Differential Inhibition of Target Gene Expression by Human microRNAs

Abstract
:1. Introduction
2. Material and Methods
2.1. Target Selection
2.2. Cell Cultures
2.3. Molecular Cloning
2.4. Transfection and Reporter Assay
2.5. Bioinformatics and Statistical Analyses
2.6. miRNA Overexpression and RNA-seq
3. Results
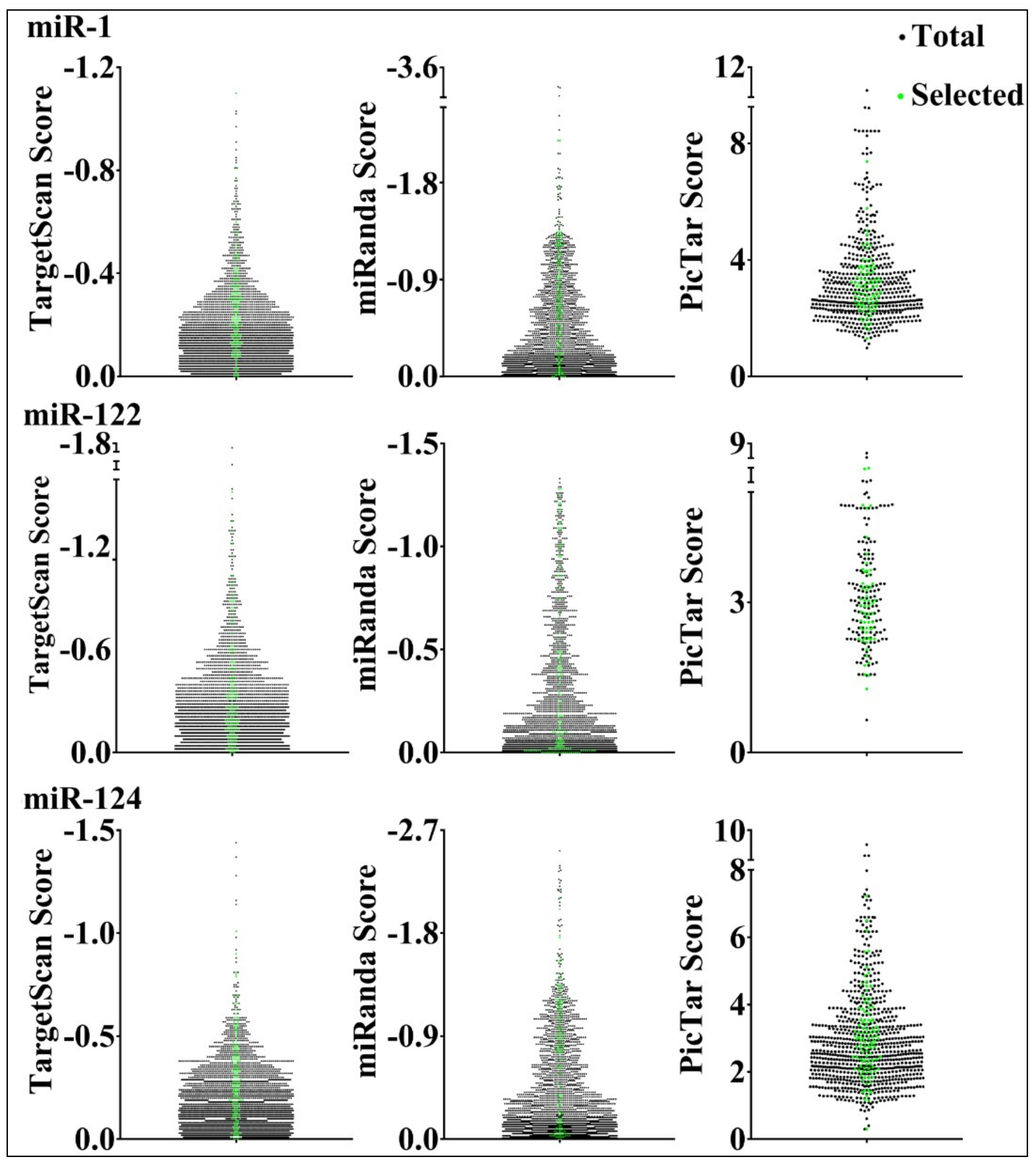
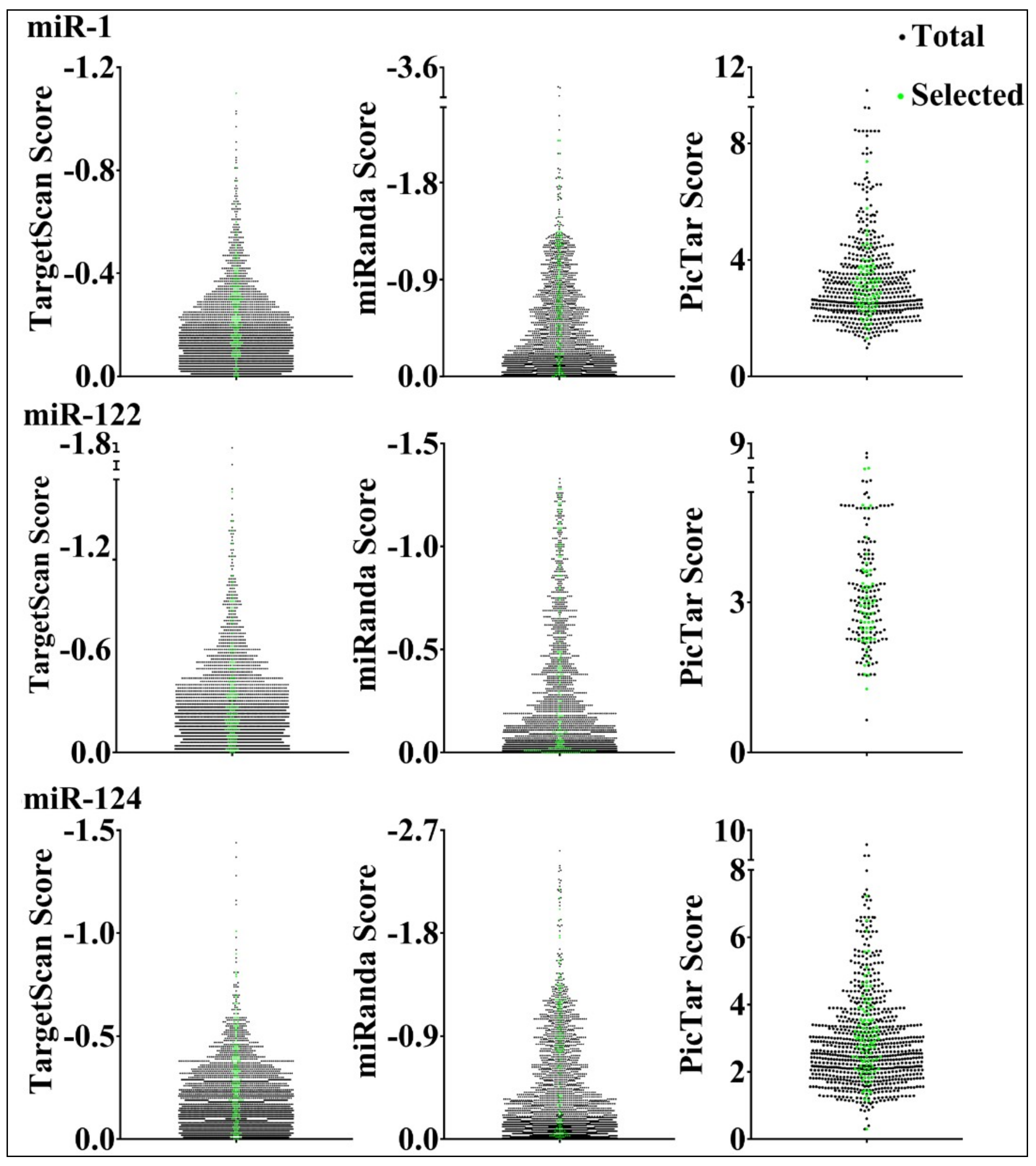
3.1. Construction of Target Reporter Gene Libraries
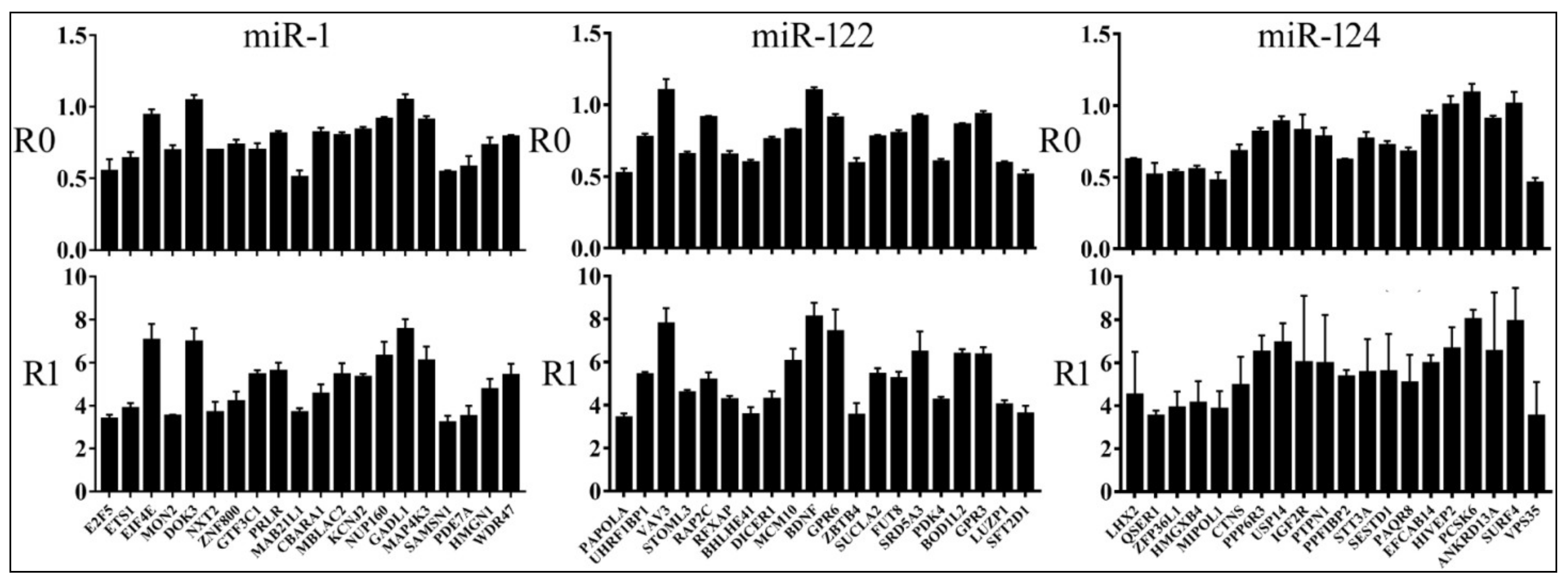
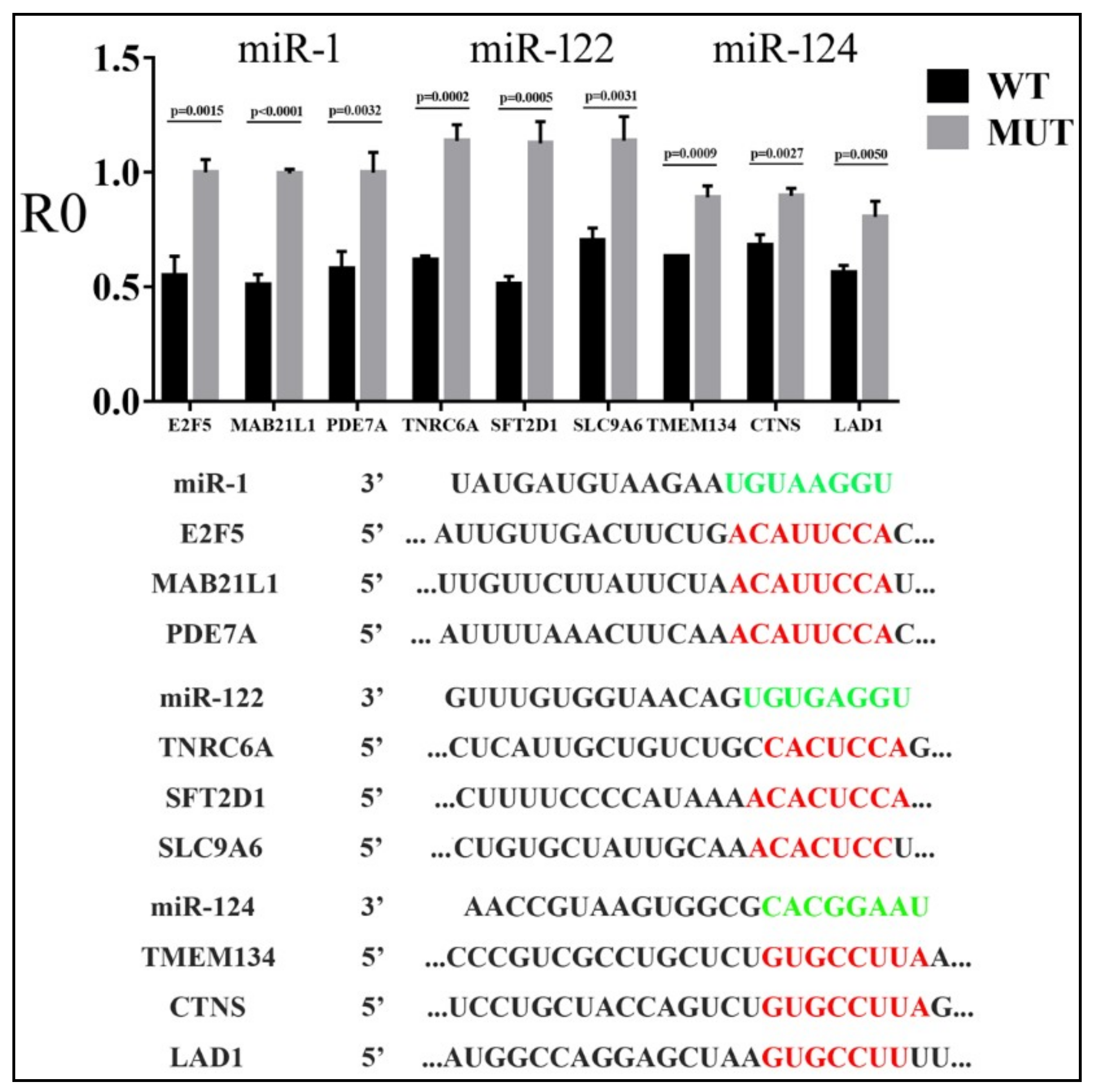
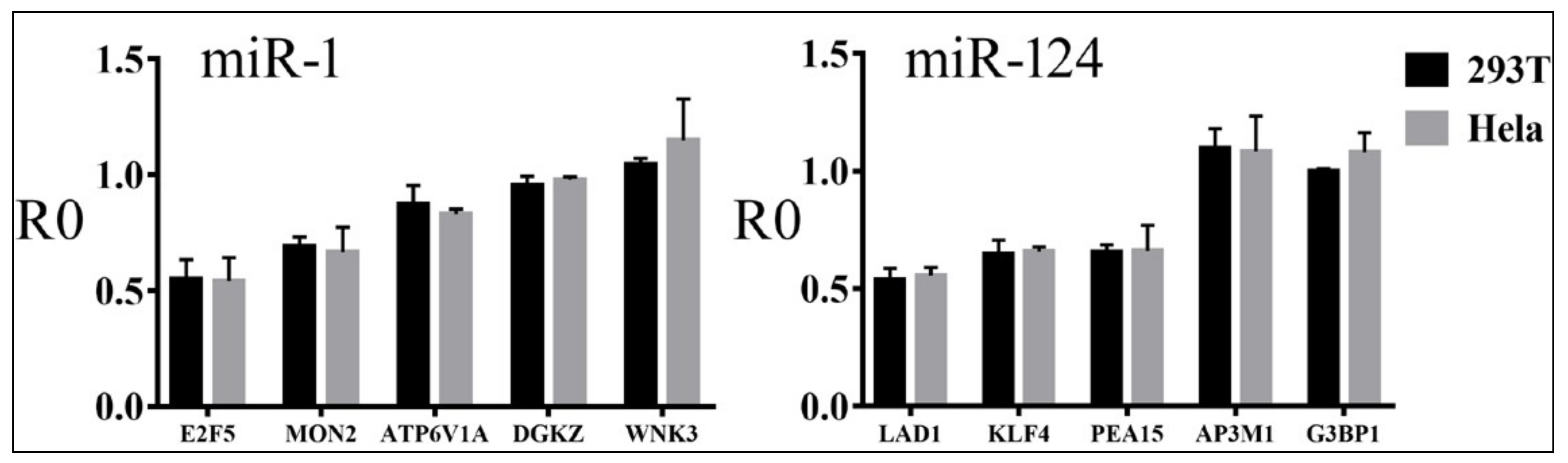
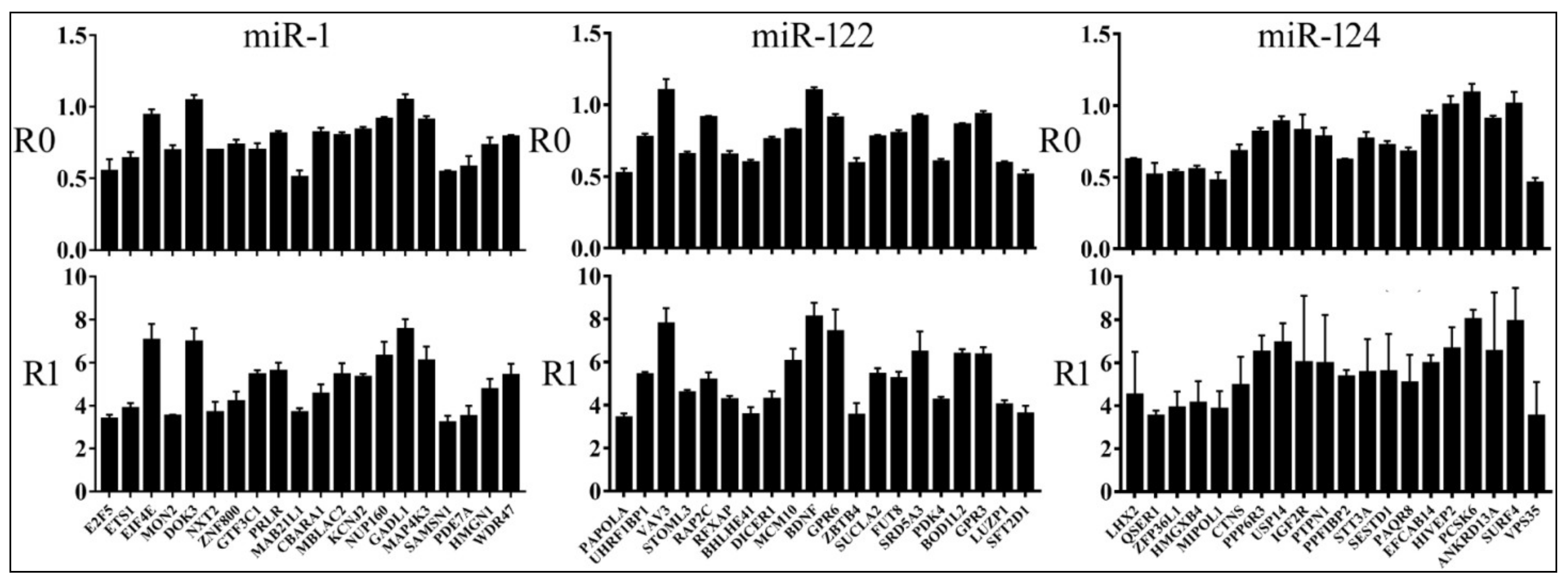
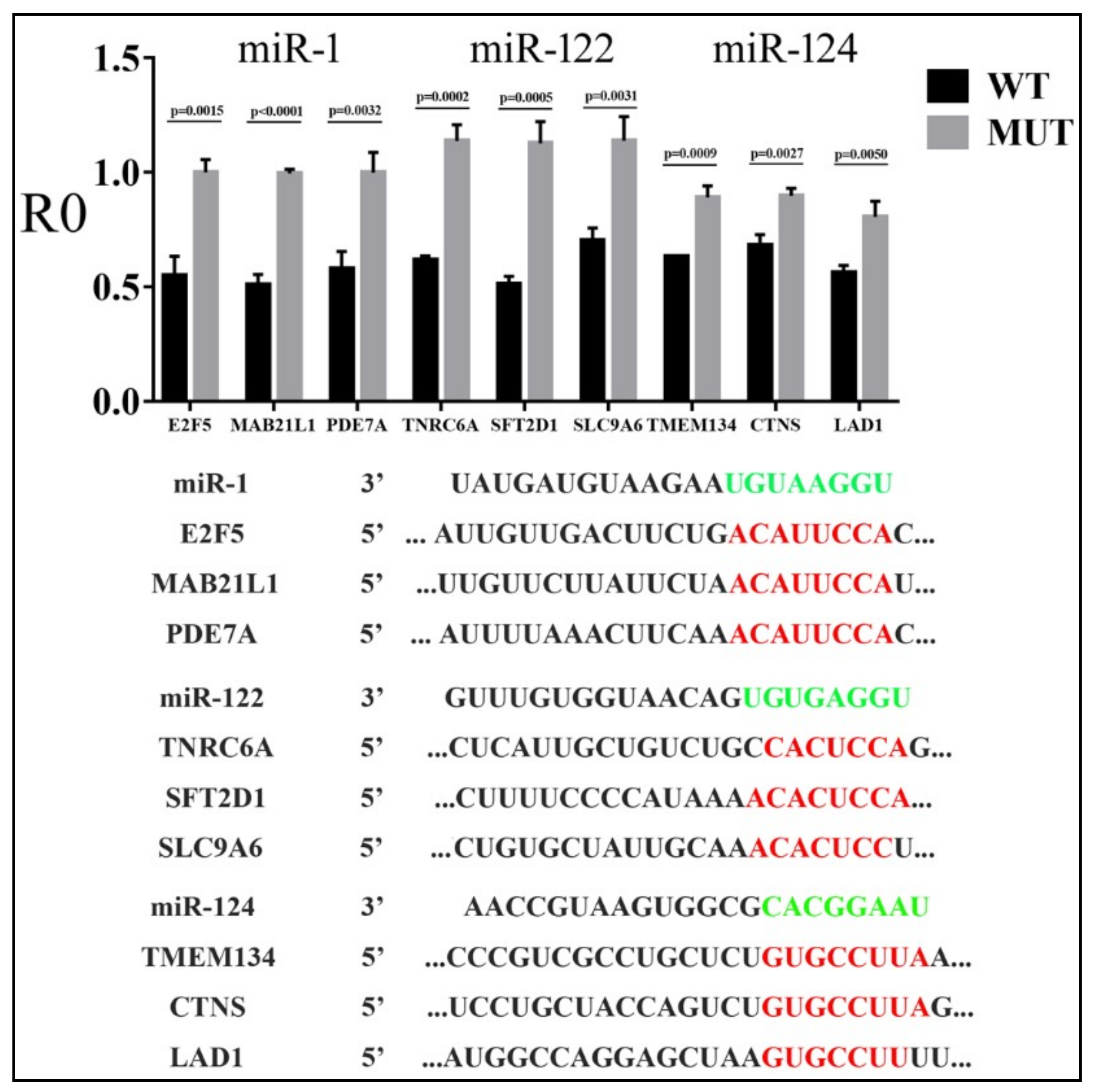
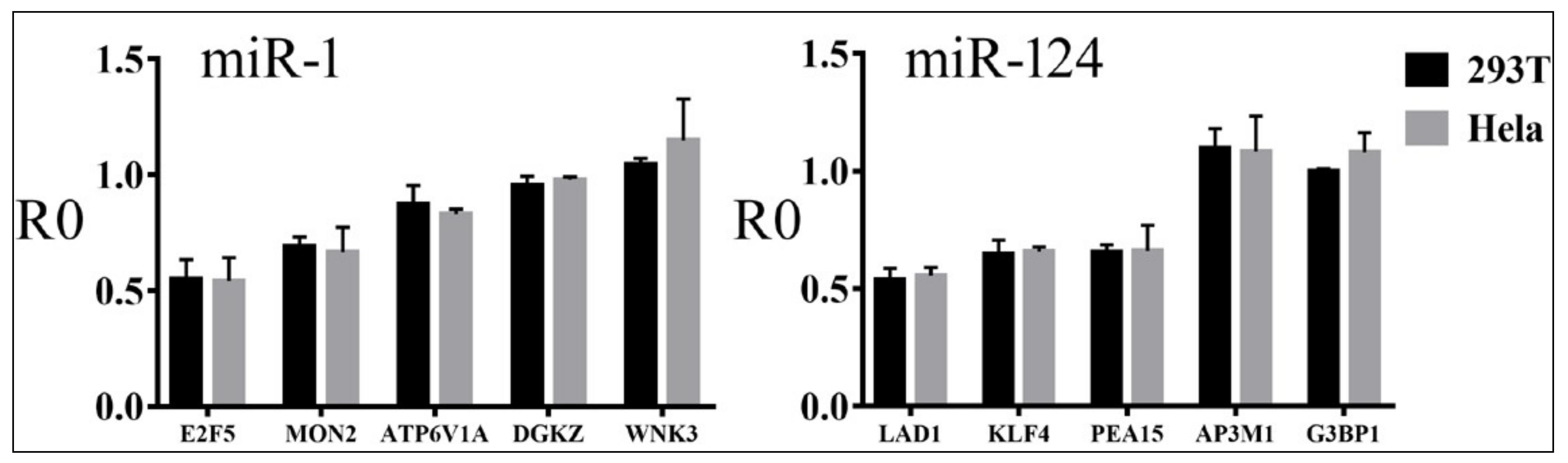
3.2. Reporter Assays to Measure Target Repression by miRNAs
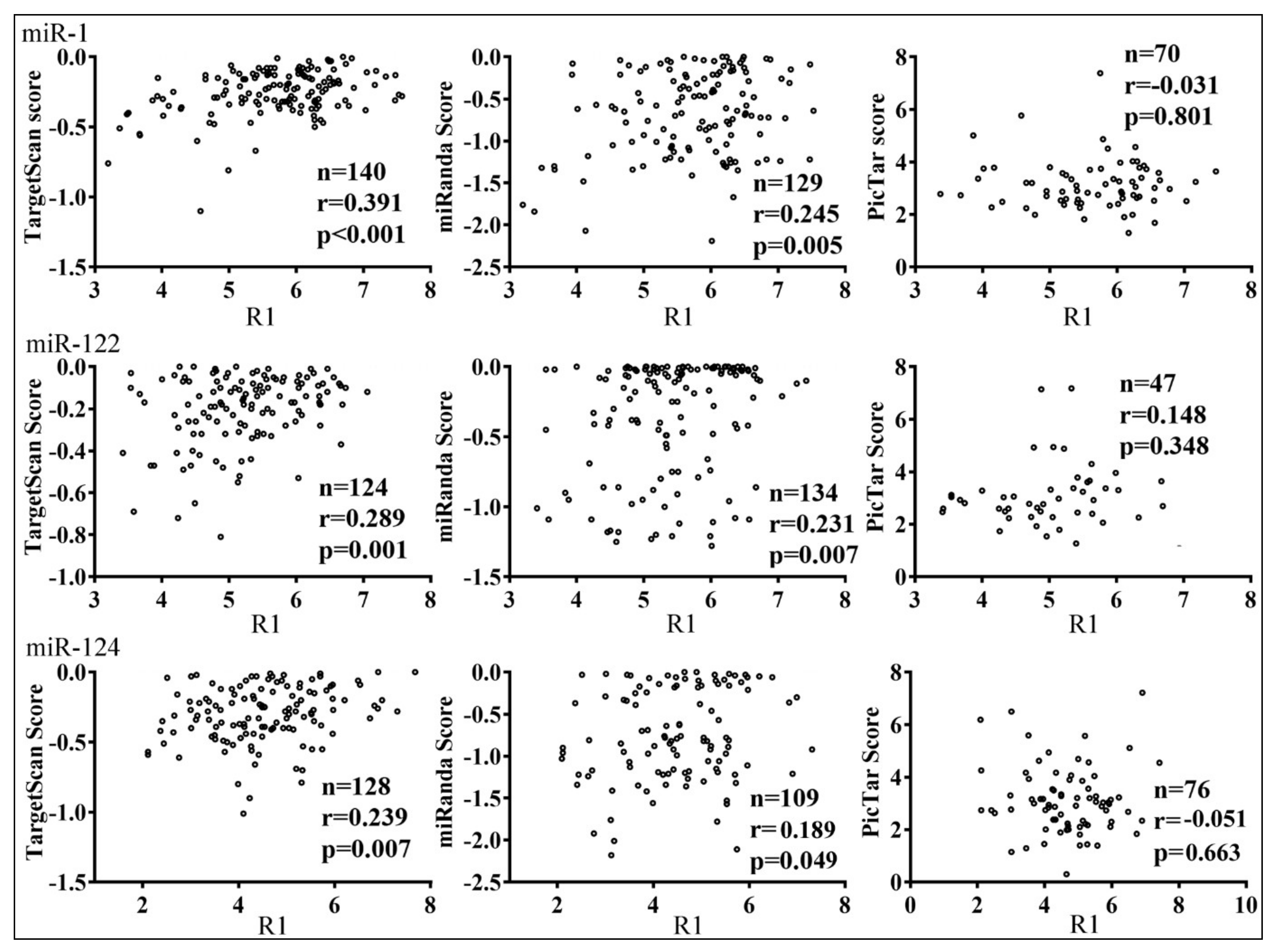
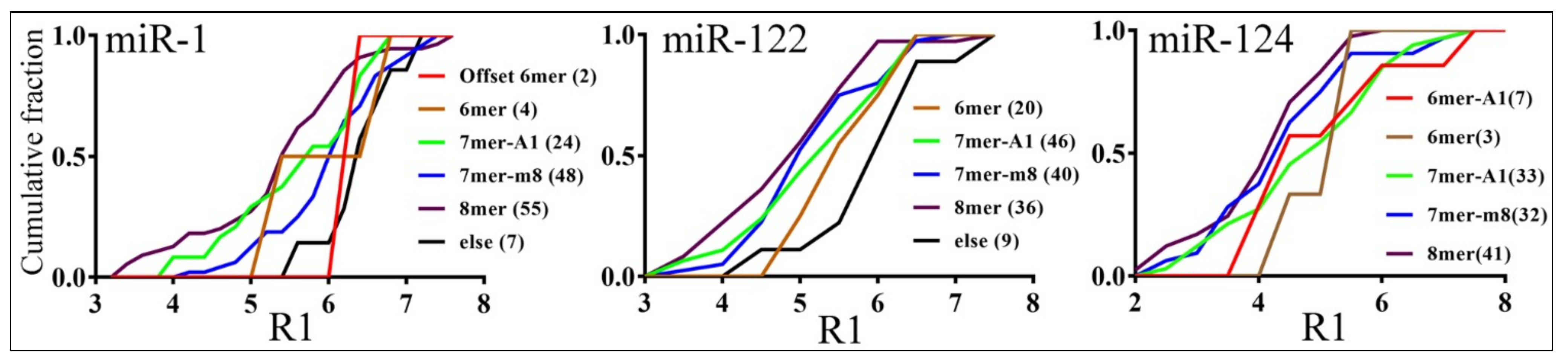
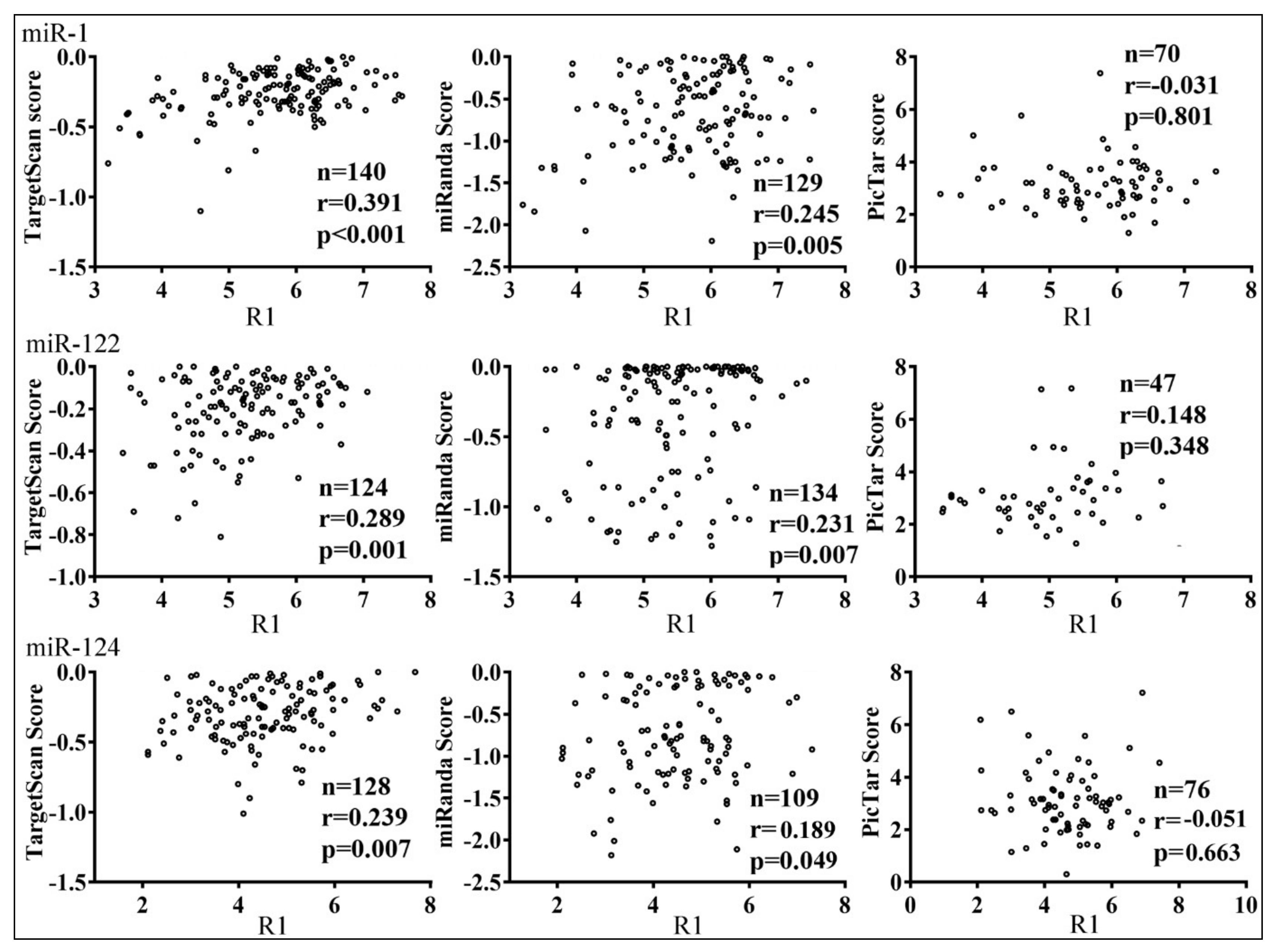
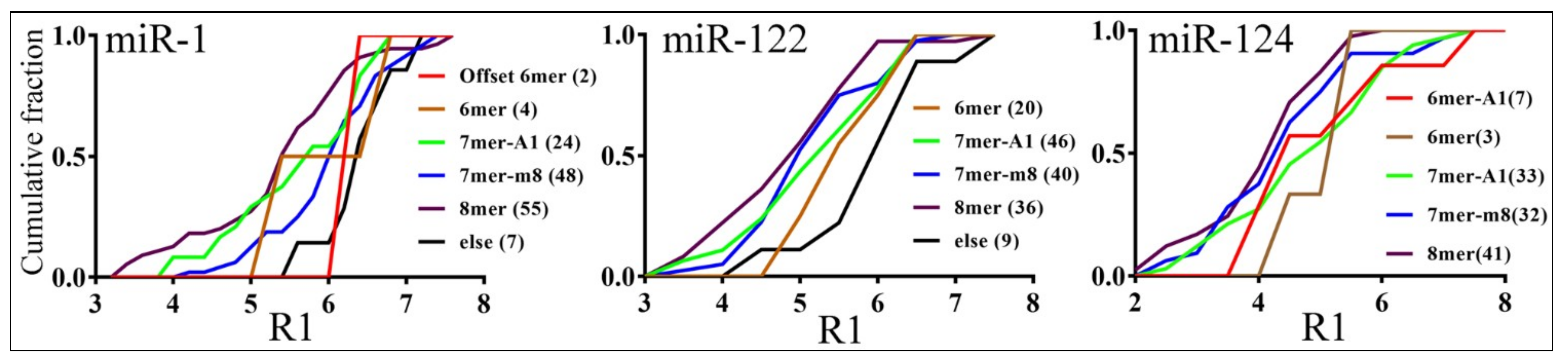
3.3. Parameters that Correlated with Target Repression by miRNAs
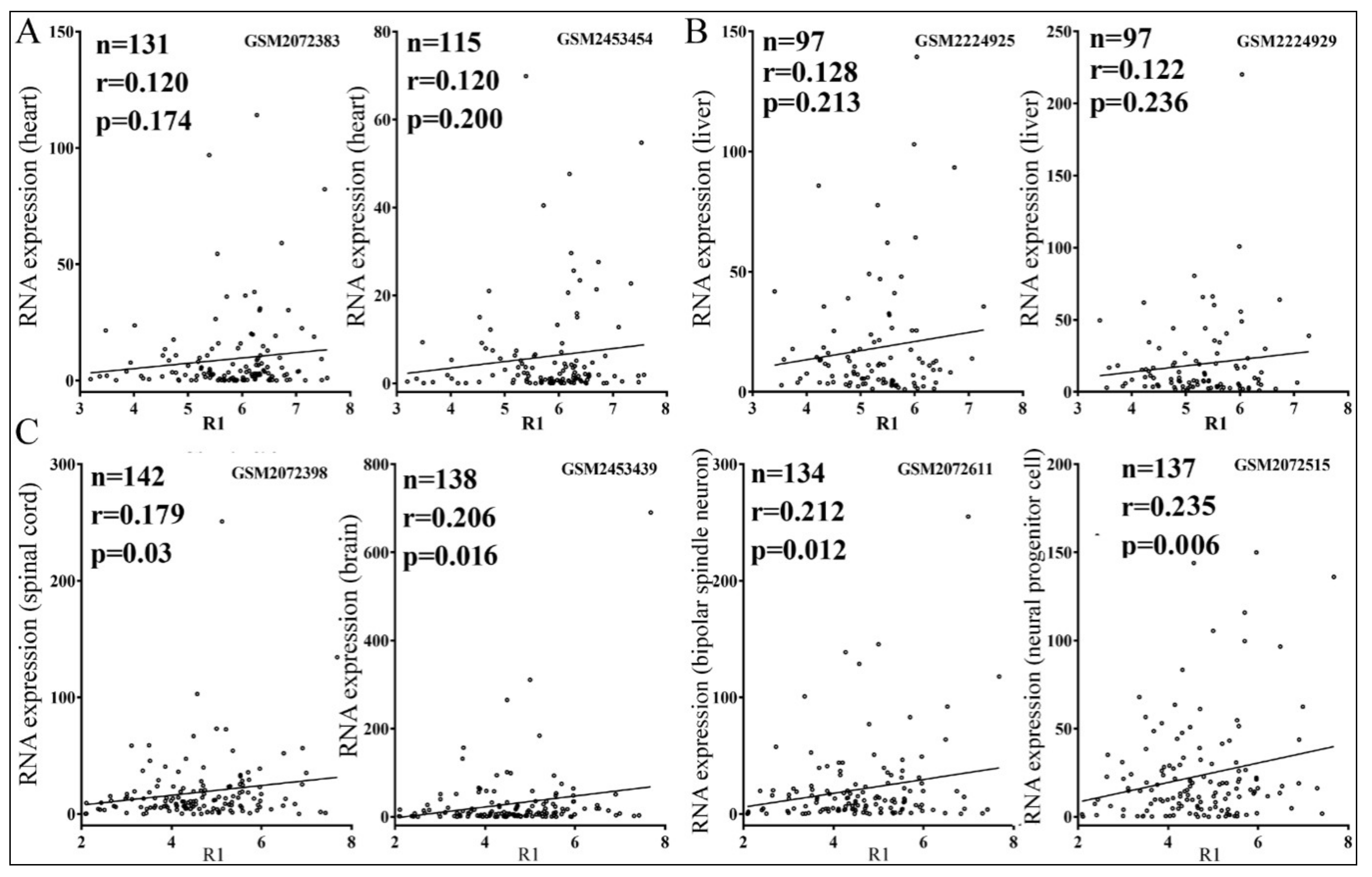
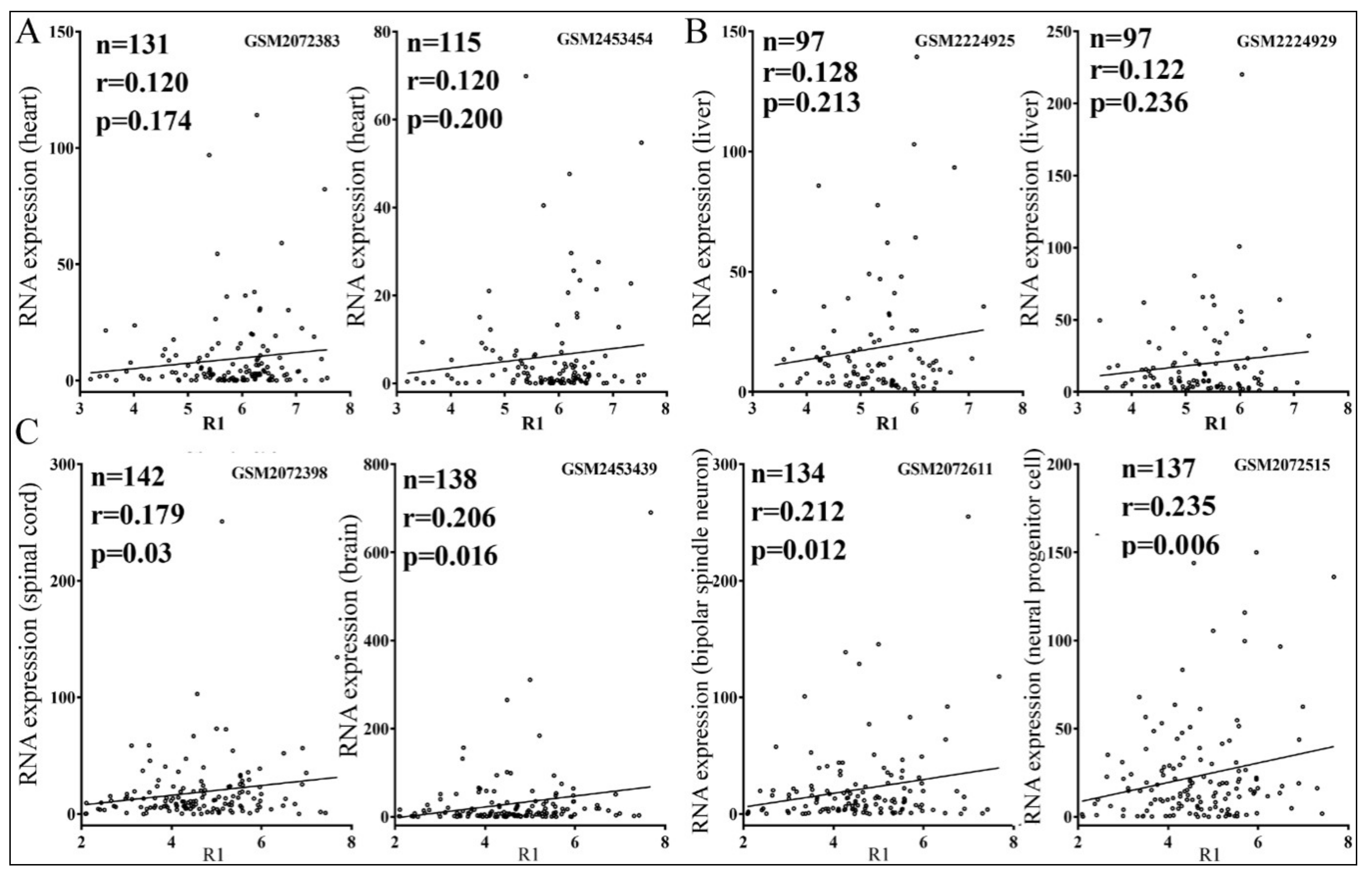
3.4. Relationships between Reporter Inhibition and Endogenous Gene Expression
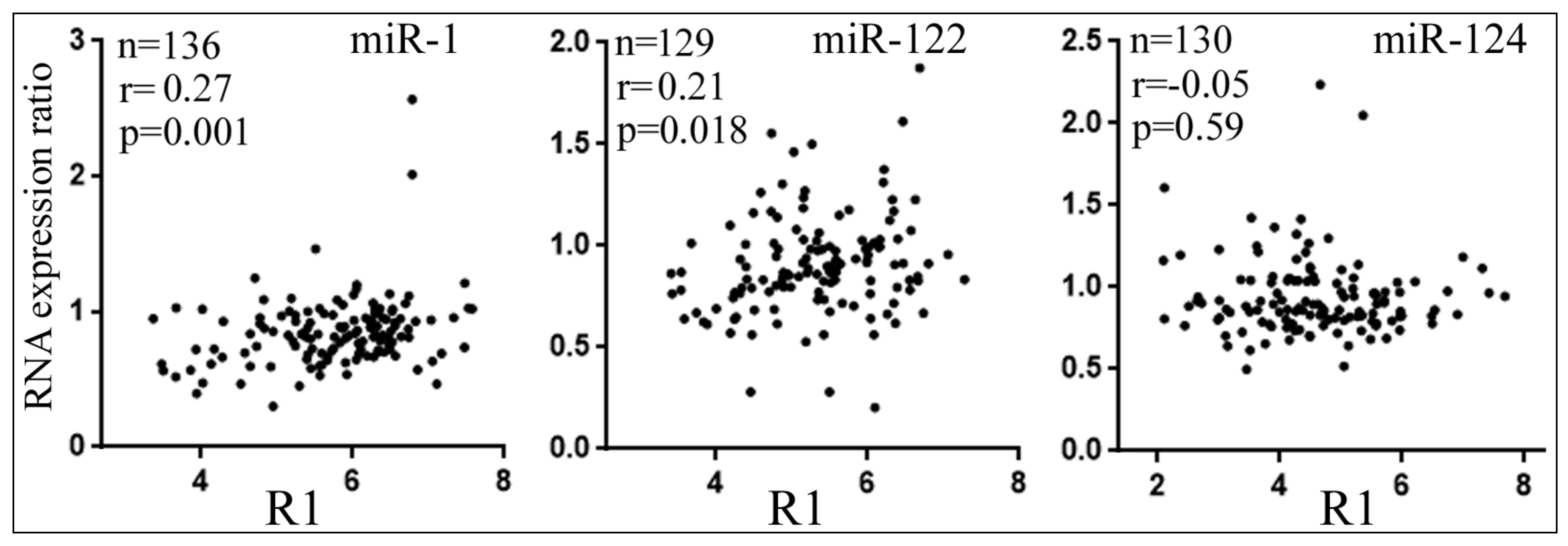
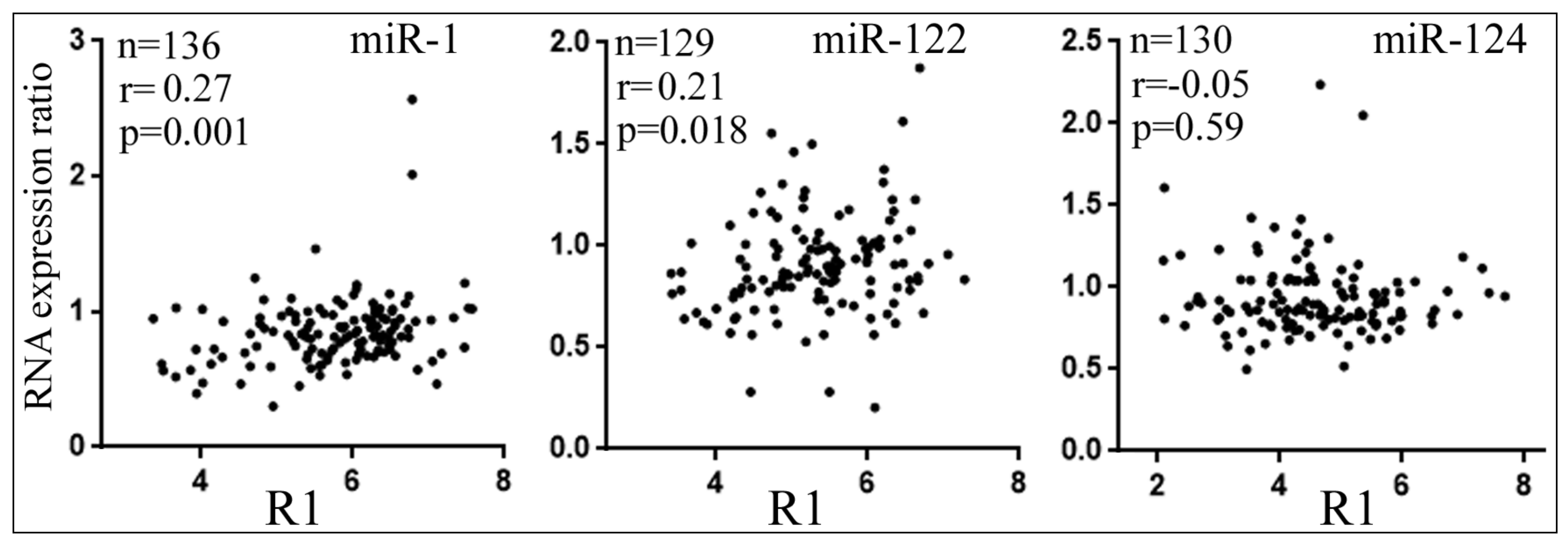
3.5. Differential Reduction of Target mRNA Expression by miRNAs
