematopoiesis and Promotes Susceptibility to Leukemic Transformation. J. Exp. Med. 2018, 215, 1729–1747. [Google Scholar] [CrossRef]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of Function of ASXL1 Truncating Protein in the Pathogenesis of Myeloid Malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Boultwood, J.; Perry, J.; Pellagatti, A.; Fernandez-Mercado, M.; Fernandez-Santamaria, C.; Calasanz, M.J.; Larrayoz, M.J.; Garcia-Delgado, M.; Giagounidis, A.; Malcovati, L.; et al. Frequent Mutation of the Polycomb-Associated Gene ASXL1 in the Myelodysplastic Syndromes and in Acute Myeloid Leukemia. Leukemia 2010, 24, 1062–1065. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Trouplin, V.; Adélaïde, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of Polycomb-associated Gene ASXL1 in Myelodysplastic Syndromes and Chronic Myelomonocytic Leukaemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Pardanani, A.; Patel, J.; Wadleigh, M.; Lasho, T.; Heguy, A.; Beran, M.; Gilliland, D.G.; Levine, R.L.; Tefferi, A. Concomitant Analysis of EZH2 and ASXL1 Mutations in Myelofibrosis, Chronic Myelomonocytic Leukaemia and Blast-Phase Myeloproliferative Neoplasms. Leukemia 2011, 25, 1200–1202. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Brecqueville, M.; Devillier, R.; Murati, A.; Mozziconacci, M.J.; Birnbaum, D. Mutations in ASXL1 Are Associated with Poor Prognosis across the Spectrum of Malignant Myeloid Diseases. J. Hematol. Oncol. 2012, 5. [Google Scholar] [CrossRef]
- Lehnertz, B.; Zhang, Y.W.; Boivin, I.; Mayotte, N.; Tomellini, E.; Chagraoui, J.; Lavallée, V.P.; Hébert, J.; Sauvageau, G. H3K27M/I Mutations Promote Context-Dependent Transformation in Acute Myeloid Leukemia with RUNX1 Alterations. Blood 2017, 130, 2204–2214. [Google Scholar] [CrossRef]
- Safaei, S.; Baradaran, B.; Hagh, M.F.; Alivand, M.R.; Talebi, M.; Gharibi, T.; Solali, S. Double Sword Role of EZH2 in Leukemia. Biomed. Pharmacother. 2018, 98, 626–635. [Google Scholar] [CrossRef]
- Xu, F.; Liu, L.; Chang, C.; He, Q.; Wu, L.; Zhang, Z. Genomic Loss of EZH2 Leads to Epigenetic Modifications and Overexpression of the HOX Gene Clusters in Myelodysplastic Syndrome. Oncotarget 2016, 7, 8119–8130. [Google Scholar] [CrossRef][Green Version]
- Sashida, G.; Harada, H.; Matsui, H.; Oshima, M.; Yui, M.; Harada, Y.; Tanaka, S.; Mochizuki-Kashio, M.; Wang, C.; Saraya, A.; et al. Ezh2 Loss Promotes Development of Myelodysplastic Syndrome but Attenuates Its Predisposition to Leukaemic Transformation. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Huet, S.; Xerri, L.; Tesson, B.; Mareschal, S.; Taix, S.; Mescam-Mancini, L.; Sohier, E.; Carrère, M.; Lazarovici, J.; Casasnovas, O. EZH2 Alterations in Follicular Lymphoma: Biological and Clinical Correlations. Blood Cancer J. 2017, 7. [Google Scholar] [CrossRef]
