Emerging Role of l-Dopa Decarboxylase in Flaviviridae Virus Infections


 and
and
Abstract
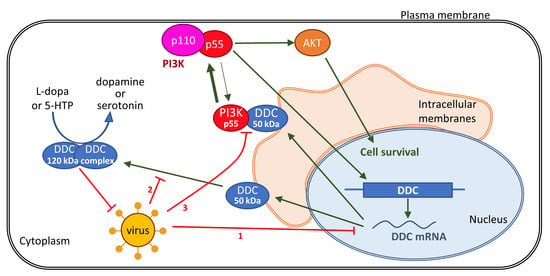
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
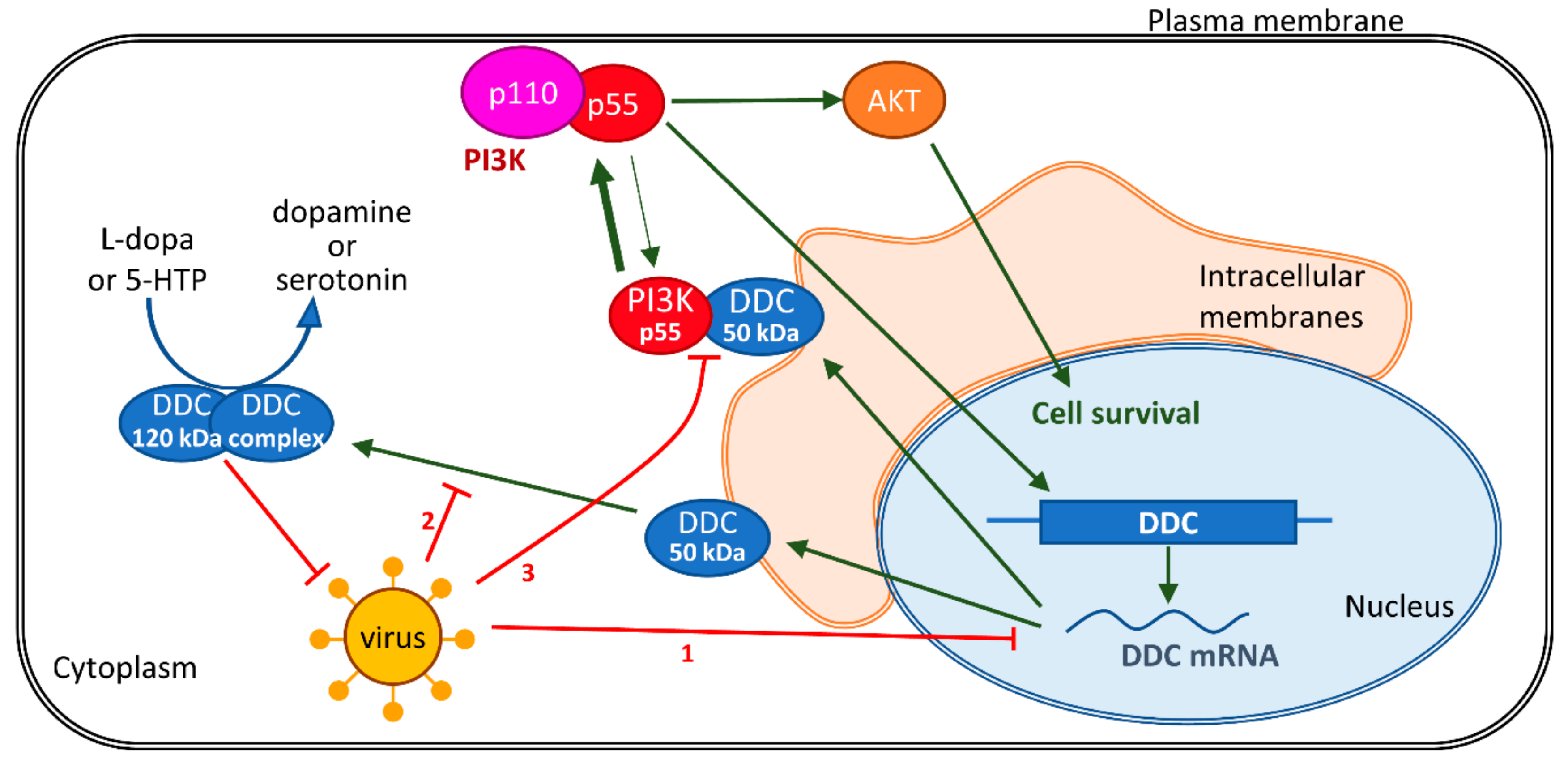{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Human Liver Biopsy RNA Samples
2.3. Viruses and Plasmid Constructs
2.4. In Vitro Transcription
2.5. Transfection Assays
2.6. Preparation of Virus Stocks and Infection Assays
2.7. Virus Titration in Cell Culture Supernatants
2.8. Gel Electrophoresis and Western Blot Analysis
2.9. Protein Immunoprecipitation under Non-Reducing Conditions
2.10. Luciferase Assays
2.11. Measurement of Intracellular ATP Levels
2.12. Subcellular Fractionation
2.13. Indirect Immunofluorescence
2.14. RNA Quantification by Reverse Transcription-Quantitative PCR (RT-qPCR)
2.15. Chemicals
2.16. Statistical Analysis
3. Results
3.1. Downregulation of l-Dopa Decarboxylase (DDC) by DENV and HCV Viruses
3.2. Effect of DENV-/HCV-Infection on DDC Subcellular Localization and DDC-PI3K Interaction
3.3. Effect of Overexpression and Chemical Inhibition of DDC on Viral Replication and Infectivity
3.4. The Role of PI3K in DENV- and HCV-Mediated DDC Regulation
3.5. Inverse Correlation Between DDC mRNA and HCV Replication In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindstrom, P.; Sehlin, J. Mechanisms underlying the effects of 5-hydroxytryptamine and 5-hydroxytryptophan in pancreatic islets. A proposed role for L-aromatic amino acid decarboxylase. Endocrinology 1983, 112, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Dyck, L.E.; Yang, C.R.; Boulton, A.A. The biosynthesis of p-tyramine, m-tyramine, and beta-phenylethylamine by rat striatal slices. J. Neurosci. Res. 1983, 10, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.A.; Juorio, A.V.; Boulton, A.A. 2-Phenylethylamine: A modulator of catecholamine transmission in the mammalian central nervous system? J. Neurochem. 1990, 55, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Christenson, J.G.; Dairman, W.; Udenfriend, S. Preparation and properties of a homogeneous aromatic L-amino acid decarboxylase from hog kidney. Arch. Biochem. Biophys. 1970, 141, 356–367. [Google Scholar] [CrossRef]
