
Identifying Cancers Impacted by CDK8/19


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Analysis of Tumor Cell Line Dependency on CDK8/CDK19/CCNC
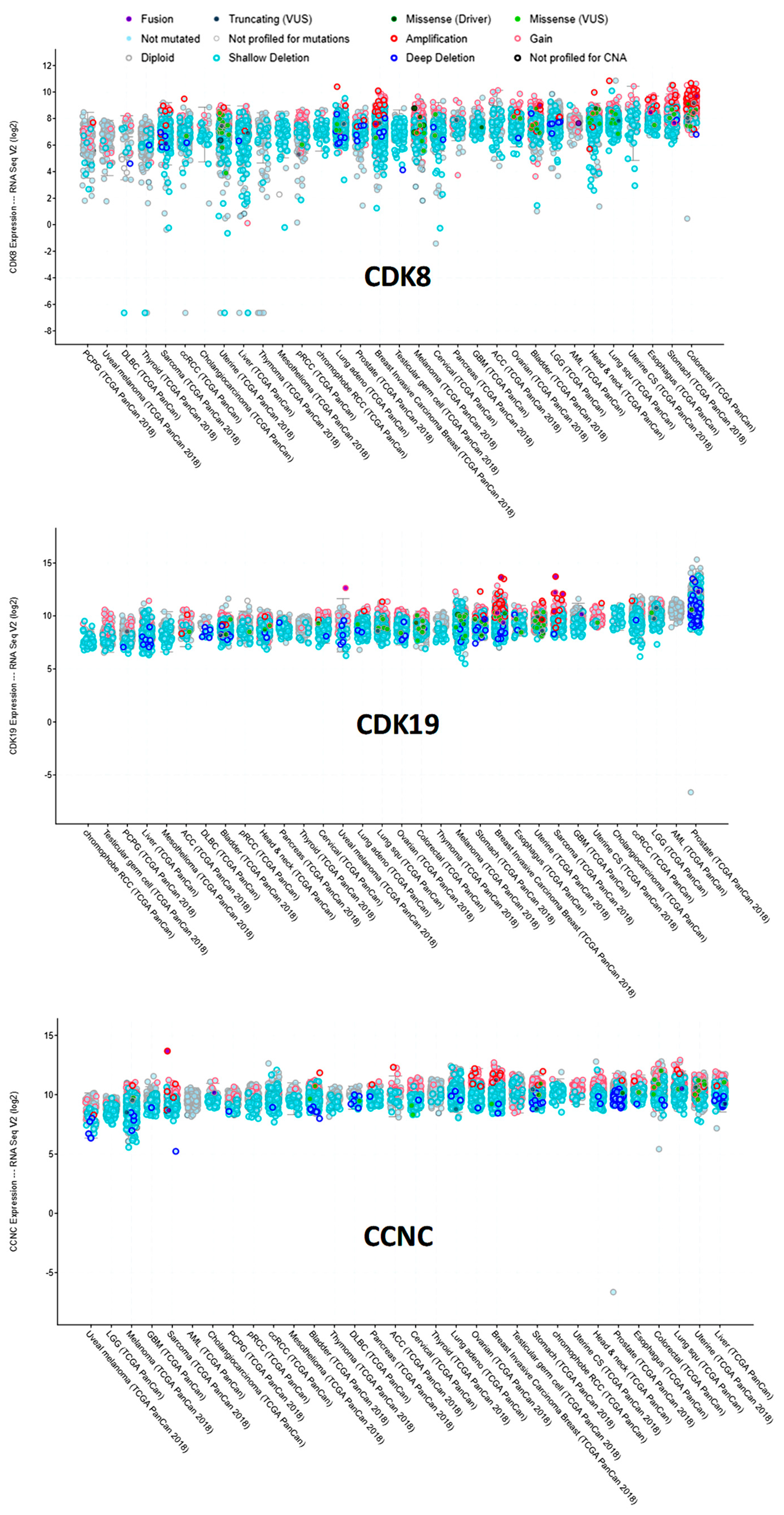
3.2. Analysis of CDK8/CDK19/CCNC Alterations and Expression in Clinical Cancers
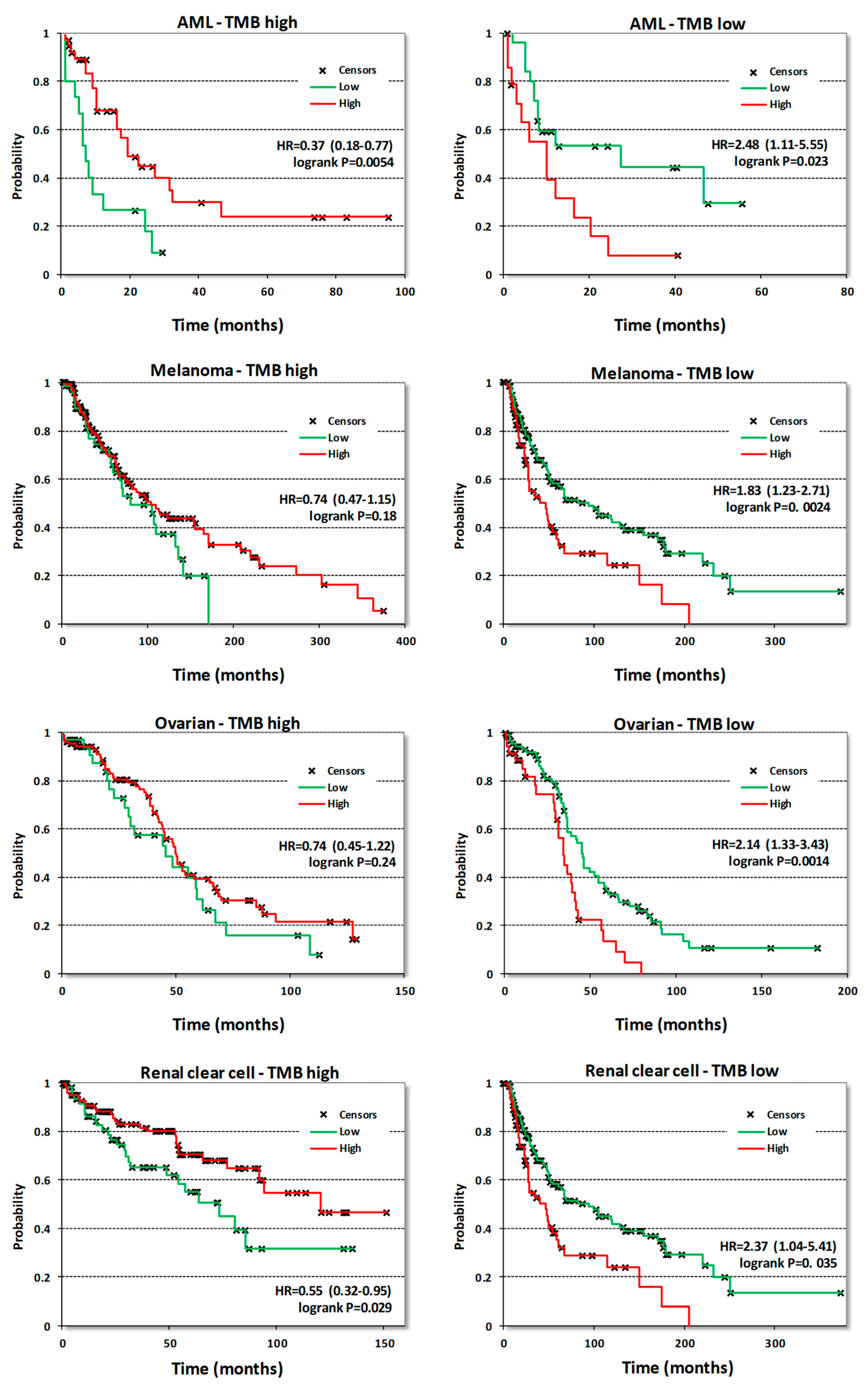
3.3. Analysis of Survival Correlations for CDK8/CDK19/CCNC
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.S.; McCleland, M.L.; Truong, T.; Lau, S.; Modrusan, Z.; Soukup, T.M.; Roose-Girma, M.; Blackwood, E.M.; Firestein, R. CDK8 Maintains Tumor Dedifferentiation and Embryonic Stem Cell Pluripotency. Cancer Res. 2012, 72, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chen, M.; Hughes, D.; Chumanevich, A.A.; Altilia, S.; Kaza, V.; Lim, C.-U.; Kiaris, H.; Mythreye, K.; Pena, M.M.; et al. CDK8 Selectively Promotes the Growth of Colon Cancer Metastases in the Liver by Regulating Gene Expression of TIMP3 and Matrix Metalloproteinases. Cancer Res. 2018, 78, 6594–6606. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.C.; Farmaki, E.; Altilia, S.; Schools, G.P.; West, D.K.; Chen, M.; Chang, B.-D.; Puzyrev, A.T.; Lim, C.-U.; Rokow-Kittell, R.; et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl. Acad. Sci. USA 2012, 109, 13799–13804. [Google Scholar] [CrossRef] [PubMed]
- Broude, E.V.; Győrffy, B.; Chumanevich, A.A.; Chen, M.; McDermott, M.S.J.; Shtutman, M.; Catroppo, J.F.; Roninson, I.B. Expression of CDK8 and CDK8-interacting Genes as Potential Biomarkers in Breast Cancer. Curr. Cancer Drug Targets 2015, 15, 739–749. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.S.; Chumanevich, A.A.; Lim, C.-U.; Liang, J.; Chen, M.; Altilia, S.; Oliver, D.; Rae, J.M.; Shtutman, M.; Kiaris, H.; et al. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget 2017, 8, 12558–12575. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Li, C.-F.; Zhang, X.; Gong, Z.; Chan, C.-H.; Lee, S.-W.; Jin, G.; Rezaeian, A.-H.; Han, F.; Wang, J.; et al. Skp2-MacroH2A1-CDK8 axis orchestrates G2/M transition, polyploidy and tumourigenesis. Nat. Commun. 2015, 6, 6641. [Google Scholar] [CrossRef]
