Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation


{kind=link}
{kind=link}
Abstract
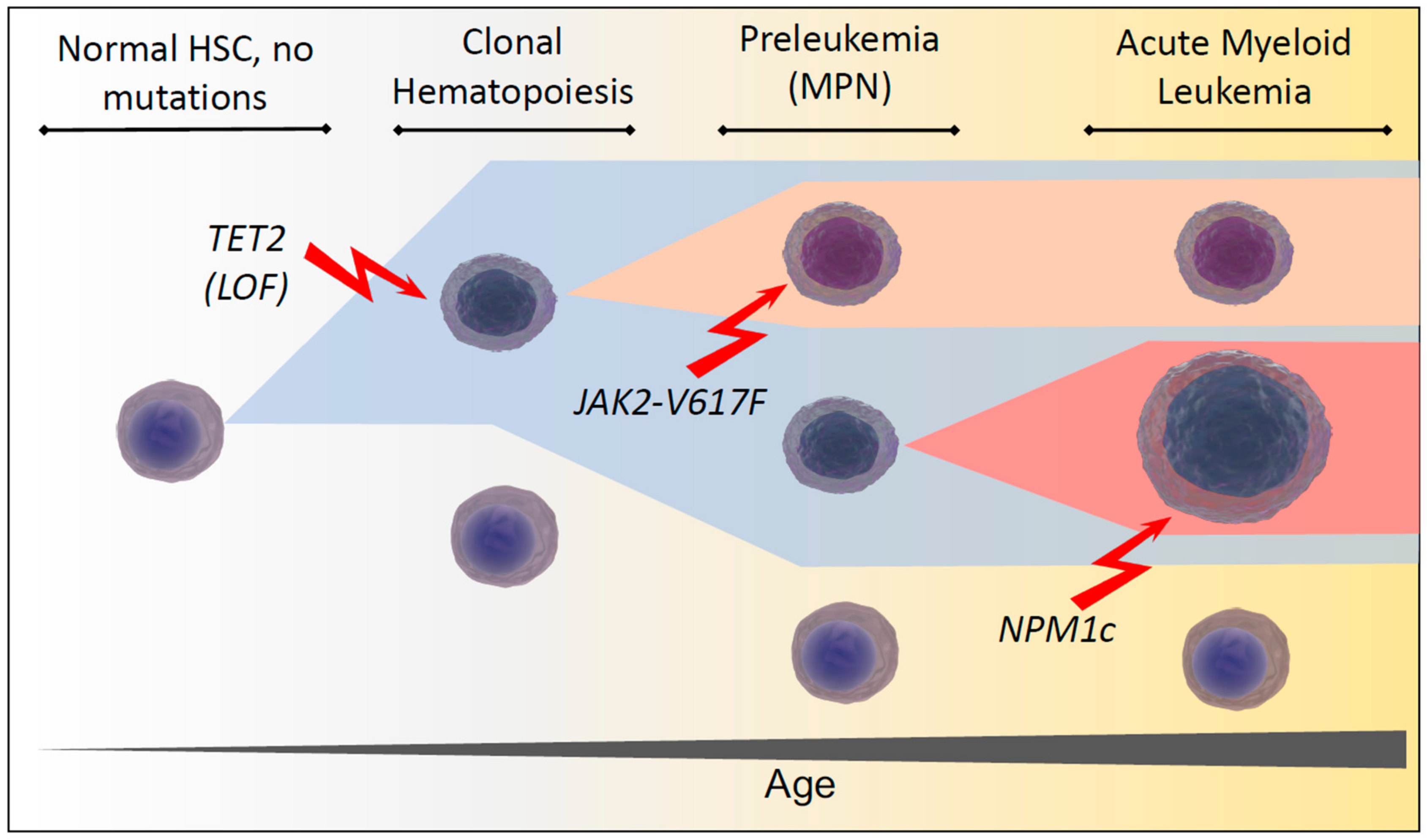
1. Development of Clonal Hematopoiesis During Aging
2. Consequences of Deregulated JAK2 Signaling in Hematopoiesis and Aging, CHIP-Driven Inflammation and Consequences of JAK2-Mutations
2.1. Dysfunctional JAK-Signaling and JAK-Inhibition in the Adaptive and Innate Immune System
2.2. Chronic Inflammation and JAK Signaling in Aging and Age-Related Degenerative Diseases
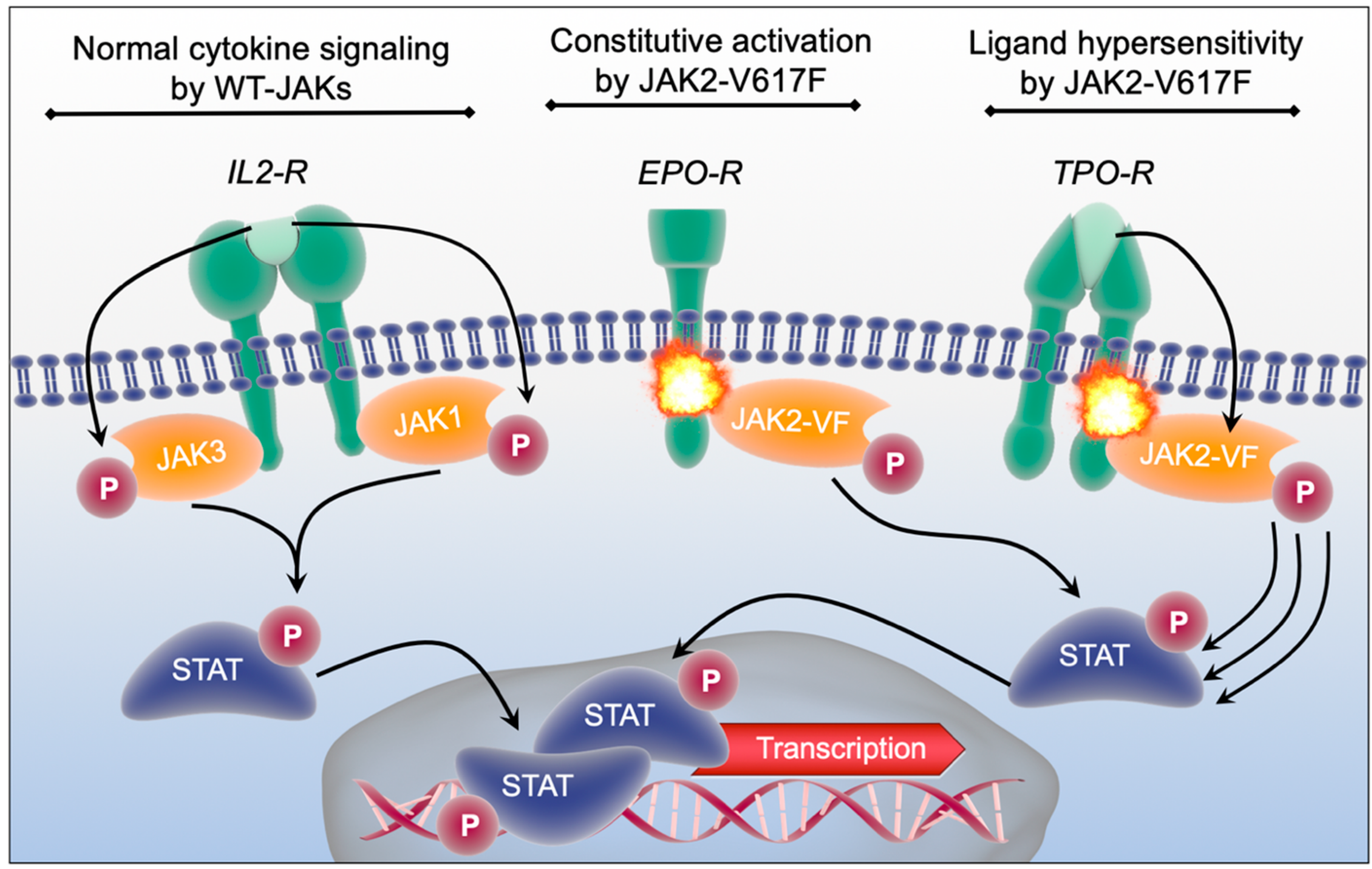
2.3. Pathophysiology of JAK2 in Malignant Transformation and Myeloproliferative Neoplasia
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beerman, I.; Bhattacharya, D.; Zandi, S.; Sigvardsson, M.; Weissman, I.L.; Bryder, D.; Rossi, D.J. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc. Natl. Acad. Sci. USA 2010, 107, 5465–5470. [Google Scholar] [CrossRef] [PubMed]
- Dykstra, B.; Olthof, S.; Schreuder, J.; Ritsema, M.; de Haan, G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J. Exp. Med. 2011, 208, 2691–2703. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Mio, R.; Mattioli, J.; Brais, E.; Blais, N.; Lalonde, Y.; Maragh, M.; Gilliland, D.G. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 1996, 88, 59–65. [Google Scholar] [PubMed]
- Gale, R.E.; Wheadon, H.; Boulos, P.; Linch, D.C. Tissue specificity of X-chromosome inactivation patterns. Blood 1994, 83, 2899–2905. [Google Scholar] [PubMed]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef]
- Young, A.L.; Challen, G.A.; Birmann, B.M.; Druley, T.E. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun. 2016. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Park, N.; Chi, J.; Collord, G.; Moreno, T.; Ponstingl, H.; Dias, J.; Gerasimou, P.; Melanthiou, K.; Prokopiou, C.; et al. JAK2 V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Adv. 2017, 1, 968–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389. [Google Scholar] [CrossRef] [PubMed]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 restrains inflammatory gene expression in macrophages. Exp. Hematol. 2017, 55, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Kotzin, J.J.; Ramdas, B.; Chen, S.; Nelanuthala, S.; Palam, L.R.; Pandey, R.; Mali, R.S.; Liu, Y.; Kelley, M.R.; et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018. [Google Scholar] [CrossRef]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Ma, H.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47, 284–297. [Google Scholar] [CrossRef]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a restrains mast cell inflammatory responses. Proc. Natl. Acad. Sci. USA 2017. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Li, Y.Z.; An, H.T.; Wang, Y.L.; Chen, S.H.; Qian, Y.J.; Wang, K.; Zhen, J.L.; Fan, Z.; Gong, X.L.; et al. The E3 Ubiquitin Ligase c-Cbl Inhibits Microglia-Mediated CNS Inflammation by Regulating PI3K/Akt/NF-kappaB Pathway. CNS Neurosci. Ther. 2016, 22, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, C.; Flavell, R.A. c-Cbl deficiency leads to diminished lymphocyte development and functions in an age-dependent manner. Proc. Natl. Acad. Sci. USA 2010, 107, 8316–8321. [Google Scholar] [CrossRef]
- Murakami, T.; Ruengsinpinya, L.; Nakamura, E.; Takahata, Y.; Hata, K.; Okae, H.; Taniguchi, S.; Takahashi, M.; Nishimura, R. Cutting Edge: G Protein Subunit beta 1 Negatively Regulates NLRP3 Inflammasome Activation. J. Immunol. 2019, 202, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Murray, F.; Koide, N.; Goldstone, J.; Dann, S.M.; Chen, J.; Bertin, S.; Fu, G.; Weinstein, L.S.; Chen, M.; et al. Divergent requirement for Gαs and cAMP in the differentiation and inflammatory profile of distinct mouse Th subsets. J. Clin. Investig. 2012, 122, 963–973. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef]
- Koschmieder, S.; Mughal, T.I.; Hasselbalch, H.C.; Barosi, G.; Valent, P.; Kiladjian, J.J.; Jeryczynski, G.; Gisslinger, H.; Jutzi, J.S.; Pahl, H.L.; et al. Myeloproliferative neoplasms and inflammation: Whether to target the malignant clone or the inflammatory process or both. Leukemia 2016, 30, 1018–1024. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 2013. [Google Scholar] [CrossRef]