- Navada, S.C.; Steinmann, J.; Lübbert, M.; Silverman, L.R. Clinical Development of Demethylating Agents in Hematology. J. Clin. Investig. 2014, 124, 40–46. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Clinical Epigenetics: Seizing Opportunities for Translation. Nat. Rev. Genet. 2018, 20, 109–127. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International Phase 3 Study of Azacitidine vs. Conventional Care Regimens in Older Patients with Newly Diagnosed AML with > 30% Blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Stahl, M.; Zeidan, A.M.; Komrokji, R.S.; Sekeres, M.A.; Gore, S.D.; Steensma, D.P. A Call for Action: Increasing Enrollment of Untreated Patients with Higher-Risk Myelodysplastic Syndromes in First-Line Clinical Trials. Cancer 2017, 123, 3662–3672. [Google Scholar]
- Stahl, M.; Davidoff, A.J.; Zeidan, A.M.; Huntington, S.; DeVeaux, M.; Ma, X.; Podoltsev, N.; Wang, R.; Gore, S.D.; Giri, S. Counseling Patients with Higher-Risk MDS Regarding Survival with Azacitidine Therapy: Are We Using Realistic Estimates? Blood Cancer J. 2018, 8, 4–7. [Google Scholar]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of Molecular Mutations on Treatment Response to DNMT Inhibitors in Myelodysplasia and Related Neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in Human Disease and Prospects for Epigenetic Therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Insinga, A.; Monestiroli, S.; Ronzoni, S.; Gelmetti, V.; Marchesi, F.; Viale, A.; Altucci, L.; Nervi, C.; Minucci, S.; Pelicci, P.G. Inhibitors of Histone Deacetylases Induce Tumor-Selective Apoptosis through Activation of the Death Receptor Pathway. Nat. Med. 2005, 11, 71–76. [Google Scholar] [CrossRef]
- Nebbioso, A.; Clarke, N.; Voltz, E.; Germain, E.; Ambrosino, C.; Bontempo, P.; Alvarez, R.; Schiavone, E.M.; Ferrara, F.; Bresciani, F.; et al. Tumor-Selective Action of HDAC Inhibitors Involves TRAIL Induction in Acute Myeloid Leukemia Cells. Nat. Med. 2005, 11, 77–84. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Lee, J.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone Deacetylase Inhibitor Induces DNA Damage, Which Normal but Not Transformed Cells Can Repair. PNAS 2010, 107, 14639–14644. [Google Scholar] [CrossRef]
- Robert, C.; Rassool, F.V. Roles of DNA Damage and Repair. In Histone Deacetylase Inhibitors as Cancer Therapeutics; Academic Press: San Diego, CA, USA, 2012; Volume 116, pp. 87–129. [Google Scholar]
- Quintas-Cardama, A.; Santos, F.P.S.; Garcia-Manero, G. Histone Deacetylase Inhibitors for the Treatment of Myelodysplastic Syndrome and Acute Myeloid Leukemia. Leukemia 2011, 25, 226–235. [Google Scholar] [CrossRef]
- Cameron, E.E.; Bachman, K.E.; Myöhänen, S.; Herman, J.G.; Baylin, S.B. Synergy of Demethylation and Histone Deacetylase Inhibition in the Re-Expression of Genes Silenced in Cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals Corporation. Farydak (panobinostat) [prescribing information]. Available online: https://us.farydak.com/ accessed on 22-07-2019 (accessed on 22 July 2019).