- Bragelmann, J.; Klumper, N.; Offermann, A.; von Massenhausen, A.; Bohm, D.; Deng, M.; Queisser, A.; Sanders, C.; Syring, I.; Merseburger, A.S.; et al. Pan-Cancer Analysis of the Mediator Complex Transcriptome Identifies CDK19 and CDK8 as Therapeutic Targets in Advanced Prostate Cancer. Clin Cancer Res. 2017, 23, 1829–1840. [Google Scholar] [CrossRef]
- Xu, W.; Wang, Z.; Zhang, W.; Qian, K.; Li, H.; Kong, D.; Li, Y.; Tang, Y. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/β-catenin signaling pathway. Cancer Lett. 2015, 356, 613–627. [Google Scholar] [CrossRef]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menéndez, S.; Vardabasso, C.; Leroy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef]
- Pelish, H.E.; Liau, B.B.; Nitulescu, I.I.; Tangpeerachaikul, A.; Poss, Z.C.; Da Silva, D.H.; Caruso, B.T.; Arefolov, A.; Fadeyi, O.; Christie, A.L.; et al. Mediator Kinase Inhibition Further Activates Super-Enhancer Associated Genes in AML. Nature 2015, 526, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Rzymski, T.; Mikula, M.; Żyłkiewicz, E.; Dreas, A.; Wiklik, K.; Gołas, A.; Wójcik, K.; Masiejczyk, M.; Wróbel, A.; Dolata, I.; et al. SEL120-34A is a novel CDK8 inhibitor active in AML cells with high levels of serine phosphorylation of STAT1 and STAT5 transactivation domains. Oncotarget 2017, 8, 33779–33795. [Google Scholar] [CrossRef] [PubMed]
- Philip, S.; Kumarasiri, M.; Teo, T.; Yu, M.; Wang, S. Cyclin-Dependent Kinase 8: A New Hope in Targeted Cancer Therapy? J. Med. Chem. 2018, 61, 5073–5092. [Google Scholar] [CrossRef] [PubMed]
- Serrao, A.; Jenkins, L.M.; Chumanevich, A.A.; Horst, B.; Liang, J.; Gatza, M.L.; Lee, N.Y.; Roninson, I.B.; Broude, E.V.; Mythreye, K. Mediator kinase CDK8/CDK19 drives YAP1-dependent BMP4-induced EMT in cancer. Oncogene 2018, 37, 4792–4808. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Nakata, D.; Kakoi, Y.; Kunitomo, M.; Murai, S.; Ebara, S.; Hata, A.; Hara, T. CDK8/19 inhibition induces premature G1/S transition and ATR-dependent cell death in prostate cancer cells. Oncotarget 2018, 9, 13474–13487. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.D.; Donner, A.J.; Espinosa, J.M. CDK8: A positive regulator of transcription. Transcription 2010, 1, 4–12. [Google Scholar] [CrossRef]