- Adam, W.R.; Culvenor, A.J.; Hall, J.; Jarrott, B.; Wellard, R.M. Aromatic L-amino acid decarboxylase: Histochemical localization in rat kidney and lack of effect of dietary potassium or sodium loading on enzyme distribution. Clin. Exp. Pharmacol. Phys. 1986, 13, 47–53. [Google Scholar] [CrossRef]
- Lovenberg, W.; Weissbach, H.; Udenfriend, S. Aromatic L-amino acid decarboxylase. J. Biol. Chem. 1962, 237, 89–93. [Google Scholar] [PubMed]
- Rahman, M.K.; Nagatsu, T.; Kato, T. Determination of aromatic L-amino acid decarboxylase in serum of various animals by high-performance liquid chromatography with electrochemical detection. Life Sci. 1981, 28, 485–492. [Google Scholar] [CrossRef]
- Dominici, P.; Tancini, B.; Barra, D.; Voltattorni, C.B. Purification and characterization of rat-liver 3,4-dihydroxyphenylalanine decarboxylase. Eur. J. Biochem. 1987, 169, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Mizuguchi, H.; Kagamiyama, H. Rat liver aromatic L-amino acid decarboxylase: Spectroscopic and kinetic analysis of the coenzyme and reaction intermediates. Biochemistry 1993, 32, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Kojima, K.; Togari, A.; Kato, Y.; Parvez, S.; Parvez, H.; Nagatsu, T. Simple purification of aromatic L-amino acid decarboxylase from human pheochromocytoma using high-performance liquid chromatography. Anal. Biochem. 1985, 150, 408–414. [Google Scholar] [CrossRef]
- Maneckjee, R.; Baylin, S.B. Use of radiolabeled monofluoromethyl-Dopa to define the subunit structure of human L-Dopa decarboxylase. Biochemistry 1983, 22, 6058–6063. [Google Scholar] [CrossRef] [PubMed]
- Mappouras, D.G.; Stiakakis, J.; Fragoulis, E.G. Purification and characterization of L-dopa decarboxylase from human kidney. Mol. Cell Biochem. 1990, 94, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Nishigaki, I.; Ichinose, H.; Tamai, K.; Nagatsu, T. Purification of aromatic L-amino acid decarboxylase from bovine brain with a monoclonal antibody. Biochem. J. 1988, 252, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Shirota, K.; Fujisawa, H. Purification and characterization of aromatic L-amino acid decarboxylase from rat kidney and monoclonal antibody to the enzyme. J. Neurochem. 