- Boulay, J.L.; O’Shea, J.J.; Paul, W.E. Molecular phylogeny within type I cytokines and their cognate receptors. Immunity 2003, 19, 159–163. [Google Scholar] [CrossRef]
- Drachman, J.G.; Millett, K.M.; Kaushansky, K. Thrombopoietin signal transduction requires functional JAK2, not TYK2. J. Biol. Chem. 1999, 274, 13480–13484. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nature Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Marrero, M.B.; Schieffer, B.; Paxton, W.G.; Heerdt, L.; Berk, B.C.; Delafontaine, P.; Bernstein, K.E. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995, 375, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Witthuhn, B.A.; Silvennoinen, O.; Miura, O.; Lai, K.S.; Cwik, C.; Liu, E.T.; Ihle, J.N. Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells. Nature 1994, 370, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.M.; Johnston, J.A.; Noguchi, M.; Kawamura, M.; Bacon, C.M.; Friedmann, M.; Berg, M.; McVicar, D.W.; Witthuhn, B.A.; Silvennoinen, O.; et al. Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: Implications for XSCID and XCID. Science 1994, 266, 1042–1045. [Google Scholar] [CrossRef]
- Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Yi, T.; Tang, B.; Miura, O.; Ihle, J.N. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell 1993, 74, 227–236. [Google Scholar] [CrossRef]
- Neubauer, H.; Cumano, A.; Muller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef]
- Rodig, S.J.; Meraz, M.A.; White, J.M.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; King, K.L.; Sheehan, K.C.; Yin, L.; Pennica, D.; et al. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef]
- Casanova, J.L.; Holland, S.M.; Notarangelo, L.D. Inborn errors of human JAKs and STATs. Immunity 2012, 36, 515–528. [Google Scholar] [CrossRef]
- Larner, A.C.; David, M.; Feldman, G.M.; Igarashi, K.; Hackett, R.H.; Webb, D.S.; Sweitzer, S.M.; Petricoin, E.F., 3rd; Finbloom, D.S. Tyrosine phosphorylation of DNA binding proteins by multiple cytokines. Science 1993, 261, 1730–1733. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Ziemiecki, A.; Wilks, A.F.; Harpur, A.G.; Sadowski, H.B.; Gilman, M.Z.; Darnell, J.E. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature 1993, 366, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.X.; Migone, T.S.; Tsang, M.; Friedmann, M.; Weatherbee, J.A.; Zhou, L.; Yamauchi, A.; Bloom, E.T.; Mietz, J.; John, S.; et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity 1995, 2, 331–339. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Gadina, M.; Schreiber, R.D. Cytokine signaling in 2002: New surprises in the Jak/Stat pathway. Cell 2002. [Google Scholar] [CrossRef]
- Jang, Y.N.; Baik, E.J. JAK-STAT pathway and myogenic differentiation. JAKSTAT 2013. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Masse, A.; James, C.; Teyssandier, I.; Lecluse, Y.; Larbret, F.; Ugo, V.; Saulnier, P.; Koscielny, S.; Le Couedic, J.P.; et al. The JAK2 617V>F mutation triggers erythropoietin hypersensitivity and terminal erythroid amplification in primary cells from patients with polycythemia vera. Blood 2007, 110, 1013–1021. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nature Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Damsky, W.; King, B.A. JAK inhibitors in dermatology: The promise of a new drug class. J. Am. Acad. Dermatol. 2017, 76, 736–744. [Google Scholar] [CrossRef]
- Pardanani, A.; Vannucchi, A.M.; Passamonti, F.; Cervantes, F.; Barbui, T.; Tefferi, A. JAK inhibitor therapy for myelofibrosis: Critical assessment of value and limitations. Leukemia 2011, 25, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Molecular pathways: Molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Bonelli, M.; Gadina, M.; O’Shea, J.J. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nature Rev. Rheumatol. 2016, 12, 25–36. [Google Scholar] [CrossRef]
