Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review


Abstract
1. Introduction
2. Osteoarthritis (OA)
2.1. Characteristics and Pathology
Genetic Factors in OA
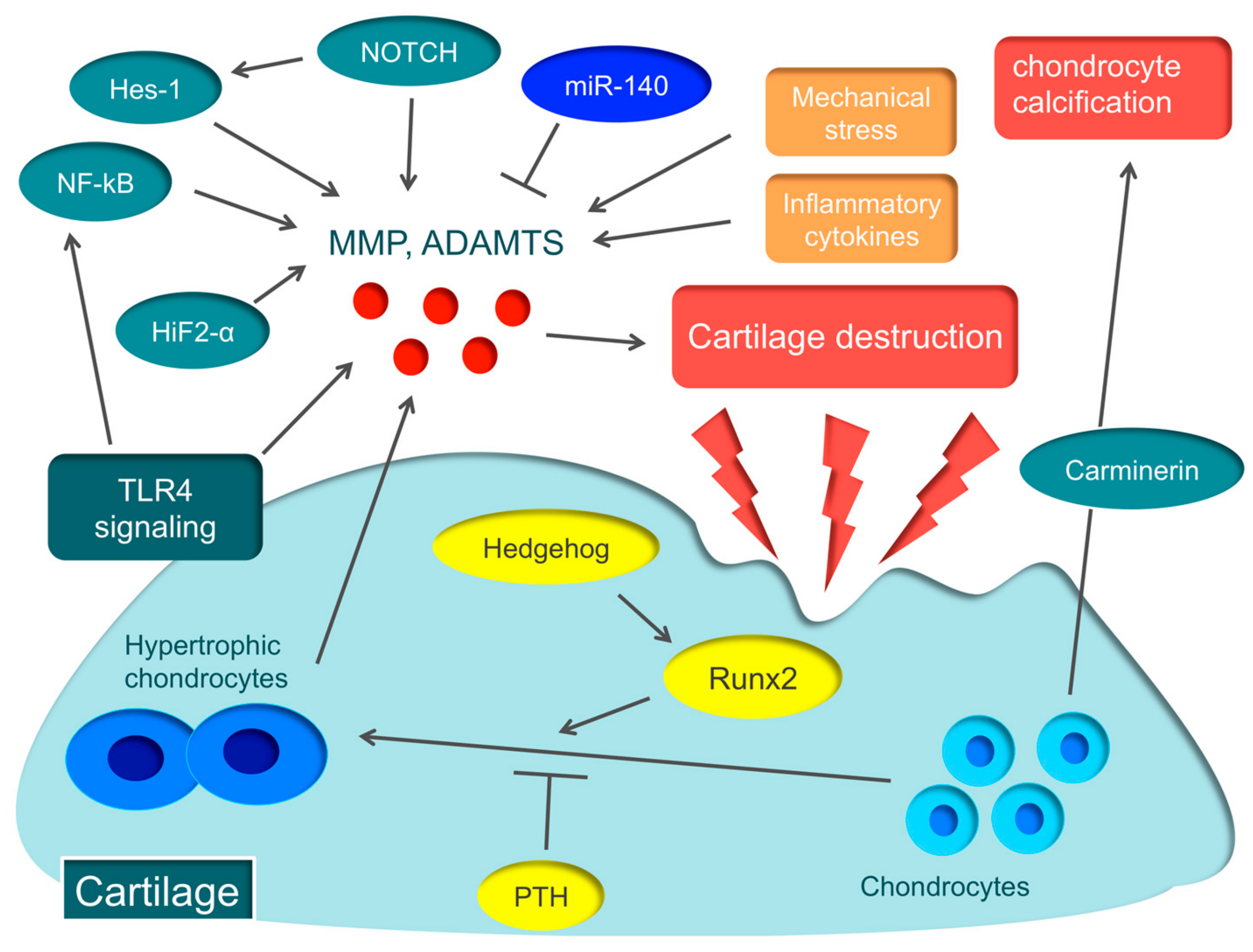
2.2. Cartilage Destruction
2.2.1. ECM Degradation
2.2.2. ECM Degradation by MMP
2.2.3. ECM Degradation by ADAMTS
2.2.4. The Role of Toll-like Receptors (TLRs)
2.3. Molecular Mechanisms Associated with OA
2.4. Present Therapeutic Strategy for OA
2.5. Novel OA Therapeutic Strategies
2.5.1. Anti-Inflammatory Cytokines Therapy
2.5.2. Bisphosphonate
2.5.3. Strontium Ranelate
2.5.4. Anti-Nerve Growth Factor (NGF) Antibody
2.5.5. Therapies Targeting TLR Signaling
2.5.6. miRNA
2.5.7. Cell-Based Therapies
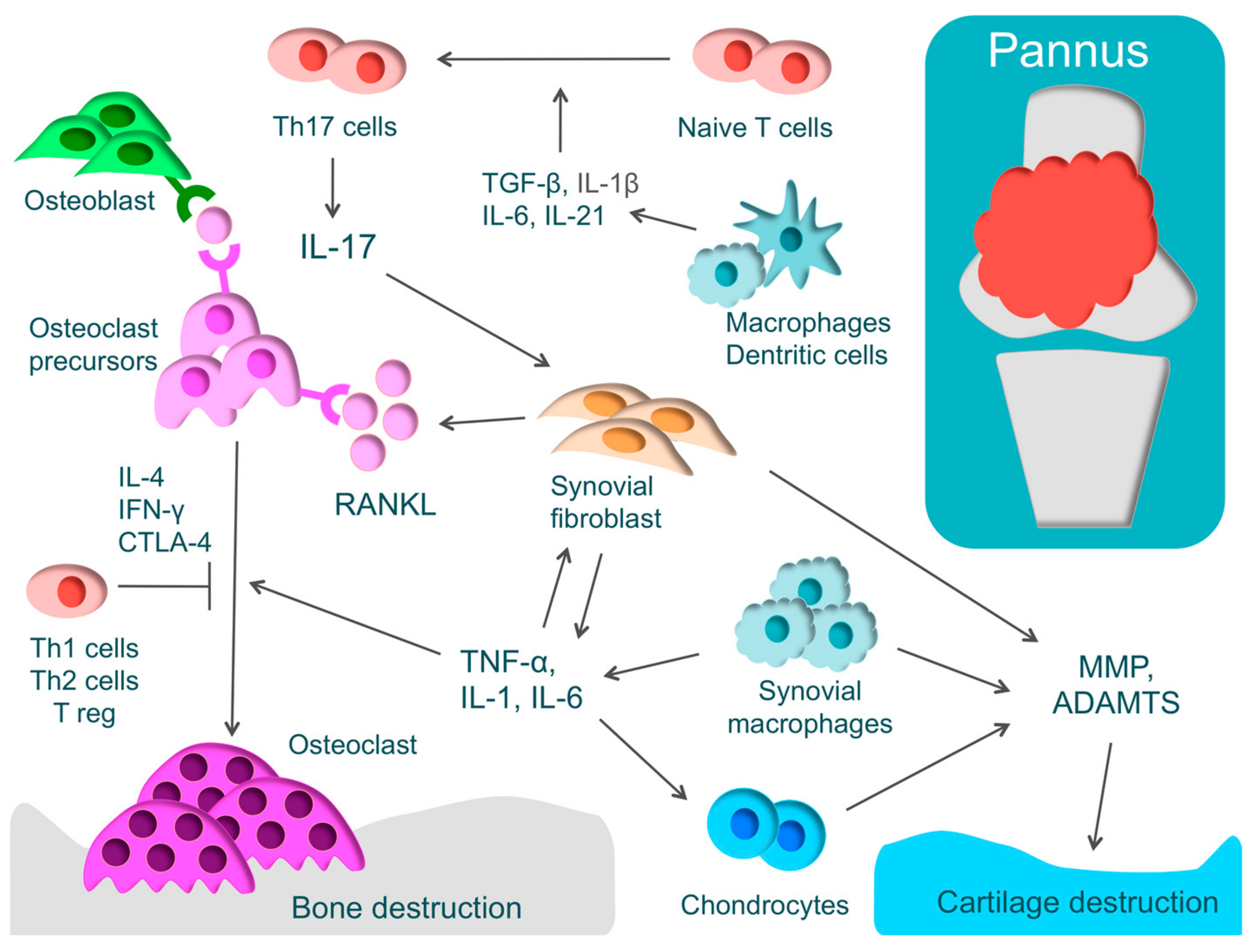
3. Rheumatoid Arthritis
3.1. Characteristics and Pathology
Genetic Factors in RA
3.2. Mechanism of Bone and Cartilage Destruction
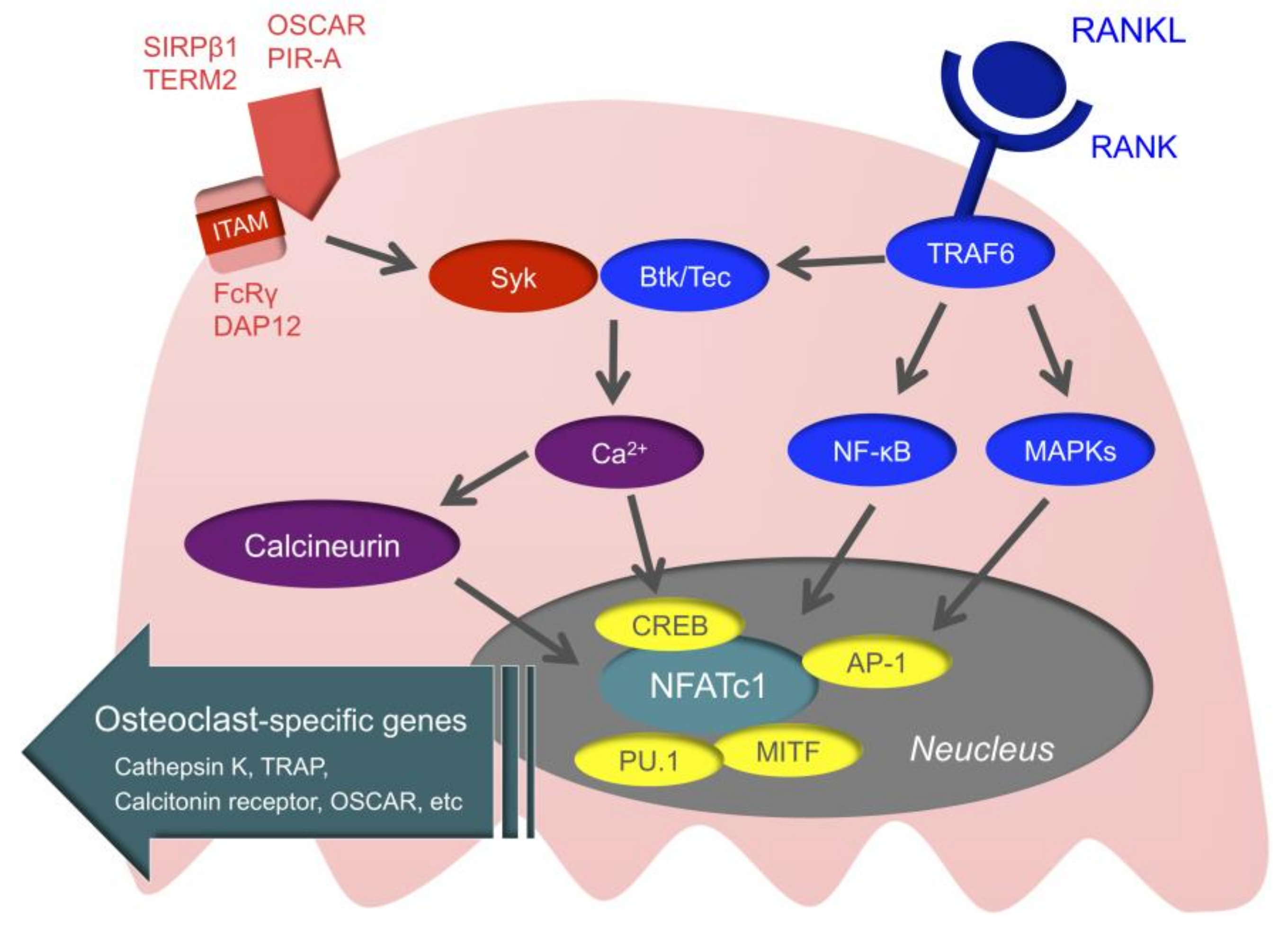
3.3. Intracellular Signals of Osteoclasts
3.4. The Current Therapeutic Strategy for RA
3.4.1. TNF inhibitors
3.4.2. IL-6 Inhibitors
3.4.3. Janus Kinase (JAK) Inhibitors
3.4.4. T-cell Activation Inhibitors
3.5. Novel Therapeutic Approches for RA
3.5.1. Btk Inhibitors
3.5.2. Syk Inhibitors
3.5.3. Phosphoinositide 3-Kinase (PI3K) Inhibitors
3.5.4. MicroRNA (miRNA)
3.5.5. Histone Deacetylase (HDAC) Inhibitors
4. Psoriatic Arthritis (PsA)
4.1. Characteristics and Pathology
Genetic and Environmental Factors in PsA
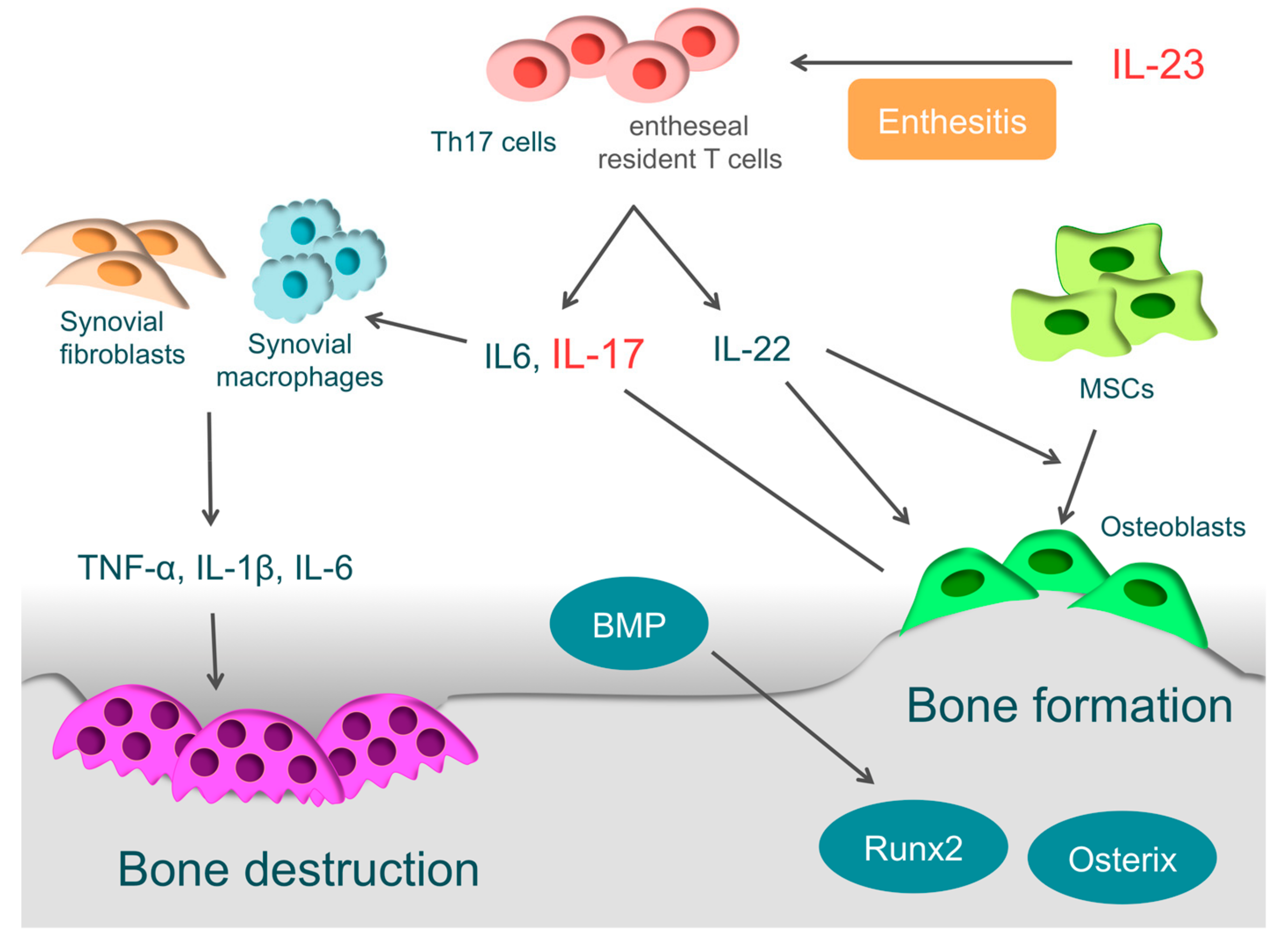
4.2. Enthesitis and IL-23
4.3. Bone Destruction in PsA
4.4. Bone Formation in PsA
4.5. Present Therapeutic Strategy for PsA
4.5.1. TNF-α Inhibitors
4.5.2. Anti-IL-23/IL-17 Therapy
4.6. Novel Therapeutic Targets in PsA
4.6.1. Using Appropriate Biological Agents
4.6.2. Novel Therapeutic Agents
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADMTS | a disintegrin and metalloproteinase with thrombospondin motifs |
| AP−1 | activator protein1 |
| Btk | Bruton’s tyrosine kinase |
| CCL20 | chemokine C-C motif ligand 20 |
| CREB | cyclic adenosine monophosphate-response element-binding protein |
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 |
| DAMP | damage-associated molecular patterns |
| DAP12 | DNAX-activating protein of 12 |
| DDS | drug delivery system |
| DMARDs | disease modifying anti rheumatic drugs |
| ECM | extracellular matrix |
| FcRγ | Fc receptor common gamma subunit |
| HDAC | histone deacetylase |
| Hes1 | hairy and enhancer of split 1 |
| HIF-2α | hypoxia inducible factor -2α |
| ITAM | immunoreceptor tyrosine-based activation motif |
| JAK | janus kinase |
| M-CSF | macrophage colony-stimulating factor |
| MITF | microphthalmia-associated transcription factor |
| MMP | matrix metalloproteinase |
| MTX | methotrexate |
| NF-κB | nuclear factor-kappa B |
| NFATc1 | nuclear factor of activated T-cell c1 |
| NGF | nerve growth factor |
| NSAIDs | non-steroidal anti-inflammatory drugs |
| OA | osteoarthritis |
| OPG | Osteoprotegerin |
| OSCAR | osteoclast associated receptor |
| PAMP | pathogen-associated molecular patterns |
| PI3K | phosphoinositide 3-kinase |
| PIP3 | phosphatidylinositol 3-phosphate |
| PIR-A | paired immunoglobulin-like receptor-A |
| PsA | psoriatic arthritis |
| RA | rheumatoid arthritis |
| RANKL | receptor activator of nuclear factor kappa B ligand |
| Runx2 | runt-related transcription factor 2 |
| SIRPβ1 | signal regulatory protein beta 1 |
| SpA | spondyloarthritis |
| Syk | spleen tyrosine kinase |
| Tec | tyrosine kinase expressed in hepatocellular carcinoma |
| TGF-β | transforming growth factor-β |
| TLR | Toll like receptor |
| TNF | tumor necrosis factor |
| TLR | Toll like receptor |
| TRAF | TNF receptor-associated factor |
| TRAP | tartrate resistant acid phosphatase |
| TREM2 | triggering receptor expressed on myeloid cells 2 |
| Tyk | tyrosine kinase |
References
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Glyn-Jones, S.; Palmer, A.J.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Veale, D.J.; Fearon, U. The pathogenesis of psoriatic arthritis. Lancet 2018, 391, 2273–2284. [Google Scholar] [CrossRef]
- Hunter, D.J.; Schofield, D.; Callander, E. The individual and socioeconomic impact of osteoarthritis. Nat. Rev. Rheumatol. 2014, 10, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- Gabriel, S.E.; Michaud, K. Epidemiological studies in incidence, prevalence, mortality, and comorbidity of the rheumatic diseases. Arthritis Res. Ther. 2009, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Furneri, G.; Mantovani, L.G.; Belisari, A.; Mosca, M.; Cristiani, M.; Bellelli, S.; Cortesi, P.A.; Turchetti, G. Systematic literature review on economic implications and pharmacoeconomic issues of rheumatoid arthritis. Clin. Exp. Rheumatol. 2012, 30 (Suppl. 73), S72–S84. [Google Scholar]
- Piscitelli, P.; Iolascon, G.; Di Tanna, G.; Bizzi, E.; Chitano, G.; Argentiero, A.; Neglia, C.; Giolli, L.; Distante, A.; Gimigliano, R.; et al. Socioeconomic burden of total joint arthroplasty for symptomatic hip and knee osteoarthritis in the Italian population: A 5-year analysis based on hospitalization records. Arthritis Care Res. 2012, 64, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- March, L.M.; Bachmeier, C.J. Economics of osteoarthritis: A global perspective. Baillieres Clin. Rheumatol. 1997, 11, 817–834. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology and the effects of the immune system on bone. Nat. Rev. Rheumatol. 2009, 5, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Danks, L.; Takayanagi, H. Immunology and bone. J. Biochem. 2013, 154, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.; Englund, M.; Struglics, A.; Lohmander, L.S. Interleukin-6 and tumor necrosis factor alpha in synovial fluid are associated with progression of radiographic knee osteoarthritis in subjects with previous meniscectomy. Osteoarthritis Cartilage 2015, 23, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Akamine, Y.; Kakudo, K.; Kondo, M.; Ota, K.; Muroi, Y.; Yoshikawa, H.; Nakata, K. Prolonged matrix metalloproteinase-3 high expression after cyclic compressive load on human synovial cells in three-dimensional cultured tissue. Int. J. Oral Maxillofac. Surg. 2012, 41, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Kurz, B.; Lemke, A.K.; Fay, J.; Pufe, T.; Grodzinsky, A.J.; Schunke, M. Pathomechanisms of cartilage destruction by mechanical injury. Ann. Anat. 2005, 187, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Saklatvala, J. Tumour necrosis factor alpha stimulates resorption and inhibits synthesis of proteoglycan in cartilage. Nature 1986, 322, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef] [PubMed]
- Spector, T.D.; Cicuttini, F.; Baker, J.; Loughlin, J.; Hart, D. Genetic influences on osteoarthritis in women: A twin study. BMJ 1996, 312, 940–943. [Google Scholar] [CrossRef]