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetics Modifications as Therapeutic Targets. Nat. Biotechnol. 2010, 28, 1069–1078. [Google Scholar] [CrossRef]
- Bowers, E.M.; Yan, G.; Mukherjee, C.; Orry, A.; Wang, L.; Holbert, M.A.; Crump, N.T.; Hazzalin, C.A.; Liszczak, G.; Yuan, H.; et al. Virtual Ligand Screening of the P300/CBP Histone Acetyltransferase: Identification of a Selective Small Molecule Inhibitor. Chem. Biol. 2010, 17, 471–482. [Google Scholar] [CrossRef]
- Gao, X.; Lin, J.; Ning, Q.; Gao, L.; Yao, Y.; Zhou, J.; Li, Y.; Wang, L.; Yu, L. A Histone Acetyltransferase P300 Inhibitor C646 Induces Cell Cycle Arrest and Apoptosis Selectively in AML1-ETO-Positive AML Cells. PLoS ONE 2013, 2. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.-C.; Toulmonde, M.; Michot, J.-M.; Lucchesi, C.; Varga, A.; Coindre, J.-M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 Inhibitor, in Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma and Advanced Solid Tumours: A First-in-Human, Open-Label, Phase 1 Study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Wouters, B.J.; Delwel, R. Epigenetics and Approaches to Targeted Epigenetic Therapy in Acute Myeloid Leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef]
- Galkin, M.; Jonas, B.A. Enasidenib in the Treatment of Relapsed/Refractory Acute Myeloid Leukemia: An Evidence-Based Review of Its Place in Therapy. Core Evid. 2019, 14, 3–17. [Google Scholar] [CrossRef]
- Nassereddine, S.; Lap, C.J.; Haroun, F.; Tabbara, I. The Role of Mutant IDH1 and IDH2 Inhibitors in the Treatment of Acute Myeloid Leukemia. Ann. Hematol. 2017, 96, 1983–1991. [Google Scholar] [CrossRef]
- Goldman, S.L.; Hassan, C.; Khunte, M.; Soldatenko, A.; Jong, Y.; Afshinnekoo, E.; Mason, C.E. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a Selective Small Molecule Inhibitor of the CBP/P300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef]
- Magliulo, D.; Bernardi, R.; Messina, S. Lysine-Specific Demethylase 1A as a Promising Target in Acute Myeloid Leukemia. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Nakagawa, M.; Fujita, S.; Katsumoto, T.; Yamagata, K.; Ogawara, Y.; Hattori, A.; Kagiyama, Y.; Honma, D.; Araki, K.; Inoue, T.; et al. Dual Inhibition of Enhancer of Zeste Homolog 1/2 Overactivates WNT Signaling to Deplete Cancer Stem Cells in Multiple Myeloma. Cancer Sci. 2018, 110, 194–208. [Google Scholar] [CrossRef]
- Fujita, S.; Honma, D.; Adachi, N.; Araki, K.; Takamatsu, E.; Katsumoto, T.; Yamagata, K.; Akashi, K.; Aoyama, K.; Iwama, A.; et al. Dual Inhibition of EZH1/2 Breaks the Quiescence of Leukemia Stem Cells in Acute Myeloid Leukemia. Leukemia 2018, 32, 855–864. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L Inhibitor Pinometostat Reduces H3K79 Methylation and Has Modest Clinical Activity in Adult Acute Leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Ren, C.; Smith, S.G.; Yap, K.; Li, S.; Li, J.; Mezei, M.; Rodriguez, Y.; Vincek, A.; Aguilo, F.; Walsh, M.J.; et al. Structure-Guided Discovery of Selective Antagonists for the Chromodomain of Polycomb Repressive Protein CBX7. ACS Med. Chem. Lett. 2016, 7, 601–605. [Google Scholar] [CrossRef]
- Stuckey, J.I.; Dickson, B.M.; Cheng, N.; Liu, Y.; Norris, J.L.; Cholensky, S.H.; Tempel, W.; Qin, S.; Huber, K.G.; Sagum, C.; et al. A Cellular Chemical Probe Targeting the Chromodomains of Polycomb Repressive Complex 1. Nat. Chem. Biol. 2016, 12, 180–187. [Google Scholar] [CrossRef]
- Ren, C.; Morohashi, K.; Plotnikov, A.N.; Jakoncic, J.; Smith, S.G.; Li, J.; Zeng, L.; Rodriguez, Y.; Stojanoff, V.; Walsh, M.; et al. Small Molecule Modulators of Methyl-Lysine Binding for the CBX7 Chromodomain. Chem. Biol. 2015, 22, 161–168. [Google Scholar] [CrossRef]