1988, 51, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Siow, Y.L.; Dakshinamurti, K. Purification of dopa decarboxylase from bovine striatum. Mol. Cell Biochem. 1990, 94, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.D.; Juorio, A.V.; Li, X.M.; Boulton, A.A. Aromatic L-amino acid decarboxylase: A neglected and misunderstood enzyme. Neurochem. Res. 1996, 21, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Guenter, J.; Lenartowski, R. Molecular characteristic and physiological role of DOPA-decarboxylase. Postep. Hig. Med. Dosw. 2016, 70, 1424–1440. [Google Scholar] [CrossRef] [PubMed]
- Bertoldi, M. Mammalian Dopa decarboxylase: Structure, catalytic activity and inhibition. Arch. Biochem. Biophys. 2014, 546, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Florou, D.; Scorilas, A.; Vassilacopoulou, D.; Fragoulis, E.G. DDC (dopa decarboxylase (aromatic L-amino acid decarboxylase)). Atlas Gen. Cytogen. Oncol. Haematol. 2010, 14, 942–950. [Google Scholar] [CrossRef][Green Version]
- Vassiliou, A.G.; Fragoulis, E.G.; Vassilacopoulou, D. Detection, purification and identification of an endogenous inhibitor of L-Dopa decarboxylase activity from human placenta. Neurochem. Res. 2009, 34, 1089–1100. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Siaterli, M.Z.; Frakolaki, E.; Gkogkosi, P.; Paspaltsis, I.; Sklaviadis, T.; Vassilacopoulou, D.; Vassilaki, N. L-Dopa decarboxylase interaction with the major signaling regulator PI3K in tissues and cells of neural and peripheral origin. Biochimie 2019, 160, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. Biosyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Diehl, N.; Schaal, H. Make yourself at home: Viral hijacking of the PI3K/Akt signaling pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.P.; Thai, A.L.; Weber, M.; Craig, I.W. Localisation of the gene for human aromatic L-amino acid decarboxylase (DDC) to chromosome 7p13-->p11 by in situ hybridisation. Cytogen. Gen. 1992, 61, 114–116. [Google Scholar] [CrossRef]
- NCBI. DDC-Dopa decarboxylase. Available online: http://www.ncbi.nlm.nih.gov/gene/1644 (accessed on 25 April 2019).