- Evangelou, E.; Kerkhof, H.J.; Styrkarsdottir, U.; Ntzani, E.E.; Bos, S.D.; Esko, T.; Evans, D.S.; Metrustry, S.; Panoutsopoulou, K.; Ramos, Y.F.; et al. A meta-analysis of genome-wide association studies identifies novel variants associated with osteoarthritis of the hip. Ann. Rheum. Dis. 2014, 73, 2130–2136. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.B. Anatomy and physiology of the mineralized tissues: Role in the pathogenesis of osteoarthrosis. Osteoarthritis Cartilage 2004, 12 (Suppl. A), S20–S30. [Google Scholar] [CrossRef]
- Xia, B.; Di, C.; Zhang, J.; Hu, S.; Jin, H.; Tong, P. Osteoarthritis pathogenesis: A review of molecular mechanisms. Calcif. Tissue Int. 2014, 95, 495–505. [Google Scholar] [CrossRef]
- Goldring, M.B. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000, 43, 1916–1926. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg. Med. Chem. 2007, 15, 2223–2268. [Google Scholar] [CrossRef]
- Murphy, G.; Nagase, H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: Destruction or repair? Nat. Clin. Pract. Rheumatol. 2008, 4, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef]
- Brocker, C.N.; Vasiliou, V.; Nebert, D.W. Evolutionary divergence and functions of the ADAM and ADAMTS gene families. Hum. Genomics 2009, 4, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef]
- Yatabe, T.; Mochizuki, S.; Takizawa, M.; Chijiiwa, M.; Okada, A.; Kimura, T.; Fujita, Y.; Matsumoto, H.; Toyama, Y.; Okada, Y. Hyaluronan inhibits expression of ADAMTS4 (aggrecanase-1) in human osteoarthritic chondrocytes. Ann. Rheum. Dis. 2009, 68, 1051–1058. [Google Scholar] [CrossRef]
- Gomez, R.; Villalvilla, A.; Largo, R.; Gualillo, O.; Herrero-Beaumont, G. TLR4 signalling in osteoarthritis--finding targets for candidate DMOADs. Nat. Rev. Rheumatol. 2015, 11, 159–170. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Wang, P.; Zhu, F.; Tong, Z.; Konstantopoulos, K. Response of chondrocytes to shear stress: Antagonistic effects of the binding partners Toll-like receptor 4 and caveolin-1. FASEB J. 2011, 25, 3401–3415. [Google Scholar] [CrossRef]
- Kim, H.A.; Cho, M.L.; Choi, H.Y.; Yoon, C.S.; Jhun, J.Y.; Oh, H.J.; Kim, H.Y. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006, 54, 2152–2163. [Google Scholar] [CrossRef]
- Campo, G.M.; Avenoso, A.; D’Ascola, A.; Prestipino, V.; Scuruchi, M.; Nastasi, G.; Calatroni, A.; Campo, S. Hyaluronan differently modulates TLR-4 and the inflammatory response in mouse chondrocytes. BioFactors 2012, 38, 69–76. [Google Scholar] [CrossRef]
- Saito, T.; Kawaguchi, H. HIF-2alpha as a possible therapeutic target of osteoarthritis. Osteoarthritis Cartilage 2010, 18, 1552–1556. [Google Scholar] [CrossRef]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef]
- Kamekura, S.; Kawasaki, Y.; Hoshi, K.; Shimoaka, T.; Chikuda, H.; Maruyama, Z.; Komori, T.; Sato, S.; Takeda, S.; Karsenty, G.; et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006, 54, 2462–2470. [Google Scholar] [CrossRef]
- Lin, A.C.; Seeto, B.L.; Bartoszko, J.M.; Khoury, M.A.; Whetstone, H.; Ho, L.; Hsu, C.; Ali, S.A.; Alman, B.A. Modulating hedgehog signaling can attenuate the severity of osteoarthritis. Nat. Med. 2009, 15, 1421–1425. [Google Scholar] [CrossRef]
- Sampson, E.R.; Hilton, M.J.; Tian, Y.; Chen, D.; Schwarz, E.M.; Mooney, R.A.; Bukata, S.V.; O’Keefe, R.J.; Awad, H.; Puzas, J.E.; et al. Teriparatide as a chondroregenerative therapy for injury-induced osteoarthritis. Sci. Transl. Med. 2011, 3, 101ra93. [Google Scholar] [CrossRef]
- Chen, L.X.; Lin, L.; Wang, H.J.; Wei, X.L.; Fu, X.; Zhang, J.Y.; Yu, C.L. Suppression of early experimental osteoarthritis by in vivo delivery of the adenoviral vector-mediated NF-kappaBp65-specific siRNA. Osteoarthritis Cartilage 2008, 16, 174–184. [Google Scholar] [CrossRef]
- Hosaka, Y.; Saito, T.; Sugita, S.; Hikata, T.; Kobayashi, H.; Fukai, A.; Taniguchi, Y.; Hirata, M.; Akiyama, H.; Chung, U.I.; et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA 2013, 110, 1875–1880. [Google Scholar] [CrossRef]
- Wang, M.; Shen, J.; Jin, H.; Im, H.J.; Sandy, J.; Chen, D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann. N. Y. Acad. Sci. 2011, 1240, 61–69. [Google Scholar] [CrossRef]
- Kamekura, S.; Hoshi, K.; Shimoaka, T.; Chung, U.; Chikuda, H.; Yamada, T.; Uchida, M.; Ogata, N.; Seichi, A.; Nakamura, K.; et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis Cartilage 2005, 13, 632–641. [Google Scholar] [CrossRef]
- Jimi, E.; Ghosh, S. Role of nuclear factor-kappaB in the immune system and bone. Immunol. Rev. 2005, 208, 80–87. [Google Scholar] [CrossRef]
- Ahmad, R.; Sylvester, J.; Ahmad, M.; Zafarullah, M. Adaptor proteins and Ras synergistically regulate IL-1-induced ADAMTS-4 expression in human chondrocytes. J. Immunol. 2009, 182, 5081–5087. [Google Scholar] [CrossRef]
- Karlsson, C.; Brantsing, C.; Egell, S.; Lindahl, A. Notch1, Jagged1, and HES5 are abundantly expressed in osteoarthritis. Cells Tissues Organs 2008, 188, 287–298. [Google Scholar] [CrossRef]
- Sugita, S.; Hosaka, Y.; Okada, K.; Mori, D.; Yano, F.; Kobayashi, H.; Taniguchi, Y.; Mori, Y.; Okuma, T.; Chang, S.H.; et al. Transcription factor Hes1 modulates osteoarthritis development in cooperation with calcium/calmodulin-dependent protein kinase 2. Proc. Natl. Acad. Sci. USA 2015, 112, 3080–3085. [Google Scholar] [CrossRef]
- Yamada, T.; Kawano, H.; Koshizuka, Y.; Fukuda, T.; Yoshimura, K.; Kamekura, S.; Saito, T.; Ikeda, T.; Kawasaki, Y.; Azuma, Y.; et al. Carminerin contributes to chondrocyte calcification during endochondral ossification. Nat. Med. 2006, 12, 665–670. [Google Scholar] [CrossRef]
- McAlindon, T.E.; Bannuru, R.R.; Sullivan, M.C.; Arden, N.K.; Berenbaum, F.; Bierma-Zeinstra, S.M.; Hawker, G.A.; Henrotin, Y.; Hunter, D.J.; Kawaguchi, H.; et al. OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthritis Cartilage 2014, 22, 363–388. [Google Scholar] [CrossRef]
- Rutjes, A.W.; Juni, P.; da Costa, B.R.; Trelle, S.; Nuesch, E.; Reichenbach, S. Viscosupplementation for osteoarthritis of the knee: A systematic review and meta-analysis. Ann. Intern. Med. 2012, 157, 180–191. [Google Scholar] [CrossRef]
- Bannuru, R.R.; Natov, N.S.; Dasi, U.R.; Schmid, C.H.; McAlindon, T.E. Therapeutic trajectory following intra-articular hyaluronic acid injection in knee osteoarthritis--meta-analysis. Osteoarthritis Cartilage 2011, 19, 611–619. [Google Scholar] [CrossRef]
- Raynauld, J.P.; Buckland-Wright, C.; Ward, R.; Choquette, D.; Haraoui, B.; Martel-Pelletier, J.; Uthman, I.; Khy, V.; Tremblay, J.L.; Bertrand, C.; et al. Safety and efficacy of long-term intraarticular steroid injections in osteoarthritis of the knee: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2003, 48, 370–377. [Google Scholar] [CrossRef]
- Bastow, E.R.; Byers, S.; Golub, S.B.; Clarkin, C.E.; Pitsillides, A.A.; Fosang, A.J. Hyaluronan synthesis and degradation in cartilage and bone. Cell. Mol. Life Sci. 2008, 65, 395–413. [Google Scholar] [CrossRef]