- Fant, C.B.; Taatjes, D.J. Regulatory functions of the Mediator kinases CDK8 and CDK19. Transcription 2019, 10, 76–90. [Google Scholar] [CrossRef]
- Chen, M.; Liang, J.; Ji, H.; Yang, Z.; Altilia, S.; Hu, B.; Schronce, A.; McDermott, M.S.J.; Schools, G.P.; Lim, C.-U.; et al. CDK8/19 Mediator kinases potentiate induction of transcription by NFκB. Proc. Natl. Acad. Sci. USA 2017, 114, 10208–10213. [Google Scholar] [CrossRef]
- Boehm, J.S.; Golub, T.R. An ecosystem of cancer cell line factories to support a cancer dependency map. Nat. Rev. Genet. 2015, 16, 373–374. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef]
- Gyorffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szallasi, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2010, 123, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.M.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Lánczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef]
- Firestein, R.; Shima, K.; Nosho, K.; Irahara, N.; Baba, Y.; Bojarski, E.; Giovannucci, E.L.; Hahn, W.C.; Fuchs, C.S.; Ogino, S. CDK8 Expression in 470 Colorectal Cancers in Relation to β-Catenin Activation, Other Molecular Alterations and Patient Survival. Int. J. Cancer 2010, 126, 2863–2873. [Google Scholar] [CrossRef]
- Croce, S.; Chibon, F. MED12 and uterine smooth muscle oncogenesis: State of the art and perspectives. Eur. J. Cancer 2015, 51, 1603–1610. [Google Scholar] [CrossRef]
- Huang, S.; Hölzel, M.; Knijnenburg, T.; Schlicker, A.; Roepman, P.; Mc Dermott, U.; Garnett, M.; Grernrum, W.; Sun, C.; Prahallad, A.; et al. MED12 Controls the Response to Multiple Cancer Drugs through Regulation of TGF-β Receptor Signaling. Cell 2012, 151, 937–950. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type/Gene | CDK8 | CDK19 | CCNC | |||||
|---|---|---|---|---|---|---|---|---|
| n | HR | logrank p | HR | logrank p | HR | logrank p | ||