- Berekmeri, A.; Mahmood, F.; Wittmann, M.; Helliwell, P. Tofacitinib for the treatment of psoriasis and psoriatic arthritis. Expert Rev. Clin. Immunol 2018, 14, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, S.A.; McInnes, I.B. JAK Inhibitors in Rheumatology: Implications for Paediatric Syndromes? Curr. Rheumatol. Rep. 2018. [Google Scholar] [CrossRef]
- Nishanth, G.; Wolleschak, D.; Fahldieck, C.; Fischer, T.; Mullally, A.; Perner, F.; Schnoder, T.M.; Just, S.; Heidel, F.H.; Schluter, D. Gain of function in Jak2(V617F)-positive T-cells. Leukemia 2017, 31, 1000–1003. [Google Scholar] [CrossRef]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef]
- Stijnis, C.; Kroes, W.G.; Balkassmi, S.; Marijt, E.W.; van Rossum, A.P.; Bakker, E.; Vlasveld, L.T. No evidence for JAK2(V617F) mutation in monoclonal B cells in 2 patients with polycythaemia vera and concurrent monoclonal B cell disorder. Acta Haematologica 2012, 128, 183–186. [Google Scholar] [CrossRef]
- Prestipino, A.; Emhardt, A.J.; Aumann, K.; O’Sullivan, D.; Gorantla, S.P.; Duquesne, S.; Melchinger, W.; Braun, L.; Vuckovic, S.; Boerries, M.; et al. Oncogenic JAK2(V617F) causes PD-L1 expression, mediating immune escape in myeloproliferative neoplasms. Sci. Transl. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Cattaneo, M.; Rambaldi, A.; Squizzato, A. Ruxolitinib-associated infections: A systematic review and meta-analysis. Am.J. Haematol. 2018, 93, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Keohane, C.; Kordasti, S.; Seidl, T.; Perez Abellan, P.; Thomas, N.S.; Harrison, C.N.; McLornan, D.P.; Mufti, G.J. JAK inhibition induces silencing of T Helper cytokine secretion and a profound reduction in T regulatory cells. Br. J. Haematol. 2015, 171, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Parampalli Yajnanarayana, S.; Stubig, T.; Cornez, I.; Alchalby, H.; Schonberg, K.; Rudolph, J.; Triviai, I.; Wolschke, C.; Heine, A.; Brossart, P.; et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br. J. Haematol. 2015, 169, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Massa, M.; Rosti, V.; Campanelli, R.; Fois, G.; Barosi, G. Rapid and long-lasting decrease of T-regulatory cells in patients with myelofibrosis treated with ruxolitinib. Leukemia 2014, 28, 449–451. [Google Scholar] [CrossRef]
- Xing, L.; Dai, Z.; Jabbari, A.; Cerise, J.E.; Higgins, C.A.; Gong, W.; de Jong, A.; Harel, S.; DeStefano, G.M.; Rothman, L.; et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat. Med. 2014, 20, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.P.; Iwata, S.; Nakayamada, S.; Sakata, K.; Yamaoka, K.; Tanaka, Y. Tofacitinib, a JAK inhibitor, inhibits human B cell activation in vitro. Ann. Rheum. Dis. 2014, 73, 2213–2215. [Google Scholar] [CrossRef]
- Rizzi, M.; Lorenzetti, R.; Fischer, K.; Staniek, J.; Janowska, I.; Troilo, A.; Strohmeier, V.; Erlacher, M.; Kunze, M.; Bannert, B.; et al. Impact of tofacitinib treatment on human B-cells in vitro and in vivo. J. Autoimm. 2017, 77, 55–66. [Google Scholar] [CrossRef]
- Heine, A.; Held, S.A.; Daecke, S.N.; Wallner, S.; Yajnanarayana, S.P.; Kurts, C.; Wolf, D.; Brossart, P. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122, 1192–1202. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.; Miller, C.B.; Silver, R.T.; Talpaz, M.; et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J. Hematol. Oncol. 2017. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.; Miller, C.B.; Silver, R.T.; et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: Results of a median 2-year follow-up of COMFORT-I. Haematologica 2013, 98, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Spoerl, S.; Mathew, N.R.; Bscheider, M.; Schmitt-Graeff, A.; Chen, S.; Mueller, T.; Verbeek, M.; Fischer, J.; Otten, V.; Schmickl, M.; et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood 2014, 123, 3832–3842. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Cooper, M.L.; Alahmari, B.; Ritchey, J.; Collins, L.; Holt, M.; DiPersio, J.F. Pharmacologic blockade of JAK1/JAK2 reduces GvHD and preserves the graft-versus-leukemia effect. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Perner, F.; Schnoder, T.M.; Ranjan, S.; Wolleschak, D.; Ebert, C.; Pils, M.C.; Frey, S.; Polanetzki, A.; Fahldieck, C.; Schonborn, U.; et al. Specificity of JAK-kinase inhibition determines impact on human and murine T-cell function. Leukemia 2016, 30, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Gotlib, J.R.; Jamieson, C.; Cortes, J.E.; Talpaz, M.; Stone, R.M.; Silverman, M.H.; Gilliland, D.G.; Shorr, J.; Tefferi, A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 2011, 29, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Tefferi, A.; Jamieson, C.; Gabrail, N.Y.; Lebedinsky, C.; Gao, G.; Liu, F.; Xu, C.; Cao, H.; Talpaz, M. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015. [Google Scholar] [CrossRef]
- Verstovsek, S.; Talpaz, M.; Ritchie, E.; Wadleigh, M.; Odenike, O.; Jamieson, C.; Stein, B.; Uno, T.; Mesa, R.A. A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis. Leukemia 2017, 31, 393–402. [Google Scholar] [CrossRef]
- Verstovsek, S.; Tam, C.S.; Wadleigh, M.; Sokol, L.; Smith, C.C.; Bui, L.A.; Song, C.; Clary, D.O.; Olszynski, P.; Cortes, J.; et al. Phase I evaluation of XL019, an oral, potent, and selective JAK2 inhibitor. Leukemia Res. 2014, 38, 316–322. [Google Scholar] [CrossRef][Green Version]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017. [Google Scholar] [CrossRef]
- Zhang, Y.; Liang, R.; Chen, C.W.; Mallano, T.; Dees, C.; Distler, A.; Reich, A.; Bergmann, C.; Ramming, A.; Gelse, K.; et al. JAK1-dependent transphosphorylation of JAK2 limits the antifibrotic effects of selective JAK2 inhibitors on long-term treatment. Ann. Rheum. Dis. 2017, 76, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.O.; Talpaz, M.; Gupta, V.; Foltz, L.M.; Savona, M.R.; Paquette, R.; Turner, A.R.; Coughlin, P.; Winton, E.; Burn, T.C.; et al. Primary analysis of a phase II open-label trial of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Haematologica 2017, 102, 327–335. [Google Scholar] [CrossRef] [PubMed]
- De Vries, L.C.S.; Ludbrook, V.J.; Hicks, K.J.; D’Haens, G.R. GSK2586184, a JAK1 selective inhibitor, in two patients with ulcerative colitis. BMJ Case Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Kremer, J.; Ponce, L.; Cseuz, R.; Reshetko, O.V.; Stanislavchuk, M.; Greenwald, M.; Van der Aa, A.; Vanhoutte, F.; Tasset, C.; et al. Filgotinib (GLPG0634/GS-6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: Results from a randomised, dose-finding study (DARWIN 2). Ann. Rheum. Dis. 2017, 76, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Smolen, J.S.; Weinblatt, M.E.; Burmester, G.R.; Meerwein, S.; Camp, H.S.; Wang, L.; Othman, A.A.; Khan, N.; Pangan, A.L.; et al. Efficacy and Safety of ABT-494, a Selective JAK-1 Inhibitor, in a Phase IIb Study in Patients With Rheumatoid Arthritis and an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2016, 68, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Kahl, L.; Patel, J.; Layton, M.; Binks, M.; Hicks, K.; Leon, G.; Hachulla, E.; Machado, D.; Staumont-Salle, D.; Dickson, M.; et al. Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus 2016, 25, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Ludbrook, V.J.; Hicks, K.J.; Hanrott, K.E.; Patel, J.S.; Binks, M.H.; Wyres, M.R.; Watson, J.; Wilson, P.; Simeoni, M.; Schifano, L.A.; et al. Investigation of selective JAK1 inhibitor GSK2586184 for the treatment of psoriasis in a randomized placebo-controlled phase IIa study. Br. J. Dermatol. 2016, 174, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.; Coates, L.C.; Helliwell, P.S.; Stanislavchuk, M.; Rychlewska-Hanczewska, A.; Dudek, A.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Harrison, P.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active psoriatic arthritis (EQUATOR): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2367–2377. [Google Scholar] [CrossRef]