- Zhang, W.; Moskowitz, R.W.; Nuki, G.; Abramson, S.; Altman, R.D.; Arden, N.; Bierma-Zeinstra, S.; Brandt, K.D.; Croft, P.; Doherty, M.; et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage 2008, 16, 137–162. [Google Scholar] [CrossRef]
- Wang, J. Efficacy and safety of adalimumab by intra-articular injection for moderate to severe knee osteoarthritis: An open-label randomized controlled trial. J. Int. Med. Res. 2018, 46, 326–334. [Google Scholar] [CrossRef]
- Verbruggen, G.; Wittoek, R.; Vander Cruyssen, B.; Elewaut, D. Tumour necrosis factor blockade for the treatment of erosive osteoarthritis of the interphalangeal finger joints: A double blind, randomised trial on structure modification. Ann. Rheum. Dis. 2012, 71, 891–898. [Google Scholar] [CrossRef]
- Maksymowych, W.P.; Russell, A.S.; Chiu, P.; Yan, A.; Jones, N.; Clare, T.; Lambert, R.G. Targeting tumour necrosis factor alleviates signs and symptoms of inflammatory osteoarthritis of the knee. Arthritis Res. Ther. 2012, 14, R206. [Google Scholar] [CrossRef]
- Fidelix, T.S.; Macedo, C.R.; Maxwell, L.J.; Fernandes Moca Trevisani, V. Diacerein for osteoarthritis. Cochrane Database Syst. Rev. 2014, Cd005117. [Google Scholar] [CrossRef]
- Chevalier, X.; Goupille, P.; Beaulieu, A.D.; Burch, F.X.; Bensen, W.G.; Conrozier, T.; Loeuille, D.; Kivitz, A.J.; Silver, D.; Appleton, B.E. Intraarticular injection of anakinra in osteoarthritis of the knee: A multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2009, 61, 344–352. [Google Scholar] [CrossRef]
- Cohen, S.B.; Proudman, S.; Kivitz, A.J.; Burch, F.X.; Donohue, J.P.; Burstein, D.; Sun, Y.N.; Banfield, C.; Vincent, M.S.; Ni, L.; et al. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 2011, 13, R125. [Google Scholar] [CrossRef]
- Laslett, L.L.; Dore, D.A.; Quinn, S.J.; Boon, P.; Ryan, E.; Winzenberg, T.M.; Jones, G. Zoledronic acid reduces knee pain and bone marrow lesions over 1 year: A randomised controlled trial. Ann. Rheum. Dis. 2012, 71, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Adami, S.; Fracassi, E.; Viapiana, O.; Orsolini, G.; Povino, M.R.; Idolazzi, L.; Gatti, D. Effects of intra-articular clodronate in the treatment of knee osteoarthritis: Results of a double-blind, randomized placebo-controlled trial. Rheumatol. Int. 2015, 35, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Saviola, G.; Abdi-Ali, L.; Povino, M.R.; Campostrini, L.; Sacco, S.; Carbonare, L.D. Intramuscular clodronate in erosive osteoarthritis of the hand is effective on pain and reduces serum COMP: A randomized pilot trial-The ER.O.D.E. study (ERosive Osteoarthritis and Disodium-clodronate Evaluation). Clin. Rheumatol. 2017, 36, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Hiasa, M.; Ichikawa, R.; Hasuzawa, N.; Kadowaki, A.; Iwatsuki, K.; Shima, K.; Endo, Y.; Kitahara, Y.; Inoue, T.; et al. Identification of a vesicular ATP release inhibitor for the treatment of neuropathic and inflammatory pain. Proc. Natl. Acad. Sci. USA 2017, 201704847. [Google Scholar] [CrossRef] [PubMed]
- Valenti, M.T.; Mottes, M.; Biotti, A.; Perduca, M.; Pisani, A.; Bovi, M.; Deiana, M.; Cheri, S.; Dalle Carbonare, L. Clodronate as a Therapeutic Strategy against Osteoarthritis. Int. J. Mol. Sci. 2017, 18, E2696. [Google Scholar] [CrossRef]
- Hamdy, N.A. Strontium ranelate improves bone microarchitecture in osteoporosis. Rheumatology (Oxford) 2009, 48 (Suppl. 4), iv9–iv13. [Google Scholar] [CrossRef]
- Henrotin, Y.; Labasse, A.; Zheng, S.X.; Galais, P.; Tsouderos, Y.; Crielaard, J.M.; Reginster, J.Y. Strontium ranelate increases cartilage matrix formation. J. Bone Miner. Res. 2001, 16, 299–308. [Google Scholar] [CrossRef]
- Reginster, J.Y.; Badurski, J.; Bellamy, N.; Bensen, W.; Chapurlat, R.; Chevalier, X.; Christiansen, C.; Genant, H.; Navarro, F.; Nasonov, E.; et al. Efficacy and safety of strontium ranelate in the treatment of knee osteoarthritis: Results of a double-blind, randomised placebo-controlled trial. Ann. Rheum. Dis. 2013, 72, 179–186. [Google Scholar] [CrossRef]
- Reginster, J.Y. Cardiac concerns associated with strontium ranelate. Expert Opin. Drug Saf. 2014, 13, 1209–1213. [Google Scholar] [CrossRef]
- Denk, F.; Bennett, D.L.; McMahon, S.B. Nerve Growth Factor and Pain Mechanisms. Ann. Rev. Neurosci. 2017, 40, 307–325. [Google Scholar] [CrossRef]
- Pecchi, E.; Priam, S.; Gosset, M.; Pigenet, A.; Sudre, L.; Laiguillon, M.C.; Berenbaum, F.; Houard, X. Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: Possible involvement in osteoarthritis pain. Arthritis Res. Ther. 2014, 16, R16. [Google Scholar] [CrossRef] [PubMed]
- Levi-Montalcini, R. The nerve growth factor 35 years later. Science 1987, 237, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Lane, N.E.; Schnitzer, T.J.; Birbara, C.A.; Mokhtarani, M.; Shelton, D.L.; Smith, M.D.; Brown, M.T. Tanezumab for the treatment of pain from osteoarthritis of the knee. N. Engl. J. Med. 2010, 363, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Schnitzer, T.J.; Ekman, E.F.; Spierings, E.L.; Greenberg, H.S.; Smith, M.D.; Brown, M.T.; West, C.R.; Verburg, K.M. Efficacy and safety of tanezumab monotherapy or combined with non-steroidal anti-inflammatory drugs in the treatment of knee or hip osteoarthritis pain. Ann. Rheum. Dis. 2015, 74, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Tiseo, P.J.; Kivitz, A.J.; Ervin, J.E.; Ren, H.; Mellis, S.J. Fasinumab (REGN475), an antibody against nerve growth factor for the treatment of pain: Results from a double-blind, placebo-controlled exploratory study in osteoarthritis of the knee. Pain 2014, 155, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.M.; Avenoso, A.; D’Ascola, A.; Scuruchi, M.; Prestipino, V.; Nastasi, G.; Calatroni, A.; Campo, S. Adenosine A2A receptor activation and hyaluronan fragment inhibition reduce inflammation in mouse articular chondrocytes stimulated with interleukin-1beta. FEBS J. 2012, 279, 2120–2133. [Google Scholar] [CrossRef] [PubMed]
- De Seny, D.; Cobraiville, G.; Charlier, E.; Neuville, S.; Esser, N.; Malaise, D.; Malaise, O.; Calvo, F.Q.; Relic, B.; Malaise, M.G. Acute-phase serum amyloid a in osteoarthritis: Regulatory mechanism and proinflammatory properties. PLoS ONE 2013, 8, e66769. [Google Scholar] [CrossRef]
- Juarranz, Y.; Gutierrez-Canas, I.; Arranz, A.; Martinez, C.; Abad, C.; Leceta, J.; Pablos, J.L.; Gomariz, R.P. VIP decreases TLR4 expression induced by LPS and TNF-alpha treatment in human synovial fibroblasts. Ann. N. Y. Acad. Sci. 2006, 1070, 359–364. [Google Scholar] [CrossRef]
- Iacono, A.; Gomez, R.; Sperry, J.; Conde, J.; Bianco, G.; Meli, R.; Gomez-Reino, J.J.; Smith, A.B., 3rd; Gualillo, O. Effect of oleocanthal and its derivatives on inflammatory response induced by lipopolysaccharide in a murine chondrocyte cell line. Arthritis Rheum. 2010, 62, 1675–1682. [Google Scholar] [CrossRef]
- Ahn, S.I.; Lee, J.K.; Youn, H.S. Inhibition of homodimerization of toll-like receptor 4 by 6-shogaol. Mol. Cells 2009, 27, 211–215. [Google Scholar] [CrossRef]