- Albert, V.R.; Lee, M.R.; Bolden, A.H.; Wurzburger, R.J.; Aguanno, A. Distinct promoters direct neuronal and nonneuronal expression of rat aromatic L-amino acid decarboxylase. Proc. Natl. Acad. Sci. USA 1992, 89, 12053–12057. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Sumi-Ichinose, C.; Ohye, T.; Hagino, Y.; Fujita, K.; Nagatsu, T. Tissue-specific alternative splicing of the first exon generates two types of mRNAs in human aromatic L-amino acid decarboxylase. Biochemistry 1992, 31, 11546–11550. [Google Scholar] [CrossRef]
- Jahng, J.W.; Wessel, T.C.; Houpt, T.A.; Son, J.H.; Joh, T.H. Alternate promoters in the rat aromatic L-amino acid decarboxylase gene for neuronal and nonneuronal expression: An in situ hybridization study. J. Neurochem. 1996, 66, 14–19. [Google Scholar] [CrossRef]
- Sumi-Ichinose, C.; Hasegawa, S.; Ichinose, H.; Sawada, H.; Kobayashi, K.; Sakai, M.; Fujii, T.; Nomura, H.; Nomura, T.; Nagatsu, I.; et al. Analysis of the alternative promoters that regulate tissue-specific expression of human aromatic L-amino acid decarboxylase. J. Neurochem. 1995, 64, 514–524. [Google Scholar] [CrossRef]
- Chalatsa, I.; Nikolouzou, E.; Fragoulis, E.G.; Vassilacopoulou, D. L-Dopa decarboxylase expression profile in human cancer cells. Mol. Biol. Rep. 2011, 38, 1005–1011. [Google Scholar] [CrossRef]
- Kokkinou, I.; Nikolouzou, E.; Hatzimanolis, A.; Fragoulis, E.G.; Vassilacopoulou, D. Expression of enzymatically active L-DOPA decarboxylase in human peripheral leukocytes. Blood Cell Mol. Dis. 2009, 42, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Siaterli, M.Z.; Vassilacopoulou, D.; Fragoulis, E.G. Cloning and expression of human placental L-Dopa decarboxylase. Neurochem. Res. 2003, 28, 797–803. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, K.L.; Harmon, S.; Moffat, M.; Uhland-Smith, A.; Wong, S. The human aromatic L-amino acid decarboxylase gene can be alternatively spliced to generate unique protein isoforms. J. Neurochem. 1995, 65, 2409–2416. [Google Scholar] [CrossRef] [PubMed]
- Vassilacopoulou, D.; Sideris, D.C.; Vassiliou, A.G.; Fragoulis, E.G. Identification and characterization of a novel form of the human L-dopa decarboxylase mRNA. Neurochem. Res. 2004, 29, 1817–1823. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, G.A.; Sourkes, T.L. Purification and properties of hog-kidney 3,4-dihydroxyphenylalanine decarboxylase. Can. J. Biochem. 1972, 50, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Voltattorni, C.B.; Minelli, A.; Vecchini, P.; Fiori, A.; Turano, C. Purification and characterization of 3,4-dihydroxyphenylalanine decarboxyase from pig kidney. Eur. J. Biochem. 1979, 93, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Rubi, B.; Maechler, P. Minireview: New roles for peripheral dopamine on metabolic control and tumor growth: let’s seek the balance. Endocrinology 2010, 151, 5570–5581. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Li, J.; Luo, Z.; Zhang, S.; Xue, S.; Wang, K.; Shi, Y.; Zhang, C.; Chen, H.; Li, Z. Roles of dopamine receptors and their antagonist thioridazine in hepatoma metastasis. OncoTargets Ther. 2015, 8, 1543–1552. [Google Scholar] [CrossRef]