4. Discussion
Supplementary Material
Author Contributions
Funding
Conflicts of Interest
References
- Ambros, V. MicroRNAs and developmental timing. Curr. Opin. Genet. Dev. 2011, 21, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. Metazoan microRNAs. Cell 2018, 173, 20–50. [Google Scholar] [CrossRef] [PubMed]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Vella, M.C.; Choi, E.Y.; Lin, S.Y.; Reinert, K.; Slack, F.J. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3’UTR. Genes. Dev. 2004, 18, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals, determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Shin, C.; Nam, J.W.; Farh, K.K.; Chiang, H.R.; Shkumatava, A.; Bartel, D.P. Expanding the microRNA targeting code, functional sites with centered pairing. Mol. Cell 2010, 38, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Ecsedi, M.; Rausch, M.; Großhans, H. The let-7 microRNA directs vulval development through a single target. Dev. Cell 2015, 32, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Loeb, G.B.; Khan, A.A.; Canner, D.; Hiatt, J.B.; Shendure, J.; Darnell, R.B.; Leslie, C.S.; Rudensky, A.Y. Transcriptome-wide miR-155 binding map reveals widespread noncanonical microRNA targeting. Mol. Cell 2012, 48, 760–770. [Google Scholar] [CrossRef]
- Helwak, A.; Kudla, G.; Dudnakova, T.; Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013, 153, 654–665. [Google Scholar] [CrossRef]
- Grosswendt, S.; Filipchyk, A.; Manzano, M.; Klironomos, F.; Schilling, M.; Herzog, M.; Gottwein, E.; Rajewsky, N. Unambiguous identification of miRNA, target site interactions by different types of ligation reactions. Mol. Cell 2014, 54, 1042–1054. [Google Scholar] [CrossRef]
- Denzler, R.; McGeary, S.E.; Title, A.C.; Agarwal, V.; Bartel, D.P.; Stoffel, M. Impact of microRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol. Cell 2016, 64, 565–579. [Google Scholar] [CrossRef]
- Krutzfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Farh, K.K.; Grimson, A.; Jan, C.; Lewis, B.P.; Johnston, W.K.; Lim, L.P.; Burge, C.B.; Bartel, D.P. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 2005, 310, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; Rajewsky, N. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Slutskin, V.I.; Weingarten-Gabbay, S.; Nir, R.; Weinberger, A.; Segal, E. Unraveling the determinants of microRNA mediated regulation using a massively parallel reporter assay. Nat. Commun. 2018, 9, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.H.; Tu, S.J.; Lee, W.H.; et al. miRTarBase update 2018, a resource for experimentally validated microRNA-target interactions. Nucl. Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Arvey, A.; Larsson, E.; Sander, C.; Leslie, C.S.; Marks, D.S. Target mRNA abundance dilutes microRNA and siRNA activity. Mol. Syst. Bio. 2010, 6, 363. [Google Scholar] [CrossRef]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011, 18, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Erhard, F.; Haas, J.; Lieber, D.; Malterer, G.; Jaskiewicz, L.; Zavolan, M.; Dolken, L.; Zimmer, R. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014, 24, 906–919. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Invest. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.C.; Hsu, S.D.; Hsu, C.S.; Lai, T.C.; Chen, S.J.; Shen, R.; Huang, Y.; Chen, H.C.; Lee, C.H.; Tsai, T.F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 2012, 122, 2884–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, T.; Arend, K.C.; Li, Y.; Yamane, D.; McGivern, D.R.; Kato, T.; Wakita, T.; Moorman, N.J.; Lemon, S.M. miR-122 stimulates Hepatitis C Virus RNA synthesis by altering the balance of viral RNAs engaged in replication versus translation. Cell Host Microbe 2015, 17, 217–228. [Google Scholar] [CrossRef]
- Kutsche, L.K.; Gysi, D.M.; Fallmann, J.; Lenk, K.; Petri, R.; Swiersy, A.; Klapper, S.D.; Pircs, K.; Khattak, S.; Stadler, P.F.; et al. Combined experimental and system-level analyses reveal the complex regulatory network of miR-124 during human neurogenesis. Cell Syst. 2018, 7, 438–452. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Cho, S.; Jang, I.; Jun, Y.; Yoon, S.; Ko, M.; Kwon, Y.; Choi, I.; Chang, H.; Ryu, D.; Lee, B.; et al. MiRGator v3.0, a microRNA portal for deep sequencing, expression profiling and mRNA targeting. Nucl. Acids Res. 2013, 41, D252–D257. [Google Scholar] [CrossRef]
- Zeng, Y.; Cullen, B.R. Sequence requirements for microRNA processing and function in human cells. RNA 2003, 9, 112–123. [Google Scholar] [CrossRef]
- Zeng, Y.; Yi, R.; Cullen, B.R. Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 2005, 24, 138–148. [Google Scholar] [CrossRef]
- Zuker, M.; Stiegler, P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucl. Acid Res. 1981, 9, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Rehmsmeier, M.; Steffen, P.; Hoechsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes RNA. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses, 2018 update. Nucl. Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protocols 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Tokar, T.; Pastrello, C.; Rossos, A.E.M.; Abovsky, M.; Hauschild, A.C.; Tsay, M.; Lu, R.; Jurisica, I. mirDIP 4.1-integrative database of human microRNA target predictions. Nucl. Acids Res. 2018, 46, D360–D370. [Google Scholar] [CrossRef] [PubMed]