- Baltimore, D.; Boldin, M.P.; O’Connell, R.M.; Rao, D.S.; Taganov, K.D. MicroRNAs: New regulators of immune cell development and function. Nat. Immunol. 2008, 9, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Nugent, M. MicroRNAs: Exploring new horizons in osteoarthritis. Osteoarthritis Cartilage 2016, 24, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Mirzamohammadi, F.; Papaioannou, G.; Kobayashi, T. MicroRNAs in cartilage development, homeostasis, and disease. Curr. Osteoporos. Rep. 2014, 12, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Nakasa, T.; Otsuki, S.; Grogan, S.P.; Higashiyama, R.; Inoue, A.; Kato, Y.; Sato, T.; Lotz, M.K.; Asahara, H. MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum. 2009, 60, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Malizos, K.N.; Oikonomou, P.; Tsezou, A. Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS ONE 2008, 3, e3740. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.C.; Gao, J.Q. Exosomes as novel bio-carriers for gene and drug delivery. Int. J. Pharm. 2017, 521, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Nakasa, T.; Mochizuki, Y.; Ishikawa, M.; Miyaki, S.; Shibuya, H.; Yamasaki, K.; Adachi, N.; Asahara, H.; Ochi, M. Induction of apoptosis in the synovium of mice with autoantibody-mediated arthritis by the intraarticular injection of double-stranded MicroRNA-15a. Arthritis Rheum. 2009, 60, 2677–2683. [Google Scholar] [CrossRef] [PubMed]
- Brittberg, M.; Lindahl, A.; Nilsson, A.; Ohlsson, C.; Isaksson, O.; Peterson, L. Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N. Engl. J. Med. 1994, 331, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Connock, M.; Pink, J.; Shyangdan, D.; Clar, C.; Royle, P.; Court, R.; Biant, L.C.; Metcalfe, A.; Waugh, N. Autologous chondrocyte implantation in the knee: Systematic review and economic evaluation. Health Technol. Assess. 2017, 21, 1–294. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Markovic, B.S.; Fellabaum, C.; Arsenijevic, A.; Volarevic, V. Mesenchymal stem cell-based therapy of osteoarthritis: Current knowledge and future perspectives. Biomed. Pharmacother. 2019, 109, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Kristjansson, B.; Honsawek, S. Mesenchymal stem cells for cartilage regeneration in osteoarthritis. World J. Orthop. 2017, 8, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Okumachi, E.; Lee, S.Y.; Niikura, T.; Iwakura, T.; Dogaki, Y.; Waki, T.; Takahara, S.; Ueha, T.; Sakai, Y.; Kuroda, R.; et al. Comparative analysis of rat mesenchymal stem cells derived from slow and fast skeletal muscle in vitro. Int. Orthop. 2015, 39, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.; Zhang, W.; James, I.B.; Allbright, K.; Havis, E.; Bliley, J.M.; Almadori, A.; Schweizer, R.; Plock, J.A.; Washington, K.M.; et al. Characteristics and Immunomodulating Functions of Adipose-Derived and Bone Marrow-Derived Mesenchymal Stem Cells Across Defined Human Leukocyte Antigen Barriers. Front. Immunol. 2018, 9, 1642. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Dilisio, M.F.; Dietz, N.E.; Agrawal, D.K. Recent strategies in cartilage repair: A systemic review of the scaffold development and tissue engineering. J. Biomed. Mater. Res. A 2017, 105, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.G.; Kwon, O.R.; Kim, Y.S.; Choi, Y.J.; Tak, D.H. Adipose-Derived Mesenchymal Stem Cells With Microfracture Versus Microfracture Alone: 2-Year Follow-up of a Prospective Randomized Trial. Arthroscopy 2016, 32, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Vangsness, C.T., Jr.; Farr, J., 2nd; Boyd, J.; Dellaero, D.T.; Mills, C.R.; LeRoux-Williams, M. Adult human mesenchymal stem cells delivered via intra-articular injection to the knee following partial medial meniscectomy: A randomized, double-blind, controlled study. J. Bone Joint Surg. Am. 2014, 96, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Audia, S.; Janikashvili, N.; Ciudad, M.; Trad, M.; Fraszczak, J.; Ornetti, P.; Maillefert, J.F.; Miossec, P.; Bonnotte, B. Brief report: Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2499–2503. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.; Huizinga, T.W.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.; de Vries, R.R. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Silman, A.J.; MacGregor, A.J.; Thomson, W.; Holligan, S.; Carthy, D.; Farhan, A.; Ollier, W.E. Twin concordance rates for rheumatoid arthritis: Results from a nationwide study. Br. J. Rheumatol. 1993, 32, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Kapitany, A.; Zilahi, E.; Szanto, S.; Szucs, G.; Szabo, Z.; Vegvari, A.; Rass, P.; Sipka, S.; Szegedi, G.; Szekanecz, Z. Association of rheumatoid arthritis with HLA-DR1 and HLA-DR4 in Hungary. Ann. N. Y. Acad. Sci. 2005, 1051, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Kawaguchi, T.; Stahl, E.A.; Kurreeman, F.A.; Nishida, N.; et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 2012, 44, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 2013, 92, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Gravallese, E.M.; Harada, Y.; Wang, J.T.; Gorn, A.H.; Thornhill, T.S.; Goldring, S.R. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am. Journal Pathol. 1998, 152, 943–951. [Google Scholar]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Zwerina, J. Positive regulators of osteoclastogenesis and bone resorption in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, 235. [Google Scholar] [CrossRef] [PubMed]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Feige, U.; Sarosi, I.; Bolon, B.; Tafuri, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999, 402, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W., Jr.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Takahashi, N.; Udagawa, N.; Tamura, T.; Akatsu, T.; Stanley, E.R.; Kurokawa, T.; Suda, T. Macrophage colony-stimulating factor is indispensable for both proliferation and differentiation of osteoclast progenitors. J. Clin. Investig. 1993, 91, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Redlich, K.; Hayer, S.; Ricci, R.; David, J.P.; Tohidast-Akrad, M.; Kollias, G.; Steiner, G.; Smolen, J.S.; Wagner, E.F.; Schett, G. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J. Clin. Investig. 2002, 110, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Tsuzuki Wada, T.; Aizaki, Y.; Sato, K.; Yokota, K.; Fujimoto, K.; Kim, Y.T.; Oda, H.; Kurokawa, R.; Mimura, T. Histone Methylation and STAT-3 Differentially Regulate Interleukin-6-Induced Matrix Metalloproteinase Gene Activation in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol. 2016, 68, 1111–1123. [Google Scholar]
- Wong, B.R.; Josien, R.; Lee, S.Y.; Vologodskaia, M.; Steinman, R.M.; Choi, Y. The TRAF family of signal transducers mediates NF-kappaB activation by the TRANCE receptor. J. Biol. Chem. 1998, 273, 28355–28359. [Google Scholar] [CrossRef]