- Bewersdorf, J.P.; Shallis, R.; Stahl, M.; Zeidan, A.M. Epigenetic Therapy Combinations in Acute Myeloid Leukemia: What Are the Options? Ther. Adv. Hematol. 2019, 10, 1–19. [Google Scholar] [CrossRef]
- Claes, B.; Buysschaert, I.; Lambrechts, D. Pharmaco-Epigenomics: Discovering Therapeutic Approaches and Biomarkers for Cancer Therapy. Heredity 2010, 105, 152–160. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Baer-Dubowska, W. Pharmacoepigenetics: An Element of Personalized Therapy? Expert Opin. Drug Metab. Toxicol. 2017, 13, 387–398. [Google Scholar] [CrossRef]
- Kronfol, M.M.; Dozmorov, M.G.; Huang, R.; Slattum, P.W.; McClay, J.L. The Role of Epigenomics in Personalized Medicine. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 33–45. [Google Scholar] [CrossRef]
- Stunnenberg, H.G.; Abrignani, S.; Adams, D.; de Almeida, M.; Altucci, L.; Amin, V.; Amit, I.; Antonarakis, S.E.; Aparicio, S.; Arima, T.; et al. The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell 2016, 167, 1145–1149. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Function | Effect on HSC | Reference |
|---|---|---|---|
| DNA modifications | |||
| DNMT1 | DNA methyltransferase: maintenance parental cell methylation patterns. | Required for HSC self-renewal, niche retention and progression from multipotent to myeloid progenitors. Deletion leads to lineage skewing towards myelopoiesis and defective self-renewal. | [4,5,6] |
| DNMT3A/B | DNA methyltransferase: de novo DNA methylation. | Essential for HSC self-renewal and deletion increases HSC life span. DNMT3A and DNMT3B show complementary de novo methylation patterns responsible for silencing of self-renewal genes in HSCs. | [7,8,9,10,11] |
| TET1/2 | Catalyse the oxidation of 5-methylcytosine into 5-hydroxymethylcytosine, resulting in DNA demethylation. | TET1 deficiency increases HSC self-renewal potential. TET2 deletion resulted in enhanced HSC self-renewal and enhanced myelopoiesis. TET2 mutations are mutually exclusive with gain of function mutations IDH1/2 in AML. | [12,13,14,15,16,17] |
| IDH1/2 | Isocitrate dehydrogenase 1/2 enzymes (IDH1/2), required for conversion of isocitrate into a-ketoglutarate, a TET2 cofactor. | IDH1 mutations impair histone demethylation and result in hypermethylation. TET2 function becomes disrupted and differentiation is inhibited. Mutant IDH1/2 overexpression in primary bone marrow cells disturbs haematopoietic differentiation and leads to elevated numbers of HSPCs. | [18,19,20] |
| Histone modifications | |||
| EZH1/2 | Histone lysine methyltransferase, PRC2 member. | Important for HSC maintenance. Deletion decreases self-renewal potential. EZH2 deletion alone does not compromise self-renewal, possibly due to complementary EZH1 activity. | [21,22,23,24] |
| DOT1L | H3K79 methyltransferase | Crucial for embryonic erythropoiesis. Not essential for adult haematopoiesis. | [25,26,27,28] |
| MLL proteins | H3Kmethyltransferases | MLL-fusion enriched target genes play a role in HSC function, MLL mediated activation of HOX genes has been described, MLL fusions are potent inducers of leukaemia. | [29,30,31,32] |
| SETDB1 | H3K9 methyltransferase | Essential for HSC function. | [33] |
| PMRT4/5 | Protein arginine methyltransferases | PMRT4 blocks myeloid differentiation. PMRT5 inhibits expression of differentiation associated genes, PMRT5 deletion results in HSPC exhaustion. | [34,35,36,37] |
| CBP/p300 MOZ | Histone acetyltransferases | CBP/p300 regulates self-renewal and differentiation in adult HSCs. MOZ maintains the generation and development of HSCs. | [38,39] |