- McEnerney, L.; Duncan, K.; Bang, B.R.; Elmasry, S.; Li, M.; Miki, T.; Ramakrishnan, S.K.; Shah, Y.M.; Saito, T. Dual modulation of human hepatic zonation via canonical and non-canonical Wnt pathways. Exp. Mol. Med. 2017, 49, e413. [Google Scholar] [CrossRef]
- Bosson, A.D.; Zamudio, J.R.; Sharp, P.A. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol. Cell 2014, 56, 347–359. [Google Scholar] [CrossRef]
- Stark, A.; Brennecke, J.; Bushati, N.; Russell, R.B.; Cohen, S.M. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3’UTR evolution. Cell 2005, 123, 1133–1146. [Google Scholar] [CrossRef]
- Csardi, G.; Franks, A.; Choi, D.S.; Airoldi, E.M.; Drummond, D.A. Accounting for experimental noise reveals that mRNA levels amplified by post-transcriptional processes, largely determine steady-state protein levels in yeast. PLoS Genet. 2015, 11, e1005206. [Google Scholar] [CrossRef]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR-1 | miR-122 | miR-124 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Selected | Confirmed | Ratio | Selected | Confirmed | Ratio | Selected | Confirmed | Ratio | |
| TargetScan | 165 | 140 | 84.8% | 149 | 124 | 83.2% | 174 | 128 | 73.6% |
| miRanda | 159 | 129 | 81.1% | 162 | 134 | 82.7% | 151 | 109 | 72.2% |
| PicTar | 84 | 70 | 83.3% | 57 | 47 | 82.5% | 104 | 76 | 73.1% |
| TargetScan only | 19 | 17 | 89.5% | 19 | 17 | 89.5% | 13 | 11 | 84.6% |
| miRanda only | 28 | 19 | 67.9% | 42 | 34 | 84.0% | 9 | 8 | 88.9% |
| PicTar only | 1 | 1 | 100% | 2 | 2 | 100% | 8 | 6 | 75.0% |
| All three | 64 | 52 | 81.3% | 43 | 33 | 76.7% | 67 | 48 | 71.6% |
| miRTarBase | 10 | 10 | 100% | 14 | 13 | 92.9% | 5 | 5 | 100% |
| Single MRE | 171 | 140 | 81.9% | 181 | 152 | 84.0% | 166 | 117 | 70.5% |
| Multiple MREs | 25 | 22 | 88.0% | 13 | 11 | 84.6% | 30 | 28 | 93.3% |
| Total | 196 | 162 | 82.7% | 194 | 163 | 84.0% | 196 | 145 | 74.0% |
| mRNA Properties | miR-1 | miR-122 | miR-124 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | r | p | n | r | p | n | r | p | ||
| AU% | 50 nt | 131 | −0.25 | 0.004 | 143 | −0.32 | <0.001 | 116 | 0.00 | 0.991 |
| 100 nt | 130 | −0.22 | 0.013 | 136 | −0.24 | 0.006 | 115 | 0.03 | 0.716 | |
| 200 nt | 110 | −0.23 | 0.017 | 125 | −0.25 | 0.005 | 99 | −0.07 | 0.502 | |
| 300 nt | 84 | −0.17 | 0.120 | 98 | −0.24 | 0.020 | 77 | −0.04 | 0.713 | |
| whole | 162 | −0.17 | 0.032 | 163 | −0.15 | 0.061 | 145 | 0.05 | 0.583 | |