- Lomaga, M.A.; Yeh, W.C.; Sarosi, I.; Duncan, G.S.; Furlonger, C.; Ho, A.; Morony, S.; Capparelli, C.; Van, G.; Kaufman, S.; et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13, 1015–1024. [Google Scholar] [CrossRef]
- Wagner, E.F.; Eferl, R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005, 208, 126–140. [Google Scholar] [CrossRef]
- Kobayashi, N.; Kadono, Y.; Naito, A.; Matsumoto, K.; Yamamoto, T.; Tanaka, S.; Inoue, J. Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. 2001, 20, 1271–1280. [Google Scholar] [CrossRef]
- Nakashima, T.; Hayashi, M.; Takayanagi, H. New insights into osteoclastogenic signaling mechanisms. Trends Endocrinol. Metab. 2012, 23, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Inui, M.; Inoue, K.; Kim, S.; Suematsu, A.; Kobayashi, E.; Iwata, T.; Ohnishi, H.; Matozaki, T.; Kodama, T.; et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 2004, 428, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Koga, T.; Okamoto, K.; Sakaguchi, S.; Arai, K.; Yasuda, H.; Takai, T.; Kodama, T.; Morio, T.; Geha, R.S.; et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008, 132, 794–806. [Google Scholar] [CrossRef]
- Chen, S.J.; Lin, G.J.; Chen, J.W.; Wang, K.C.; Tien, C.H.; Hu, C.F.; Chang, C.N.; Hsu, W.F.; Fan, H.C.; Sytwu, H.K. Immunopathogenic Mechanisms and Novel Immune-Modulated Therapies in Rheumatoid Arthritis. Int. J. Mol. Sci. 2019, 20, E1332. [Google Scholar] [CrossRef] [PubMed]
- Kaito, T.; Hosono, N.; Ohshima, S.; Ohwaki, H.; Takenaka, S.; Fujiwara, H.; Makino, T.; Yonenobu, K. Effect of biological agents on cervical spine lesions in rheumatoid arthritis. Spine 2012, 37, 1742–1746. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewe, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. Int. J. Mol. Sci. 2018, 19, E1442. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Billmeier, U.; Dieterich, W.; Neurath, M.F.; Atreya, R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J. Gastroenterol. 2016, 22, 9300–9313. [Google Scholar] [CrossRef] [PubMed]
- Sarzi-Puttini, P.; Atzeni, F.; Shoenfeld, Y.; Ferraccioli, G. TNF-alpha, rheumatoid arthritis, and heart failure: A rheumatological dilemma. Autoimmun. Rev. 2005, 4, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Kroesen, S.; Widmer, A.F.; Tyndall, A.; Hasler, P. Serious bacterial infections in patients with rheumatoid arthritis under anti-TNF-alpha therapy. Rheumatology (Oxford) 2003, 42, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Akira, S.; Taga, T. Interleukin-6 and its receptor: A paradigm for cytokines. Science 1992, 258, 593–597. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Ogata, A.; Kishimoto, T. A new era for the treatment of inflammatory autoimmune diseases by interleukin-6 blockade strategy. Semin. Immunol. 2014, 26, 88–96. [Google Scholar] [CrossRef]
- Hibi, M.; Murakami, M.; Saito, M.; Hirano, T.; Taga, T.; Kishimoto, T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell 1990, 63, 1149–1157. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, T.W.; Fleischmann, R.M.; Jasson, M.; Radin, A.R.; van Adelsberg, J.; Fiore, S.; Huang, X.; Yancopoulos, G.D.; Stahl, N.; Genovese, M.C. Sarilumab, a fully human monoclonal antibody against IL-6Ralpha in patients with rheumatoid arthritis and an inadequate response to methotrexate: Efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann. Rheum. Dis. 2014, 73, 1626–1634. [Google Scholar] [CrossRef]
- Gabay, C.; Msihid, J.; Zilberstein, M.; Paccard, C.; Lin, Y.; Graham, N.M.H.; Boyapati, A. Identification of sarilumab pharmacodynamic and predictive markers in patients with inadequate response to TNF inhibition: A biomarker substudy of the phase 3 TARGET study. RMD Open 2018, 4, e000607. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef]
- Yamaoka, K.; Saharinen, P.; Pesu, M.; Holt, V.E., 3rd; Silvennoinen, O.; O’Shea, J.J. The Janus kinases (Jaks). Genome Biol. 2004, 5, 253. [Google Scholar] [CrossRef]
- Fridman, J.S.; Scherle, P.A.; Collins, R.; Burn, T.C.; Li, Y.; Li, J.; Covington, M.B.; Thomas, B.; Collier, P.; Favata, M.F.; et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: Preclinical characterization of INCB028050. J. Immunol. 2010, 184, 5298–5307. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.C.; Taylor, P.C.; Drescher, E.; Schlichting, D.E.; Beattie, S.D.; Berclaz, P.Y.; Lee, C.H.; Fidelus-Gort, R.K.; Luchi, M.E.; Rooney, T.P.; et al. Safety and efficacy of baricitinib at 24 weeks in patients with rheumatoid arthritis who have had an inadequate response to methotrexate. Ann. Rheum. Dis. 2015, 74, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Dougados, M.; van der Heijde, D.; Chen, Y.C.; Greenwald, M.; Drescher, E.; Liu, J.; Beattie, S.; Witt, S.; de la Torre, I.; Gaich, C.; et al. Baricitinib in patients with inadequate response or intolerance to conventional synthetic DMARDs: Results from the RA-BUILD study. Ann. Rheum. Dis. 2017, 76, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Kremer, J.M.; Gaich, C.L.; DeLozier, A.M.; Schlichting, D.E.; Xie, L.; Stoykov, I.; Rooney, T.; Bird, P.; Sanchez Burson, J.M.; et al. Patient-reported outcomes from a randomised phase III study of baricitinib in patients with rheumatoid arthritis and an inadequate response to biological agents (RA-BEACON). Ann. Rheum. Dis. 2017, 76, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Keystone, E.C.; Taylor, P.C.; Tanaka, Y.; Gaich, C.; DeLozier, A.M.; Dudek, A.; Zamora, J.V.; Cobos, J.A.C.; Rooney, T.; Bono, S.; et al. Patient-reported outcomes from a phase 3 study of baricitinib versus placebo or adalimumab in rheumatoid arthritis: Secondary analyses from the RA-BEAM study. Ann. Rheum. Dis. 2017, 76, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Kaine, J.L. Abatacept for the treatment of rheumatoid arthritis: A review. Curr. Ther. Res. Clin. Exp. 2007, 68, 379–399. [Google Scholar] [CrossRef]
- Kremer, J.M.; Peterfy, C.; Russell, A.S.; Emery, P.; Abud-Mendoza, C.; Sibilia, J.; Becker, J.C.; Westhovens, R.; Genant, H.K. Longterm safety, efficacy, and inhibition of structural damage progression over 5 years of treatment with abatacept in patients with rheumatoid arthritis in the abatacept in inadequate responders to methotrexate trial. J. Rheumatol. 2014, 41, 1077–1087. [Google Scholar] [CrossRef][Green Version]
- Fukuyo, S.; Nakayamada, S.; Iwata, S.; Kubo, S.; Saito, K.; Tanaka, Y. Abatacept therapy reduces CD28+CXCR5+ follicular helper-like T cells in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2017, 35, 562–570. [Google Scholar]
- Takata, M.; Kurosaki, T. A role for Bruton‘s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J. Exp. Med. 1996, 184, 31–40. [Google Scholar] [CrossRef]
- De Weers, M.; Verschuren, M.C.; Kraakman, M.E.; Mensink, R.G.; Schuurman, R.K.; van Dongen, J.J.; Hendriks, R.W. The Bruton’s tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur. J. Immunol. 1993, 23, 3109–3114. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, T.; Jeong, D.; Kim, N.; Choi, Y. The tec family tyrosine kinase Btk Regulates RANKL-induced osteoclast maturation. J. Biol. Chem. 2008, 283, 11526–11534. [Google Scholar] [CrossRef]