- Zou, J.; Li, H.; Huang, Q.; Liu, X.; Qi, X.; Wang, Y.; Lu, L.; Liu, Z. Dopamine-induced SULT1A3/4 promotes EMT and cancer stemness in hepatocellular carcinoma. Tumour Biol. 2017. [Google Scholar] [CrossRef]
- Liu, X.F.; Long, H.J.; Miao, X.Y.; Liu, G.L.; Yao, H.L. Fisetin inhibits liver cancer growth in a mouse model: Relation to dopamine receptor. Oncol. Rep. 2017, 38, 53–62. [Google Scholar] [CrossRef]
- He, S.; Lin, B.; Chu, V.; Hu, Z.; Hu, X.; Xiao, J.; Wang, A.Q.; Schweitzer, C.J.; Li, Q.; Imamura, M.; et al. Repurposing of the antihistamine chlorcyclizine and related compounds for treatment of hepatitis C virus infection. Sci. Transl. Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Chen, J.; Wang, Y.; Yang, Y.; Qing, J.; Rao, Z.; Chen, X.; Lou, Z. Identification of serotonin 2A receptor as a novel HCV entry factor by a chemical biology strategy. Protein Cell 2018, 10, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Simanjuntak, Y.; Liang, J.J.; Lee, Y.L.; Lin, Y.L. Repurposing of prochlorperazine for use against dengue virus infection. J. Infect. Dis. 2015, 211, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Stein, D.A.; Shum, D.; Fischer, M.A.; Radu, C.; Bhinder, B.; Djaballah, H.; Nelson, J.A.; Fruh, K.; Hirsch, A.J. Inhibition of dengue virus replication by a class of small-molecule compounds that antagonize dopamine receptor d4 and downstream mitogen-activated protein kinase signaling. J. Virol. 2014, 88, 5533–5542. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Singh, M.; Kumar, S.; Kumar, A. The epidemiology of dengue viral infection in developing countries: A systematic review. J. Health. Res. Rep. 2017. [Google Scholar] [CrossRef]
- World Health Organization, Dengue and severe dengue, 2018. Available online: http://www.who.int/mediacentre/factsheets/fs117/en/ (accessed on 25 April 2019).
- World Mosquito Program. Available online: http://www.eliminatedengue.com/our-research/dengue-fever (accessed on 25 April 2019).
- Leong, A.S.; Wong, K.T.; Leong, T.Y.; Tan, P.H.; Wannakrairot, P. The pathology of dengue hemorrhagic fever. Semin. Diagn. Pathol. 2007, 24, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Povoa, T.F.; Alves, A.M.; Oliveira, C.A.; Nuovo, G.J.; Chagas, V.L.; Paes, M.V. The pathology of severe dengue in multiple organs of human fatal cases: Histopathology, ultrastructure and virus replication. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Reyes-del Valle, J.; Salas-Benito, J.; Soto-Acosta, R.; del Angel, R.M. Dengue Virus Cellular Receptors and Tropism. Curr. Trop. Med. Rep. 2014, 1, 36–43. [Google Scholar] [CrossRef]
- Itha, S.; Kashyap, R.; Krishnani, N.; Saraswat, V.A.; Choudhuri, G.; Aggarwal, R. Profile of liver involvement in dengue virus infection. Natl. Med. J. India 2005, 18, 127–130. [Google Scholar]
- Dissanayake, H.A.; Seneviratne, S.L. Liver involvement in dengue viral infections. Rev. Med. Virol. 2018. [Google Scholar] [CrossRef]
- World Health Organization, Hepatitis C, 2018. Available online: http://www.who.int/en/news-room/fact-sheets/detail/hepatitis-c (accessed on 25 April 2019).