- van der Heijde, D.; Baraliakos, X.; Gensler, L.S.; Maksymowych, W.P.; Tseluyko, V.; Nadashkevich, O.; Abi-Saab, W.; Tasset, C.; Meuleners, L.; Besuyen, R.; et al. Efficacy and safety of filgotinib, a selective Janus kinase 1 inhibitor, in patients with active ankylosing spondylitis (TORTUGA): Results from a randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 2378–2387. [Google Scholar] [CrossRef]
- Vermeire, S.; Schreiber, S.; Petryka, R.; Kuehbacher, T.; Hebuterne, X.; Roblin, X.; Klopocka, M.; Goldis, A.; Wisniewska-Jarosinska, M.; Baranovsky, A.; et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): Results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet 2017, 389, 266–275. [Google Scholar] [CrossRef]
- Genovese, M.C.; van Vollenhoven, R.F.; Pacheco-Tena, C.; Zhang, Y.; Kinnman, N. VX-509 (Decernotinib), an Oral Selective JAK-3 Inhibitor, in Combination With Methotrexate in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.M.; Damjanov, N.S.; Kivitz, A.J.; Legedza, A.; Hoock, T.; Kinnman, N. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.; Gordon, K.; Thaci, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.E.; Crimmins, E.M. Inflammatory exposure and historical changes in human life-spans. Science 2004, 305, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zheng, X.; Zheng, Y. Age-associated loss of lamin-B leads to systemic inflammation and gut hyperplasia. Cell 2014, 159, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Cesari, M.; Anton, S.; Marzetti, E.; Giovannini, S.; Seo, A.Y.; Carter, C.; Yu, B.P.; Leeuwenburgh, C. Molecular inflammation: Underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009, 8, 18–30. [Google Scholar] [CrossRef]
- d’Avila, J.C.; Siqueira, L.D.; Mazeraud, A.; Azevedo, E.P.; Foguel, D.; Castro-Faria-Neto, H.C.; Sharshar, T.; Chretien, F.; Bozza, F.A. Age-related cognitive impairment is associated with long-term neuroinflammation and oxidative stress in a mouse model of episodic systemic inflammation. J. Neuroinflamm. 2018. [Google Scholar] [CrossRef]
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 6995–7004. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Wu, L.W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, N.; Kizil, C.; Zocher, S.; Kroehne, V.; Kaslin, J.; Freudenreich, D.; Iltzsche, A.; Brand, M. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science 2012, 338, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; O’Brien, M.; Mau, T.; Yung, R. Toll-like receptor 4 (TLR4) deficient mice are protected from adipose tissue inflammation in aging. Aging 2017, 9, 1971–1982. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017. [Google Scholar] [CrossRef]
- Herrera, S.C.; Bach, E.A. JAK/STAT signaling in stem cells and regeneration: From Drosophila to vertebrates. Development 2019. [Google Scholar] [CrossRef]
- Elsaeidi, F.; Bemben, M.A.; Zhao, X.F.; Goldman, D. Jak/Stat signaling stimulates zebrafish optic nerve regeneration and overcomes the inhibitory actions of Socs3 and Sfpq. J. Neurosci. 2014, 34, 2632–2644. [Google Scholar] [CrossRef]
- Richmond, C.A.; Rickner, H.; Shah, M.S.; Ediger, T.; Deary, L.; Zhou, F.; Tovaglieri, A.; Carlone, D.L.; Breault, D.T. JAK/STAT-1 Signaling Is Required for Reserve Intestinal Stem Cell Activation during Intestinal Regeneration Following Acute Inflammation. Stem Cell Rep. 2018, 10, 17–26. [Google Scholar] [CrossRef]
- Doles, J.D.; Olwin, B.B. The impact of JAK-STAT signaling on muscle regeneration. Nat. Med. 2014, 20, 1094–1095. [Google Scholar] [CrossRef] [PubMed]
- La Fortezza, M.; Schenk, M.; Cosolo, A.; Kolybaba, A.; Grass, I.; Classen, A.K. JAK/STAT signalling mediates cell survival in response to tissue stress. Development 2016, 143, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Patel, P.H.; Kohlmaier, A.; Grenley, M.O.; McEwen, D.G.; Edgar, B.A. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 2009, 137, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Shen-Orr, S.S.; Furman, D.; Kidd, B.A.; Hadad, F.; Lovelace, P.; Huang, Y.W.; Rosenberg-Hasson, Y.; Mackey, S.; Grisar, F.A.; Pickman, Y.; et al. Defective Signaling in the JAK-STAT Pathway Tracks with Chronic Inflammation and Cardiovascular Risk in Aging Humans. Cell Syst. 2016, 3, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H., 3rd; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against alpha-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J. Neurosci. 2016, 36, 5144–5159. [Google Scholar] [CrossRef]