- Park, J.K.; Byun, J.Y.; Park, J.A.; Kim, Y.Y.; Lee, Y.J.; Oh, J.I.; Jang, S.Y.; Kim, Y.H.; Song, Y.W.; Son, J.; et al. HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: A potential drug for rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 91. [Google Scholar] [CrossRef]
- Kurosaki, T.; Takata, M.; Yamanashi, Y.; Inazu, T.; Taniguchi, T.; Yamamoto, T.; Yamamura, H. Syk activation by the Src-family tyrosine kinase in the B cell receptor signaling. J. Exp. Med. 1994, 179, 1725–1729. [Google Scholar] [CrossRef]
- Deng, G.M.; Kyttaris, V.C.; Tsokos, G.C. Targeting Syk in Autoimmune Rheumatic Diseases. Front. Immunol. 2016, 7, 78. [Google Scholar] [CrossRef]
- Pine, P.R.; Chang, B.; Schoettler, N.; Banquerigo, M.L.; Wang, S.; Lau, A.; Zhao, F.; Grossbard, E.B.; Payan, D.G.; Brahn, E. Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clin. Immunol. 2007, 124, 244–257. [Google Scholar] [CrossRef]
- Genovese, M.C.; Kavanaugh, A.; Weinblatt, M.E.; Peterfy, C.; DiCarlo, J.; White, M.L.; O’Brien, M.; Grossbard, E.B.; Magilavy, D.B. An oral Syk kinase inhibitor in the treatment of rheumatoid arthritis: A three-month randomized, placebo-controlled, phase II study in patients with active rheumatoid arthritis that did not respond to biologic agents. Arthritis Rheum. 2011, 63, 337–345. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602. [Google Scholar] [CrossRef]
- Zhang, H.G.; Wang, Y.; Xie, J.F.; Liang, X.; Liu, D.; Yang, P.; Hsu, H.C.; Ray, R.B.; Mountz, J.D. Regulation of tumor necrosis factor alpha-mediated apoptosis of rheumatoid arthritis synovial fibroblasts by the protein kinase Akt. Arthritis Rheum. 2001, 44, 1555–1567. [Google Scholar] [CrossRef]
- Toyama, S.; Tamura, N.; Haruta, K.; Karakida, T.; Mori, S.; Watanabe, T.; Yamori, T.; Takasaki, Y. Inhibitory effects of ZSTK474, a novel phosphoinositide 3-kinase inhibitor, on osteoclasts and collagen-induced arthritis in mice. Arthritis Res. Ther. 2010, 12, R92. [Google Scholar] [CrossRef]
- Chen, X.M.; Huang, Q.C.; Yang, S.L.; Chu, Y.L.; Yan, Y.H.; Han, L.; Huang, Y.; Huang, R.Y. Role of Micro RNAs in the Pathogenesis of Rheumatoid Arthritis: Novel Perspectives Based on Review of the Literature. Medicine 2015, 94, e1326. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Bluml, S.; Bonelli, M.; Niederreiter, B.; Puchner, A.; Mayr, G.; Hayer, S.; Koenders, M.I.; van den Berg, W.B.; Smolen, J.; Redlich, K. Essential role of microRNA-155 in the pathogenesis of autoimmune arthritis in mice. Arthritis Rheum. 2011, 63, 1281–1288. [Google Scholar] [CrossRef]
- Stanczyk, J.; Pedrioli, D.M.; Brentano, F.; Sanchez-Pernaute, O.; Kolling, C.; Gay, R.E.; Detmar, M.; Gay, S.; Kyburz, D. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008, 58, 1001–1009. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Alivernini, S.; Ballantine, L.E.; Asquith, D.L.; Millar, N.L.; Gilchrist, D.S.; Reilly, J.; Ierna, M.; Fraser, A.R.; Stolarski, B.; et al. MicroRNA-155 as a proinflammatory regulator in clinical and experimental arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 11193–11198. [Google Scholar] [CrossRef]
- Li, X.; Tian, F.; Wang, F. Rheumatoid arthritis-associated microRNA-155 targets SOCS1 and upregulates TNF-alpha and IL-1beta in PBMCs. Int. J. Mol. Sci. 2013, 14, 23910–23921. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Korchynskyi, O.; Tak, P.P.; Reedquist, K.A. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann. Rheum. Dis. 2012, 71, 424–431. [Google Scholar] [CrossRef]
- Vojinovic, J.; Damjanov, N.; D’Urzo, C.; Furlan, A.; Susic, G.; Pasic, S.; Iagaru, N.; Stefan, M.; Dinarello, C.A. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011, 63, 1452–1458. [Google Scholar] [CrossRef]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; Van Baarsen, I.M.; Ferguson, B.S.; Garcia, S.; Malvar Fernandez, B.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef]
- Kane, D.; Stafford, L.; Bresnihan, B.; FitzGerald, O. A prospective, clinical and radiological study of early psoriatic arthritis: An early synovitis clinic experience. Rheumatology (Oxford) 2003, 42, 1460–1468. [Google Scholar] [CrossRef]
- Goldring, S.R. Differential mechanisms of de-regulated bone formation in rheumatoid arthritis and spondyloarthritis. Rheumatology (Oxford) 2016, 55 (Suppl. 2), ii56–ii60. [Google Scholar] [CrossRef]
- Raychaudhuri, S.P.; Raychaudhuri, S.K. IL-23/IL-17 axis in spondyloarthritis-bench to bedside. Clin. Rheumatol. 2016, 35, 1437–1441. [Google Scholar] [CrossRef]
- Ritchlin, C.T.; Krueger, J.G. New therapies for psoriasis and psoriatic arthritis. Curr. Opin. Rheumatol. 2016, 28, 204–210. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Zhou, L.; Littman, D.R. Transcriptional regulation of Th17 cell differentiation. Semin. Immunol. 2007, 19, 409–417. [Google Scholar] [CrossRef]
- Iwakura, Y.; Nakae, S.; Saijo, S.; Ishigame, H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol. Rev. 2008, 226, 57–79. [Google Scholar] [CrossRef]
- Palmer, M.T.; Weaver, C.T. Immunology: Narcissistic helpers. Nature 2007, 448, 416–418. [Google Scholar] [CrossRef]
- Benham, H.; Norris, P.; Goodall, J.; Wechalekar, M.D.; FitzGerald, O.; Szentpetery, A.; Smith, M.; Thomas, R.; Gaston, H. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res. Ther. 2013, 15, R136. [Google Scholar] [CrossRef]
- Kirkham, B.W.; Kavanaugh, A.; Reich, K. Interleukin-17A: A unique pathway in immune-mediated diseases: Psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology 2014, 141, 133–142. [Google Scholar] [CrossRef]
- Pacifici, R. The Role of IL-17 and TH17 Cells in the Bone Catabolic Activity of PTH. Front. Immunol. 2016, 7, 57. [Google Scholar] [CrossRef]
- Stuart, P.E.; Nair, R.P.; Tsoi, L.C.; Tejasvi, T.; Das, S.; Kang, H.M.; Ellinghaus, E.; Chandran, V.; Callis-Duffin, K.; Ike, R.; et al. Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. American J. Hum. Genet. 2015, 97, 816–836. [Google Scholar] [CrossRef]
- Myers, A.; Kay, L.J.; Lynch, S.A.; Walker, D.J. Recurrence risk for psoriasis and psoriatic arthritis within sibships. Rheumatology (Oxford) 2005, 44, 773–776. [Google Scholar] [CrossRef]
- FitzGerald, O.; Haroon, M.; Giles, J.T.; Winchester, R. Concepts of pathogenesis in psoriatic arthritis: Genotype determines clinical phenotype. Arthritis Res. Ther. 2015, 17, 115. [Google Scholar] [CrossRef]
- O’Rielly, D.D.; Rahman, P. Genetics of psoriatic arthritis. Best Pract. Res. Clin. Rheumatol. 2014, 28, 673–685. [Google Scholar] [CrossRef]
- Griffiths, C.E.; Barker, J.N. Pathogenesis and clinical features of psoriasis. Lancet 2007, 370, 263–271. [Google Scholar] [CrossRef]