- Bartenschlager, R.; Lohmann, V.; Penin, F. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat. Rev. Microbiol. 2013, 11, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue virus- and hepatitis C virus-induced replication and assembly compartments: The enemy inside--caught in the web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Liao, C.L.; Lin, Y.L. Flavivirus activates phosphatidylinositol 3-kinase signaling to block caspase-dependent apoptotic cell death at the early stage of virus infection. J. Virol. 2005, 79, 8388–8399. [Google Scholar] [CrossRef] [PubMed]
- Martins Sde, T.; Silveira, G.F.; Alves, L.R.; Duarte dos Santos, C.N.; Bordignon, J. Dendritic cell apoptosis and the pathogenesis of dengue. Viruses 2012, 4, 2736–2753. [Google Scholar] [CrossRef] [PubMed]
- Torrentes-Carvalho, A.; Azeredo, E.L.; Reis, S.R.; Miranda, A.S.; Gandini, M.; Barbosa, L.S.; Kubelka, C.F. Dengue-2 infection and the induction of apoptosis in human primary monocytes. Mem. I. Oswaldo Cruz 2009, 104, 1091–1099. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Zou, J.; Zhang, B.; Yuan, Z. Dengue virus subgenomic RNA induces apoptosis through the Bcl-2-mediated PI3k/Akt signaling pathway. Virology 2014, 448, 15–25. [Google Scholar] [CrossRef]
- Chen, H.H.; Chen, C.C.; Lin, Y.S.; Chang, P.C.; Lu, Z.Y.; Lin, C.F.; Chen, C.L.; Chang, C.P. AR-12 suppresses dengue virus replication by down-regulation of PI3K/AKT and GRP78. Antiviral Res. 2017, 142, 158–168. [Google Scholar] [CrossRef]
- Mannova, P.; Beretta, L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell survival and viral replication. J. Virol. 2005, 79, 8742–8749. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 2005, 79, 5006–5016. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Nakao, H.; Tan, S.L.; Polyak, S.J.; Neddermann, P.; Vijaysri, S.; Jacobs, B.L.; Katze, M.G. Subversion of cell signaling pathways by hepatitis C virus nonstructural 5A protein via interaction with Grb2 and P85 phosphatidylinositol 3-kinase. J. Virol. 2002, 76, 9207–9217. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Zhang, L.; Yang, G.; Zhao, L.; Peng, F.; Tian, Y.; Xiao, X.; Chung, R.T.; Gong, G. Hepatitis C virus NS5A drives a PTEN-PI3K/Akt feedback loop to support cell survival. Liver Int. 2015, 35, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, N.; Kalliampakou, K.I.; Kotta-Loizou, I.; Befani, C.; Liakos, P.; Simos, G.; Mentis, A.F.; Kalliaropoulos, A.; Doumba, P.P.; Smirlis, D.; et al. Low oxygen tension enhances hepatitis C virus replication. J. Virol. 2013, 87, 2935–2948. [Google Scholar] [CrossRef] [PubMed]
- Frakolaki, E.; Kaimou, P.; Moraiti, M.; Kalliampakou, K.I.; Karampetsou, K.; Dotsika, E.; Liakos, P.; Vassilacopoulou, D.; Mavromara, P.; Bartenschlager, R.; et al. The Role of Tissue Oxygen Tension in Dengue Virus Replication. Cells 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Taketa, K.; Miyano, K.; Yamane, T.; Sato, J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982, 42, 3858–3863. [Google Scholar]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed]
- Friebe, P.; Boudet, J.; Simorre, J.P.; Bartenschlager, R. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J. Virol. 2005, 79, 380–392. [Google Scholar] [CrossRef]
- Werth, N.; Beerlage, C.; Rosenberger, C.; Yazdi, A.S.; Edelmann, M.; Amr, A.; Bernhardt, W.; von Eiff, C.; Becker, K.; Schafer, A.; et al. Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS ONE 2010. [Google Scholar] [CrossRef]