- Jones, R.S.; Minogue, A.M.; Fitzpatrick, O.; Lynch, M.A. Inhibition of JAK2 attenuates the increase in inflammatory markers in microglia from APP/PS1 mice. Neurobiol. Aging 2015, 36, 2716–2724. [Google Scholar] [CrossRef]
- Xu, M.; Tchkonia, T.; Kirkland, J.L. Perspective: Targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol. Res. 2016, 111, 152–154. [Google Scholar] [CrossRef]
- Cano, M.; Ayala, A.; Marotta, F.; Arguelles, S. Application of Kinase Inhibitors for Anti-aging Intervention. Curr. Pharm. Des. 2017, 23, 4351–4368. [Google Scholar] [CrossRef]
- Sidon, P.; El Housni, H.; Dessars, B.; Heimann, P. The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 2006. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Q.; Luo, J.; Xing, S.; Li, Q.; Krantz, S.B.; Fu, X.; Zhao, Z.J. JAK2(V617F): Prevalence in a large Chinese hospital population. Blood 2007, 109, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Zoi, K.; Cross, N.C. Genomics of Myeloproliferative Neoplasms. J. Clin. Oncol. 2017, 35, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Beer, P.A.; Delhommeau, F.; LeCouedic, J.P.; Dawson, M.A.; Chen, E.; Bareford, D.; Kusec, R.; McMullin, M.F.; Harrison, C.N.; Vannucchi, A.M.; et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood 2010, 115, 2891–2900. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Schaub, F.X.; Jager, R.; Looser, R.; Hao-Shen, H.; Hermouet, S.; Girodon, F.; Tichelli, A.; Gisslinger, H.; Kralovics, R.; Skoda, R.C. Clonal analysis of deletions on chromosome 20q and JAK2-V617F in MPD suggests that del20q acts independently and is not one of the predisposing mutations for JAK2-V617F. Blood 2009, 113, 2022–2027. [Google Scholar] [CrossRef]
- Schaub, F.X.; Looser, R.; Li, S.; Hao-Shen, H.; Lehmann, T.; Tichelli, A.; Skoda, R.C. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010, 115, 2003–2007. [Google Scholar] [CrossRef]
- Lundberg, P.; Takizawa, H.; Kubovcakova, L.; Guo, G.; Hao-Shen, H.; Dirnhofer, S.; Orkin, S.H.; Manz, M.G.; Skoda, R.C. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J. Exp. Med. 2014, 211, 2213–2230. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [PubMed]
- Jacquelin, S.; Straube, J.; Cooper, L.; Vu, T.; Song, A.; Bywater, M.; Baxter, E.; Heidecker, M.; Wackrow, B.; Porter, A.; et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood 2018, 132, 2707–2721. [Google Scholar] [CrossRef] [PubMed]
- Mora, B.; Giorgino, T.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; Kiladjian, J.J.; et al. Value of cytogenetic abnormalities in post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A study of the MYSEC project. Haematologica 2018, 103. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.; Faisal, M.; Busche, G.; Schlue, J.; Kreipe, H.; Lehmann, U. Fibrotic progression in Polycythemia vera is associated with early concomitant driver-mutations besides JAK2. Leukemia 2018, 32, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Sajid, Z.; Pedersen, R.K.; Gudmand-Hoeyer, J.; Ellervik, C.; Skov, V.; Kjaer, L.; Pallisgaard, N.; Kruse, T.A.; Thomassen, M.; et al. Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, 438–442. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Manshouri, T.; Patel, J.; Harris, K.; Yao, J.; Hedvat, C.; Heguy, A.; Bueso-Ramos, C.; Kantarjian, H.; Levine, R.L.; et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 lesions in leukemic transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Lasho, T.; Finke, C.; Oh, S.T.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Radich, J.; Burn, T.C.; Huber, R.; Paranagama, D.; Verstovsek, S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015, 126, 1551–1554. [Google Scholar]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef]