| ΔG of predicted secondary structure | 50 nt | 131 | −0.11 | 0.198 | 143 | −0.22 | 0.009 | 116 | −0.01 | 0.899 |
| 100 nt | 130 | −0.18 | 0.046 | 136 | −0.23 | 0.008 | 115 | 0.02 | 0.843 | |
| 200 nt | 110 | −0.20 | 0.033 | 125 | −0.27 | 0.003 | 99 | −0.03 | 0.751 | |
| 300 nt | 84 | −0.13 | 0.250 | 98 | −0.27 | 0.008 | 77 | −0.04 | 0.711 | |
| whole | 162 | −0.18 | 0.024 | 163 | −0.03 | 0.685 | 145 | −0.09 | 0.276 | |
| ΔG of miRNA and MRE hybrid | 140 | 0.23 | 0.005 | 152 | −0.01 | 0.926 | 117 | 0.03 | 0.722 | |
| Local AU content | 129 | −0.14 | 0.109 | 140 | −0.32 | <0.001 | 107 | −0.12 | 0.219 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Chen, Y.; Juma, C.A.; Yang, C.; Huang, J.; Zhang, X.; Zeng, Y. Differential Inhibition of Target Gene Expression by Human microRNAs. Cells 2019, 8, 791. https://doi.org/10.3390/cells8080791
Li P, Chen Y, Juma CA, Yang C, Huang J, Zhang X, Zeng Y. Differential Inhibition of Target Gene Expression by Human microRNAs. Cells. 2019; 8(8):791. https://doi.org/10.3390/cells8080791
Chicago/Turabian StyleLi, Peng, Yi Chen, Conslata Awino Juma, Chengyong Yang, Jinfeng Huang, Xiaoxiao Zhang, and Yan Zeng. 2019. "Differential Inhibition of Target Gene Expression by Human microRNAs" Cells 8, no. 8: 791. https://doi.org/10.3390/cells8080791