- Fischl, W.; Bartenschlager, R. High-throughput screening using dengue virus reporter genomes. Methods Mol. Biol. 2013, 1030, 205–219. [Google Scholar] [CrossRef]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.; Appel, N.; Koutsoudakis, G.; Kallis, S.; Lohmann, V.; Pietschmann, T.; Bartenschlager, R. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 2007, 81, 4591–4603. [Google Scholar] [CrossRef] [PubMed]
- Kalantzis, E.D.; Scorilas, A.; Vassilacopoulou, D. Evidence for L-Dopa Decarboxylase Involvement in Cancer Cell Cytotoxicity Induced by Docetaxel and Mitoxantrone. Curr. Pharm. Biotechnol. 2018, 19, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Woerz, I.; Meuleman, P.; Leroux-Roels, G.; Bartenschlager, R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J. Virol. 2007, 81, 13168–13179. [Google Scholar] [CrossRef] [PubMed]
- Vassilaki, N.; Friebe, P.; Meuleman, P.; Kallis, S.; Kaul, A.; Paranhos-Baccala, G.; Leroux-Roels, G.; Mavromara, P.; Bartenschlager, R. Role of the hepatitis C virus core+1 open reading frame and core cis-acting RNA elements in viral RNA translation and replication. J. Virol. 2008, 82, 11503–11515. [Google Scholar] [CrossRef] [PubMed]
- Kotta-Loizou, I.; Vassilaki, N.; Pissas, G.; Kakkanas, A.; Bakiri, L.; Bartenschlager, R.; Mavromara, P. Hepatitis C virus core+1/ARF protein decreases hepcidin transcription through an AP1 binding site. J. Gen. Virol. 2013, 94, 1528–1534. [Google Scholar] [CrossRef] [PubMed][Green Version]
- van den Hoff, M.J.; Christoffels, V.M.; Labruyere, W.T.; Moorman, A.F.; Lamers, W.H. Electrotransfection with “intracellular” buffer. Methods Mol. Biol. 1995, 48, 185–197. [Google Scholar] [CrossRef]
- Byrd, C.M.; Dai, D.; Grosenbach, D.W.; Berhanu, A.; Jones, K.F.; Cardwell, K.B.; Schneider, C.; Wineinger, K.A.; Page, J.M.; Harver, C.; et al. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob. Agents Chemother. 2013, 57, 15–25. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef]
- Vassilaki, N.; Boleti, H.; Mavromara, P. Expression studies of the HCV-1a core+1 open reading frame in mammalian cells. Virus Res. 2008, 133, 123–135. [Google Scholar] [CrossRef]
- Meier, J.; Georgatos, S.D. Type B lamins remain associated with the integral nuclear envelope protein p58 during mitosis: Implications for nuclear reassembly. EMBO J. 1994, 13, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Holden, P.; Horton, W.A. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2009. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [PubMed]
- Icy, an Open Community Platform for Bioimaging. Available online: http://icy.bioimageanalysis.com (accessed on 25 April 2019).
- Zhou, W.; Dosey, T.L.; Biechele, T.; Moon, R.T.; Horwitz, M.S.; Ruohola-Baker, H. Assessment of hypoxia inducible factor levels in cancer cell lines upon hypoxic induction using a novel reporter construct. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Cavadas, M.A.S.; Cheong, A.; Taylor, C.T. The regulation of transcriptional repression in hypoxia. Exp. Cell Res. 2017, 356, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Sims, K.L.; Davis, G.A.; Bloom, F.E. Activities of 3,4-dihydroxy-L-phenylalanine and 5-hydroxy-L-tryptophan decarboxylases in rat brain: Assay characteristics and distribution. J. Neurochem. 1973, 20, 449–464. [Google Scholar] [CrossRef]
- Chalatsa, I.; Fragoulis, E.G.; Vassilacopoulou, D. Release of membrane-associated L-dopa decarboxylase from human cells. Neurochem. Res. 2011, 36, 1426–1434. [Google Scholar] [CrossRef]
- Kokkinou, I.; Fragoulis, E.G.; Vassilacopoulou, D. The U937 macrophage cell line expresses enzymatically active L-Dopa decarboxylase. J. Neuroimmunol. 2009, 216, 51–58. [Google Scholar] [CrossRef]