| ASXL1 | Polycomb chromatin-binding protein, associates with PRC1 and PRC2. | Loss results in impaired self-renewal. | [40,41,42] |
| CBX2 | Chromobox 2, PRC1 member. Reads H3K27me3. | Impairs HSC and progenitor proliferation. | [43,44,45] |
| CBX7 | Chromobox 7, PRC1 member. Reads H3K27me3 and other trimethylated proteins. | Overexpression increases HSC self-renewal. | [45,46,47] |
| Ring1B | E3 ubiquitin-protein ligase. | Antiproliferative role in progenitor expansion. | [48] |
| BMI1/MEL-18 | Polycomb ring finger protein, PRC1 member. | Repression impairs self-renewal. Frequently overexpressed in malignant haematopoiesis. Loss enhances HSC self-renewal. | [49,50,51,52] |
| LSD1 | H3K4/9 demethylase | Overexpression of the short isoform increases self-renewal potential. Loss causes defects in long-term HSC self-renewal and impaired differentiation. | [53,54,55] |
| JARID1b | H3K4 demethylase | Described as a positive regulator of HSC self-renewal capacity. | [56,57] |
| KDM6A/UTX | H3K27 demethylase | Knock out leads to myelodysplasia, suppressed megakaryocytopoiesis and extramedullary haematopoiesis, indicating a regulatory role in haematopoiesis. | [58,59] |
| KDM6B/JMJD3 | H3K27 demethylase | Loss impairs HSC self-renewal potential following proliferative stress. | [59,60] |
| MYSM1 | Histone H2A deubiquitinase | Maintenance of HSC function. Deletion results in impaired self-renewal. | [61,62,63] |
| SIRT1/2/3/7 | NAD-dependent deacetylases | Sirt1 loss increases HSC numbers. Sirt2 and 3 are not required for HSC maintenance at young age, but knockout does compromise HSC function at old age. Sirt6-deficient HSCs exhibited impaired self-renewal ability. Sirt7 overexpression increases constitutive capacity of HSCs. | [64,65,66,67,68] |
| Type of Drug | Target Enzyme | Compound | Stage of Development | References |
|---|---|---|---|---|
| DNMTi | DNA methyltransferase | Azacitidine, decitabine | EMA and FDA approval for MDS, AML and CMML | [163] |
| IDH1/2 inhibitors | Pre-clinical and clinical | [164,165,166] | ||
| Ivosidenib (IDH1) | EMA and FDA approval for IDH1 mutated and R/R AML | |||
| Enasidenib (IDH2) | EMA and FDA approval for R/R AML | |||
| HDACi | Histone deacetylases | Panobinostat | EMA and FDA approval for MM, CTCL | [125,142,152,154] |
| Romidepsin | EMA and FDA approval for CTCL | |||
| HATi | Histone acetyltransferases | P300 inhibitors | Pre-clinical | [159,160,167] |
| HDMi | Histone demethylases | LSD1 inhibitors | Pre-clinical and clinical | [128,168] |
| HMTi | Histone methyltransferases | EZH1/2 inhibitors Tazemetostat (EZH2) | Pre-clinical and clinical | [169,170] |
| DOT1L inhibitors | Pre-clinical and clinical | [128,171] | ||
| MENIN1-MLL interaction inhibitors | Pre-clinical | [128] | ||
| HATi | Histone acetyl readers | BET-inhibitors | Pre-clinical and clinical | [163,166] |
| (myelodysplastic syndrome (MDS), relapsed/refractory (R/R) acute myeloid leukaemia (AML), chronic myelomonocytic leukaemia (CMML), multiple myeloma (MM), cutaneous T cell lymphoma (CTCL)) | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buisman, S.C.; de Haan, G. Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies. Cells 2019, 8, 868. https://doi.org/10.3390/cells8080868
Buisman SC, de Haan G. Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies. Cells. 2019; 8(8):868. https://doi.org/10.3390/cells8080868
Chicago/Turabian StyleBuisman, Sonja C., and Gerald de Haan. 2019. "Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies" Cells 8, no. 8: 868. https://doi.org/10.3390/cells8080868
APA StyleBuisman, S. C., & de Haan, G. (2019). Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies. Cells, 8(8), 868. https://doi.org/10.3390/cells8080868