| Acute myeloid leukemia | OS | 132 | 0.63 | 0.049 | 0.6 | 0.026 | 0.61 | 0.045 |
| Bladder Carcinoma | OS | 405 | 1.34 | 0.063 | 0.71 | 0.023 | 1.37 | 0.042 |
| RFS | 187 | 1.83 | 0.091 | 4.87 | 0.004 | 2.68 | 0.024 | |
| Breast cancer | OS | 1090 | 1.61 | 0.0031 | 1.43 | 0.038 | 1.62 | 0.01 |
| RFS | 255 | 1.75 | 0.0095 | 1.59 | 0.032 | 1.29 | 0.26 | |
| Colon cancer | OS | 293 | 1.34 | 0.26 | 0.73 | 0.21 | 0.64 | 0.066 |
| RFS | 109 | 2.1 | 0.14 | 0.59 | 0.27 | 0.42 | 0.079 | |
| Cutaneous melanoma | OS | 458 | 1.26 | 0.093 | 0.58 | 0.00048 | 0.59 | 0.00046 |
| Esophageal Squamous Cell Carcinoma | OS | 81 | 0.21 | 0.00053 | 0.48 | 0.072 | 0.53 | 0.11 |
| RFS | 54 | 2.57 | 0.19 | 1.49 | 0.42 | 2.06 | 0.25 | |
| Glioblastoma | OS | 152 | 0.66 | 0.022 | 1.28 | 0.24 | 0.63 | 0.019 |
| Head-neck squamous cell carcinoma | OS | 500 | 1.46 | 0.008 | 0.6 | 0.0026 | 1.53 | 0.0028 |
| RFS | 124 | 1.49 | 0.32 | 0.43 | 0.11 | 2.45 | 0.016 | |
| Kidney renal clear cell carcinoma | OS | 530 | 1.32 | 0.11 | 0.49 | 5.7 × 10−6 | 0.64 | 0.0028 |
| RFS | 117 | 0.3 | 0.017 | NA* | 0.027 | 0.16 | 0.045 | |
| Kidney renal papillary cell carcinoma | OS | 288 | 0.68 | 0.2 | 2 | 0.02 | 1.57 | 0.15 |
| RFS | 183 | 1.51 | 0.38 | 1.72 | 0.17 | 2.18 | 0.041 | |
| Liver hepatocellular carcinoma | OS | 71 | 1.68 | 0.0059 | 1.74 | 0.002 | 1.25 | 0.21 |
| RFS | 316 | 1.38 | 0.051 | 2.14 | 9.1 × 10−6 | 1.28 | 0.14 | |
| Low grade glioma | OS | 406 | 0.43 | 0.00025 | 1.43 | 0.058 | 1.91 | 0.0012 |
| RFS | 131 | 1.33 | 0.53 | 0.38 | 0.034 | 0.34 | 0.017 | |
| Lung adenocarcinoma | OS | 513 | 1.51 | 0.0092 | 0.72 | 0.07 | 1.35 | 0.1 |
| RFS | 300 | 1.53 | 0.049 | 1.34 | 0.17 | 0.83 | 0.38 | |
| Lung squamous cell carcinoma | OS | 501 | 1.11 | 0.44 | 0.76 | 0.046 | 0.86 | 0.29 |
| RFS | 300 | 1.49 | 0.14 | 2.28 | 0.015 | 1.63 | 0.14 | |
| Ovarian cancer | OS | 374 | 1.27 | 0.072 | 1.52 | 0.0015 | 0.69 | 0.023 |
| RFS | 177 | 0.7 | 0.064 | 1.49 | 0.026 | 0.89 | 0.54 | |
| Pancreatic ductal adenocarcinoma | OS | 177 | 1.27 | 0.25 | 1.2 | 0.4 | 0.8 | 0.3 |
| RFS | 69 | 2.42 | 0.1 | 5.31 | 0.013 | 1.62 | 0.25 | |