- Geyer, H.L.; Kosiorek, H.; Dueck, A.C.; Scherber, R.; Slot, S.; Zweegman, S.; Te Boekhorst, P.A.; Senyak, Z.; Schouten, H.C.; Sackmann, F.; et al. Associations between gender, disease features and symptom burden in patients with myeloproliferative neoplasms: An analysis by the MPN QOL International Working Group. Haematologica 2017, 102, 85–93. [Google Scholar] [CrossRef]
- Lubberich, R.K.; Walenda, T.; Goecke, T.W.; Strathmann, K.; Isfort, S.; Brummendorf, T.H.; Koschmieder, S.; Wagner, W. Serum of myeloproliferative neoplasms stimulates hematopoietic stem and progenitor cells. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, 123–133. [Google Scholar] [CrossRef]
- Schnoder, T.M.; Eberhardt, J.; Koehler, M.; Bierhoff, H.B.; Weinert, S.; Pandey, A.D.; Nimmagadda, S.C.; Wolleschak, D.; Johrens, K.; Fischer, T.; et al. Cell autonomous expression of CXCL-10 in JAK2V617F-mutated MPN. J. Cancer Res. Clin. Oncol. 2017, 143, 807–820. [Google Scholar] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Kantarjian, H.; Mesa, R.A.; Pardanani, A.D.; Cortes-Franco, J.; Thomas, D.A.; Estrov, Z.; Fridman, J.S.; Bradley, E.C.; Erickson-Viitanen, S.; et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N. Engl. J. Med. 2010, 363, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFalpha facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Bogeska, R.; Nikoloski, G.; Schmid, C.A.; Seeger, T.S.; Stegelmann, F.; Schwemmers, S.; Grunder, A.; Peeken, J.C.; Gothwal, M.; et al. MPN patients harbor recurrent truncating mutations in transcription factor NF-E2. J. Exp. Med. 2013, 210, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Pahl, H.L. The Hen or the Egg: Inflammatory Aspects of Murine MPN Models. Mediators Inflamm. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wehrle, J.; Seeger, T.S.; Schwemmers, S.; Pfeifer, D.; Bulashevska, A.; Pahl, H.L. Transcription factor nuclear factor erythroid-2 mediates expression of the cytokine interleukin 8, a known predictor of inferior outcome in patients with myeloproliferative neoplasms. Haematologica 2013, 98, 1073–1080. [Google Scholar] [CrossRef]
- Griesshammer, M.; Kiladjian, J.J.; Besses, C. Thromboembolic events in polycythemia vera. Ann. Hematol. 2019, 98, 1071–1082. [Google Scholar] [CrossRef]
- Chievitz, E.; Thiede, T. Complications and causes of death in polycythaemia vera. Acta Med. Scand. 1962, 172, 513–523. [Google Scholar] [CrossRef]
- Landolfi, R.; Marchioli, R.; Kutti, J.; Gisslinger, H.; Tognoni, G.; Patrono, C.; Barbui, T. European Collaboration on Low-Dose Aspirin in Polycythemia Vera, I. Efficacy and safety of low-dose aspirin in polycythemia vera. N. Engl. J. Med. 2004, 350, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular events and intensity of treatment in polycythemia vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Barbui, T.; Cervantes, F.; Harrison, C.; Kiladjian, J.J.; Kroger, N.; Thiele, J.; Buske, C.; Committee, E.G. Philadelphia chromosome-negative chronic myeloproliferative neoplasms: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Lippert, E.; Girodon, F.; Dobo, I.; Fouassier, M.; Masliah, C.; Praloran, V.; Hermouet, S. Latent myeloproliferative disorder revealed by the JAK2-V617F mutation and endogenous megakaryocytic colonies in patients with splanchnic vein thrombosis. Blood 2006, 108, 3223–3224. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Carobbio, A.; Di Lazzaro, V.; Guglielmelli, P.; Iurlo, A.; Finazzi, M.C.; Rumi, E.; Cervantes, F.; Elli, E.M.; Randi, M.L.; et al. Benefit-risk profile of cytoreductive drugs along with antiplatelet and antithrombotic therapy after transient ischemic attack or ischemic stroke in myeloproliferative neoplasms. Blood Cancer J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gugliotta, L.; Iurlo, A.; Gugliotta, G.; Tieghi, A.; Specchia, G.; Gaidano, G.; Scalzulli, P.R.; Rumi, E.; Dragani, A.; Martinelli, V.; et al. Unbiased pro-thrombotic features at diagnosis in 977 thrombocythemic patients with Philadelphia-negative chronic myeloproliferative neoplasms. Leukemia Res. 2016, 46, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Pancholy, N.; Thomas, L.; Rai, A.; Kher, A.; Peters, C.; Amin, A.; Patel, T.M.; Pancholy, S. Effect of Chronic Hematologic Malignancies on In-Hospital Outcomes of Patients With ST-Segment Elevation Myocardial Infarction. Am. J. Cardiol. 