- Thorarensen, S.M.; Lu, N.; Ogdie, A.; Gelfand, J.M.; Choi, H.K.; Love, T.J. Physical trauma recorded in primary care is associated with the onset of psoriatic arthritis among patients with psoriasis. Ann. Rheum. Dis. 2017, 76, 521–525. [Google Scholar] [CrossRef]
- Sherlock, J.P.; Joyce-Shaikh, B.; Turner, S.P.; Chao, C.C.; Sathe, M.; Grein, J.; Gorman, D.M.; Bowman, E.P.; McClanahan, T.K.; Yearley, J.H.; et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat. Med. 2012, 18, 1069–1076. [Google Scholar] [CrossRef]
- Lories, R.J.; McInnes, I.B. Primed for inflammation: Enthesis-resident T cells. Nat. Med. 2012, 18, 1018–1019. [Google Scholar] [CrossRef]
- Van Baarsen, L.G.; Lebre, M.C.; van der Coelen, D.; Aarrass, S.; Tang, M.W.; Ramwadhdoebe, T.H.; Gerlag, D.M.; Tak, P.P. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: Possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res. Ther. 2014, 16, 426. [Google Scholar] [CrossRef]
- Celis, R.; Planell, N.; Fernandez-Sueiro, J.L.; Sanmarti, R.; Ramirez, J.; Gonzalez-Alvaro, I.; Pablos, J.L.; Canete, J.D. Synovial cytokine expression in psoriatic arthritis and associations with lymphoid neogenesis and clinical features. Arthritis Res. Ther. 2012, 14, R93. [Google Scholar] [CrossRef]
- Adamopoulos, I.E.; Suzuki, E.; Chao, C.C.; Gorman, D.; Adda, S.; Maverakis, E.; Zarbalis, K.; Geissler, R.; Asio, A.; Blumenschein, W.M.; et al. IL-17A gene transfer induces bone loss and epidermal hyperplasia associated with psoriatic arthritis. Ann. Rheum. Dis. 2015, 74, 1284–1292. [Google Scholar] [CrossRef]
- Shaw, A.T.; Maeda, Y.; Gravallese, E.M. IL-17A deficiency promotes periosteal bone formation in a model of inflammatory arthritis. Arthritis Res. Ther. 2016, 18, 104. [Google Scholar] [CrossRef]
- El-Zayadi, A.A.; Jones, E.A.; Churchman, S.M.; Baboolal, T.G.; Cuthbert, R.J.; El-Jawhari, J.J.; Badawy, A.M.; Alase, A.A.; El-Sherbiny, Y.M.; McGonagle, D. Interleukin-22 drives the proliferation, migration and osteogenic differentiation of mesenchymal stem cells: A novel cytokine that could contribute to new bone formation in spondyloarthropathies. Rheumatology (Oxford) 2017, 56, 488–493. [Google Scholar] [CrossRef]
- Braun, J.; Bollow, M.; Neure, L.; Seipelt, E.; Seyrekbasan, F.; Herbst, H.; Eggens, U.; Distler, A.; Sieper, J. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum. 1995, 38, 499–505. [Google Scholar] [CrossRef]
- Lories, R.J.; Derese, I.; Ceuppens, J.L.; Luyten, F.P. Bone morphogenetic proteins 2 and 6, expressed in arthritic synovium, are regulated by proinflammatory cytokines and differentially modulate fibroblast-like synoviocyte apoptosis. Arthritis Rheum. 2003, 48, 2807–2818. [Google Scholar] [CrossRef]
- Lories, R.J.; Derese, I.; Luyten, F.P. Modulation of bone morphogenetic protein signaling inhibits the onset and progression of ankylosing enthesitis. J. Clin. Investig. 2005, 115, 1571–1579. [Google Scholar] [CrossRef]
- Gossec, L.; Smolen, J.S.; Ramiro, S.; de Wit, M.; Cutolo, M.; Dougados, M.; Emery, P.; Landewe, R.; Oliver, S.; Aletaha, D.; et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann. Rheum. Dis. 2016, 75, 499–510. [Google Scholar] [CrossRef]
- Coates, L.C.; Kavanaugh, A.; Mease, P.J.; Soriano, E.R.; Laura Acosta-Felquer, M.; Armstrong, A.W.; Bautista-Molano, W.; Boehncke, W.H.; Campbell, W.; Cauli, A.; et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis 2015 Treatment Recommendations for Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 1060–1071. [Google Scholar] [CrossRef]
- Glintborg, B.; Ostergaard, M.; Krogh, N.S.; Andersen, M.D.; Tarp, U.; Loft, A.G.; Lindegaard, H.M.; Holland-Fischer, M.; Nordin, H.; Jensen, D.V.; et al. Clinical response, drug survival, and predictors thereof among 548 patients with psoriatic arthritis who switched tumor necrosis factor alpha inhibitor therapy: Results from the Danish Nationwide DANBIO Registry. Arthritis Rheum. 2013, 65, 1213–1223. [Google Scholar] [CrossRef]
- McInnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y.; Mendelsohn, A.M.; et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 2013, 382, 780–789. [Google Scholar] [CrossRef]
- Ritchlin, C.; Rahman, P.; Kavanaugh, A.; McInnes, I.B.; Puig, L.; Li, S.; Wang, Y.; Shen, Y.K.; Doyle, M.K.; Mendelsohn, A.M.; et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the phase 3, multicentre, double-blind, placebo-controlled, randomised PSUMMIT 2 trial. Ann. Rheum. Dis. 2014, 73, 990–999. [Google Scholar]
- Deodhar, A.; Gottlieb, A.B.; Boehncke, W.H.; Dong, B.; Wang, Y.; Zhuang, Y.; Barchuk, W.; Xu, X.L.; Hsia, E.C. Efficacy and safety of guselkumab in patients with active psoriatic arthritis: A randomised, double-blind, placebo-controlled, phase 2 study. Lancet 2018, 391, 2213–2224. [Google Scholar] [CrossRef]
- Van der Heijde, D.; Landewe, R.B.; Mease, P.J.; McInnes, I.B.; Conaghan, P.G.; Pricop, L.; Ligozio, G.; Richards, H.B.; Mpofu, S. Brief Report: Secukinumab Provides Significant and Sustained Inhibition of Joint Structural Damage in a Phase III Study of Active Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 1914–1921. [Google Scholar] [CrossRef]
- Mease, P.J.; van der Heijde, D.; Ritchlin, C.T.; Okada, M.; Cuchacovich, R.S.; Shuler, C.L.; Lin, C.Y.; Braun, D.K.; Lee, C.H.; Gladman, D.D. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: Results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann. Rheum. Dis. 2017, 76, 79–87. [Google Scholar]
- Glatt, S.; Baeten, D.; Baker, T.; Griffiths, M.; Ionescu, L.; Lawson, A.D.G.; Maroof, A.; Oliver, R.; Popa, S.; Strimenopoulou, F.; et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: Evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann. Rheum. Dis. 2018, 77, 523–532. [Google Scholar] [CrossRef]
- Mease, P.J.; Genovese, M.C.; Greenwald, M.W.; Ritchlin, C.T.; Beaulieu, A.D.; Deodhar, A.; Newmark, R.; Feng, J.; Erondu, N.; Nirula, A. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N. Engl. J. Med. 2014, 370, 2295–2306. [Google Scholar] [CrossRef]
- Smolen, J.S.; Agarwal, S.K.; Ilivanova, E.; Xu, X.L.; Miao, Y.; Zhuang, Y.; Nnane, I.; Radziszewski, W.; Greenspan, A.; Beutler, A.; et al. A randomised phase II study evaluating the efficacy and safety of subcutaneously administered ustekinumab and guselkumab in patients with active rheumatoid arthritis despite treatment with methotrexate. Ann. Rheum. Dis. 2017, 76, 831–839. [Google Scholar] [CrossRef]
- Genovese, M.C.; Greenwald, M.; Cho, C.S.; Berman, A.; Jin, L.; Cameron, G.S.; Benichou, O.; Xie, L.; Braun, D.; Berclaz, P.Y.; et al. A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol. 2014, 66, 1693–1704. [Google Scholar] [CrossRef]
- Jacques, P.; Van den Bosch, F. Emerging therapies for rheumatoid arthritis. Expert Opin. Emerg. Drugs 2013, 18, 231–244. [Google Scholar] [CrossRef]
- Efficacy of Secukinumab Compared to Adalimumab in Patients With Psoriatic Arthritis (EXCEED 1). Available online: https://clinicaltrials.gov/ct2/show/NCT02745080 (accessed on 1 June 2019).