| Pheochromocytoma and Paraganglioma | OS | 178 | NA* | 0.11 | 13.46 | 0.0029 | 4.65 | 0.052 |
| RFS | 159 | 7.96 | 0.033 | 0.24 | 0.18 | 3.18 | 0.29 | |
| Prostate adenocarcinoma | OS | 494 | 4.36 | 0.019 | 0.58 | 0.4 | 0.48 | 0.35 |
| BCR | 337 | 2.46 | 0.011 | 2.09 | 0.039 | 0.44 | 0.082 | |
| Rectum adenocarcinoma | OS | 165 | 0.59 | 0.24 | 0.54 | 0.12 | 0.28 | 0.014 |
| RFS | 47 | 5.37 | 0.031 | NA* | 0.16 | 0.11 | 0.027 | |
| Sarcoma | OS | 259 | 1.55 | 0.031 | 1.41 | 0.09 | 1.79 | 0.0038 |
| RFS | 152 | 0.71 | 0.17 | 1.57 | 0.074 | 0.58 | 0.068 | |
| Stomach adenocarcinoma | OS | 375 | 0.55 | 0.0011 | 0.81 | 0.21 | 0.81 | 0.21 |
| RFS | 215 | 0.32 | 0.00037 | 1.45 | 0.3 | 0.78 | 0.46 | |
| Testicular Germ Cell Tumor | OS | 134 | 0.1 | 0.015 | NA* | 0.098 | 5.09 | 0.12 |
| RFS | 105 | 2.37 | 0.046 | 0.59 | 0.19 | 5.15 | 0.0029 | |
| Thymoma | OS | 119 | 0.28 | 0.061 | 0.07 | 0.00005 | 0.15 | 0.0037 |
| Thyroid carcinoma | OS | 502 | 0.67 | 0.43 | 0.26 | 0.055 | 2.1 | 0.14 |
| RFS | 353 | 2.17 | 0.14 | 2.19 | 0.047 | 0.4 | 0.12 | |
| Uterine corpus endometrial carcinoma | OS | 543 | 1.89 | 0.0037 | 1.66 | 0.023 | 0.63 | 0.059 |
| RFS | 422 | 1.43 | 0.22 | 1.88 | 0.038 | 1.57 | 0.1 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roninson, I.B.; Győrffy, B.; Mack, Z.T.; Shtil, A.A.; Shtutman, M.S.; Chen, M.; Broude, E.V. Identifying Cancers Impacted by CDK8/19. Cells 2019, 8, 821. https://doi.org/10.3390/cells8080821
Roninson IB, Győrffy B, Mack ZT, Shtil AA, Shtutman MS, Chen M, Broude EV. Identifying Cancers Impacted by CDK8/19. Cells. 2019; 8(8):821. https://doi.org/10.3390/cells8080821
Chicago/Turabian StyleRoninson, Igor B., Balázs Győrffy, Zachary T. Mack, Alexander A. Shtil, Michael S. Shtutman, Mengqian Chen, and Eugenia V. Broude. 2019. "Identifying Cancers Impacted by CDK8/19" Cells 8, no. 8: 821. https://doi.org/10.3390/cells8080821
APA StyleRoninson, I. B., Győrffy, B., Mack, Z. T., Shtil, A. A., Shtutman, M. S., Chen, M., & Broude, E. V. (2019). Identifying Cancers Impacted by CDK8/19. Cells, 8(8), 821. https://doi.org/10.3390/cells8080821