2019, 124, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Rosti, V.; Villani, L.; Riboni, R.; Poletto, V.; Bonetti, E.; Tozzi, L.; Bergamaschi, G.; Catarsi, P.; Dallera, E.; Novara, F.; et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood 2013, 121, 360–368. [Google Scholar] [CrossRef]
- Teofili, L.; Martini, M.; Iachininoto, M.G.; Capodimonti, S.; Nuzzolo, E.R.; Torti, L.; Cenci, T.; Larocca, L.M.; Leone, G. Endothelial progenitor cells are clonal and exhibit the JAK2(V617F) mutation in a subset of thrombotic patients with Ph-negative myeloproliferative neoplasms. Blood 2011, 117, 2700–2707. [Google Scholar] [CrossRef]
- Sozer, S.; Fiel, M.I.; Schiano, T.; Xu, M.; Mascarenhas, J.; Hoffman, R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood 2009, 113, 5246–5249. [Google Scholar] [CrossRef]
- Etheridge, S.L.; Roh, M.E.; Cosgrove, M.E.; Sangkhae, V.; Fox, N.E.; Chen, J.; Lopez, J.A.; Kaushansky, K.; Hitchcock, I.S. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Lamrani, L.; Lacout, C.; Ollivier, V.; Denis, C.V.; Gardiner, E.; Ho Tin Noe, B.; Vainchenker, W.; Villeval, J.L.; Jandrot-Perrus, M. Hemostatic disorders in a JAK2V617F-driven mouse model of myeloproliferative neoplasm. Blood 2014, 124, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, C.M.; Manning, H.; Bennett, C.; Vasquez, L.; Severin, S.; Brain, L.; Mazharian, A.; Guerrero, J.A.; Li, J.; Soranzo, N.; et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood 2013, 122, 3787–3797. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Larran, A.; Perez-Encinas, M.; Ferrer-Marin, F.; Hernandez-Boluda, J.C.; Ramirez, M.J.; Martinez-Lopez, J.; Magro, E.; Cruz, Y.; Mata, M.I.; Aragues, P.; et al. Risk of thrombosis according to need of phlebotomies in patients with polycythemia vera treated with hydroxyurea. Haematologica 2017, 102, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Finazzi, G.; Caruso, V.; Marchioli, R.; Capnist, G.; Chisesi, T.; Finelli, C.; Gugliotta, L.; Landolfi, R.; Kutti, J.; Gisslinger, H.; et al. Acute leukemia in polycythemia vera: An analysis of 1638 patients enrolled in a prospective observational study. Blood 2005, 105, 2664–2670. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, M.; Cambot, M.; Wautier, M.P.; Cassinat, B.; Chomienne, C.; Colin, Y.; Wautier, J.L.; Le Van Kim, C.; El Nemer, W. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood 2013, 121, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Edelmann, B.; Schnoeder, T.M.; Saalfeld, F.C.; Wolleschak, D.; Kliche, S.; Schraven, B.; Heidel, F.H.; Fischer, T. JAK2-V617F activates beta1-integrin-mediated adhesion of granulocytes to vascular cell adhesion molecule 1. Leukemia 2017, 31, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, B.; Gupta, N.; Schnoeder, T.M.; Oelschlegel, A.M.; Shahzad, K.; Goldschmidt, J.; Philipsen, L.; Weinert, S.; Ghosh, A.; Saalfeld, F.C.; et al. JAK2-V617F promotes venous thrombosis through beta1/beta2 integrin activation. J. Clin. Investig. 2018, 128, 4359–4371. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. https://doi.org/10.3390/cells8080854
Perner F, Perner C, Ernst T, Heidel FH. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells. 2019; 8(8):854. https://doi.org/10.3390/cells8080854
Chicago/Turabian StylePerner, Florian, Caroline Perner, Thomas Ernst, and Florian H. Heidel. 2019. "Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation" Cells 8, no. 8: 854. https://doi.org/10.3390/cells8080854
APA StylePerner, F., Perner, C., Ernst, T., & Heidel, F. H. (2019). Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells, 8(8), 854. https://doi.org/10.3390/cells8080854