- Poulikakos, P.; Vassilacopoulou, D.; Fragoulis, E.G. L-DOPA decarboxylase association with membranes in mouse brain. Neurochem. Res. 2001, 26, 479–485. [Google Scholar] [CrossRef]
- Limjindaporn, T.; Wongwiwat, W.; Noisakran, S.; Srisawat, C.; Netsawang, J.; Puttikhunt, C.; Kasinrerk, W.; Avirutnan, P.; Thiemmeca, S.; Sriburi, R.; et al. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem Biophys Res. Commun. 2009, 379, 196–200. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, G.L.; Henley, J.W.; Willoughby, E.W. L-alpha-methyldopa hydrazine (Carbidopa) combined with L-dopa in the treatment of Parkinson’s disease. Aust. NZ J. Med. 1974, 4, 373–378. [Google Scholar] [CrossRef]
- Das Gupta, V.; Gupta, A. Effect of pyridoxal 5-phosphate on carbidopa and decarboxylation of levodopa. J. Pharm. Sci. 1980, 69, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Canary, J.W. Rapid catalyst-free hydrazone ligation: Protein-pyridoxal phosphoramides. Bioconjugate Chem. 2012, 23, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Lynn-Bullock, C.P.; Welshhans, K.; Pallas, S.L.; Katz, P.S. The effect of oral 5-HTP administration on 5-HTP and 5-HT immunoreactivity in monoaminergic brain regions of rats. J. Chem. Neuroanat. 2004, 27, 129–138. [Google Scholar] [CrossRef] [PubMed]
- El-Bacha, T.; Midlej, V.; Pereira da Silva, A.P.; Silva da Costa, L.; Benchimol, M.; Galina, A.; Da Poian, A.T. Mitochondrial and bioenergetic dysfunction in human hepatic cells infected with dengue 2 virus. Biochim. Biophys. Acta 2007, 1772, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Juorio, A.V.; Boulton, A.A. NSD-1015 alters the gene expression of aromatic L-amino acid decarboxylase in rat PC12 pheochromocytoma cells. Neurochem. Res. 1993, 18, 915–919. [Google Scholar] [CrossRef]
- Janku, F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat. Rev. 2017, 59, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Chock, P.B. Implication of phosphatidylinositol 3-kinase membrane recruitment in hydrogen peroxide-induced activation of PI3K and Akt. Biochemistry 2003, 42, 2995–3003. [Google Scholar] [CrossRef]
- Cantrell, D.A. Phosphoinositide 3-kinase signalling pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar]
- Krasilnikov, M.A. Phosphatidylinositol-3 kinase dependent pathways: The role in control of cell growth, survival, and malignant transformation. Biochemistry 2000, 65, 59–67. [Google Scholar] [PubMed]
- Yuan, Y.U. Role of PI3K/Akt/mTOR signaling pathway in hepatocellular carcinoma. Linchuang Gandanbing Zazhi 2014, 30, 954–957. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frakolaki, E.; Kalliampakou, K.I.; Kaimou, P.; Moraiti, M.; Kolaitis, N.; Boleti, H.; Koskinas, J.; Vassilacopoulou, D.; Vassilaki, N. Emerging Role of l-Dopa Decarboxylase in Flaviviridae Virus Infections. Cells 2019, 8, 837. https://doi.org/10.3390/cells8080837
Frakolaki E, Kalliampakou KI, Kaimou P, Moraiti M, Kolaitis N, Boleti H, Koskinas J, Vassilacopoulou D, Vassilaki N. Emerging Role of l-Dopa Decarboxylase in Flaviviridae Virus Infections. Cells. 2019; 8(8):837. https://doi.org/10.3390/cells8080837
Chicago/Turabian StyleFrakolaki, Efseveia, Katerina I. Kalliampakou, Panagiota Kaimou, Maria Moraiti, Nikolaos Kolaitis, Haralabia Boleti, John Koskinas, Dido Vassilacopoulou, and Niki Vassilaki. 2019. "Emerging Role of l-Dopa Decarboxylase in Flaviviridae Virus Infections" Cells 8, no. 8: 837. https://doi.org/10.3390/cells8080837
APA StyleFrakolaki, E., Kalliampakou, K. I., Kaimou, P., Moraiti, M., Kolaitis, N., Boleti, H., Koskinas, J., Vassilacopoulou, D., & Vassilaki, N. (2019). Emerging Role of l-Dopa Decarboxylase in Flaviviridae Virus Infections. Cells, 8(8), 837. https://doi.org/10.3390/cells8080837