- Miyagawa, I.; Nakayamada, S.; Nakano, K.; Kubo, S.; Iwata, S.; Miyazaki, Y.; Yoshikawa, M.; Yoshinari, H.; Tanaka, Y. Precision medicine using different biological DMARDs based on characteristic phenotypes of peripheral T helper cells in psoriatic arthritis. Rheumatology (Oxford) 2019, 58, 336–344. [Google Scholar] [CrossRef]
- Mease, P.J.; Gottlieb, A.B.; Berman, A.; Drescher, E.; Xing, J.; Wong, R.; Banerjee, S. The Efficacy and Safety of Clazakizumab, an Anti-Interleukin-6 Monoclonal Antibody, in a Phase IIb Study of Adults With Active Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 2163–2173. [Google Scholar] [CrossRef]
- Mease, P.; Hall, S.; FitzGerald, O.; van der Heijde, D.; Merola, J.F.; Avila-Zapata, F.; Cieslak, D.; Graham, D.; Wang, C.; Menon, S.; et al. Tofacitinib or Adalimumab versus Placebo for Psoriatic Arthritis. N. Engl. J. Med. 2017, 377, 1537–1550. [Google Scholar] [CrossRef]
- Gladman, D.; Rigby, W.; Azevedo, V.F.; Behrens, F.; Blanco, R.; Kaszuba, A.; Kudlacz, E.; Wang, C.; Menon, S.; Hendrikx, T.; et al. Tofacitinib for Psoriatic Arthritis in Patients with an Inadequate Response to TNF Inhibitors. N. Engl. J. Med. 2017, 377, 1525–1536. [Google Scholar] [CrossRef]
- Mease, P.J.; Gottlieb, A.B.; van der Heijde, D.; FitzGerald, O.; Johnsen, A.; Nys, M.; Banerjee, S.; Gladman, D.D. Efficacy and safety of abatacept, a T-cell modulator, in a randomised, double-blind, placebo-controlled, phase III study in psoriatic arthritis. Ann. Rheum. Dis. 2017, 76, 1550–1558. [Google Scholar] [CrossRef]
- Elalouf, O.; Chandran, V. Novel Therapeutics in Psoriatic Arthritis. What Is in the Pipeline? Curr. Rheumatol. Rep. 2018, 20, 36. [Google Scholar] [CrossRef]
- Efficacy and Safety of BMS-986165 Compared With Placebo in Participants With Active Psoriatic Arthritis (PsA). Available online: https://clinicaltrials.gov/ct2/show/NCT03881059 (accessed on 1 June 2019).
- A Study to Evaluate the Efficacy and Safety of PF-06700841 in Subjects with Active Psoriatic Arthritis. Available online: https://clinicaltrials.gov/ct2/show/NCT03963401 (accessed on 1 June 2019).
- A Study to Evaluate the Safety, Mode of Action and Clinical Efficacy of GSK3050002 in Subjects with Psoriatic Arthritis. Available online: https://clinicaltrials.gov/ct2/show/NCT02671188 (accessed on 1 June 2019).
- Efficacy and Safety of Fecal Microbiota Transplantation in Peripheral Psoriatic Arthritis (FLORA). Available online: https://clinicaltrials.gov/ct2/show/NCT03058900 (accessed on 1 June 2019).
- Shaw, M.H.; Boyartchuk, V.; Wong, S.; Karaghiosoff, M.; Ragimbeau, J.; Pellegrini, S.; Muller, M.; Dietrich, W.F.; Yap, G.S. A natural mutation in the Tyk2 pseudokinase domain underlies altered susceptibility of B10.Q/J mice to infection and autoimmunity. Proc. Natl. Acad. Sci. USA 2003, 100, 11594–11599. [Google Scholar] [CrossRef]
- Papp, K.; Gordon, K.; Thaci, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Hieshima, K.; Imai, T.; Opdenakker, G.; Van Damme, J.; Kusuda, J.; Tei, H.; Sakaki, Y.; Takatsuki, K.; Miura, R.; Yoshie, O.; et al. Molecular cloning of a novel human CC chemokine liver and activation-regulated chemokine (LARC) expressed in liver. Chemotactic activity for lymphocytes and gene localization on chromosome 2. J. Biol. Chem. 1997, 272, 5846–5853. [Google Scholar] [CrossRef]
- Scher, J.U.; Littman, D.R.; Abramson, S.B. Microbiome in Inflammatory Arthritis and Human Rheumatic Diseases. Arthritis Rheumatol. 2016, 68, 35–45. [Google Scholar] [CrossRef]
- Tan, L.; Zhao, S.; Zhu, W.; Wu, L.; Li, J.; Shen, M.; Lei, L.; Chen, X.; Peng, C. The Akkermansia muciniphila is a gut microbiota signature in psoriasis. Exp. Dermatol. 2018, 27, 144–149. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism |
|---|---|
| Novel Therapeutic Agents for OA | |
| TNF-α inhibitors | Neutralize the biological activities of TNF-α by binding with soluble TNF-α and induce apoptosis, antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity in TNF-α-producing cells by binding to membrane-bound TNF-α |
| IL-1 inhibitors | Diacerein, Anakinra and AMG108 inhibit IL-1 pathways in inflammation respectively as an IL-1βinhibitor, an IL-1 receptor antagonist and an IL-1 receptor monoclonal antibody |
| Bisphosphonate | Suppresses the activity of osteoclasts, delay bone remodeling, and provides chondroprotective effects |
| Strontium ranelate | Induces cartilage formation through an ionic effect. Reduces osteoclastic bone resorption and simultaneously stimulate osteoblastic bone formation |
| Anti-NGF antibodies | Block nerve growth in intra-articular tissues and downregulate pain sensitivity |
| Therapies targeting TLR4 signaling | Inhibit TLR4 signaling by blocking TLR4 agonists, activating antagonist pathways and new inhibitory compounds |
| miR-140 | Protect cartilages by suppressing the expression of ADAMTS5 |
| Cell-based therapy | Chondrogenic potential and immunomodulatory properties of MSCs |
| Novel Therapeutic Agents for RA | |
| Btk inhibitors | Inhibit the differentiation and activation of osteoclasts by blocking the integration of RANK/RANKL signaling and ITAM signaling. Block the cytokine production and expression of co-stimulators via B cell receptors |
| Syk inhibitors | Block signal transduction for B-cell receptors, FcRγ, DAP12, and integrin with ITAM |
| PI3K inhibitors | Block activation of PI3K/Akt signaling pathway and suppress osteoclast formation and proliferation of B lymphocytes and synovial fibroblasts |
| miR-146 | Functions as a negative feedback that stops the inflammatory stimulation caused by TNF-α by targeting TRAF6 |
| miR-155 | Suppresses the production of inflammatory cytokines by targeting SHIP1 and SOCS1 |
| HDAC inhibitors | Promote the degradation of mRNA and regulate the generation of inflammatory cytokines in RA synovial fibroblasts and macrophage |
| Novel Therapeutic Agents for PsA | |
| JAK inhibitors | Block the JAK/STAT pathway that is the major signaling cascade for various pro-inflammatory cytokines |
| IL-6 inhibitors | Bind to both transmembrane IL-6 receptors and soluble IL-6 receptors to block IL-6 mediated signal transduction involving acute-phase response and activation of immune reaction |
| T cell activation inhibitors (Abatacept) | Inhibit the activity of T-cells that induce production of cytokines, autoantibodies, and inflammatory proteins |
| Tyk2 inhibitors | Block signaling downstream of the receptors for IL-12, IL-23, and type I and III interferons |
| CCL20 inhibitors | Bind to CCL20 and inhibit the movement of inflammatory cells into inflamed tissues |
| microbiota transplantation | Keeps gut homeostasis and regulate the activation of the inflammatory pathways |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tateiwa, D.; Yoshikawa, H.; Kaito, T. Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review. Cells 2019, 8, 818. https://doi.org/10.3390/cells8080818
Tateiwa D, Yoshikawa H, Kaito T. Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review. Cells. 2019; 8(8):818. https://doi.org/10.3390/cells8080818
Chicago/Turabian StyleTateiwa, Daisuke, Hideki Yoshikawa, and Takashi Kaito. 2019. "Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review" Cells 8, no. 8: 818. https://doi.org/10.3390/cells8080818
APA StyleTateiwa, D., Yoshikawa, H., & Kaito, T. (2019). Cartilage and Bone Destruction in Arthritis: Pathogenesis and Treatment Strategy: A Literature Review. Cells, 8(8), 818. https://doi.org/10.3390/cells8080818