Large Scale Profiling of Protein Isoforms Using Label-Free Quantitative Proteomics Revealed the Regulation of Nonsense-Mediated Decay in Moso Bamboo (Phyllostachys edulis)

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Extraction of Proteins from Moso Bamboo
2.3. Protein Digestion and Peptide Fraction
2.4. Mass Spectrometry Analysis
2.5. Protein Identification Using Mass Spectrometry
2.6. GO Enrichment Analysis and Pathway Analysis
2.7. Data Availability
3. Results
3.1. Extraction and Identification of Total Protein Using Phenol Extraction Combined with Acetonitrile Precipitation for Orbitrap Fusion Tribrid Mass Spectrometry
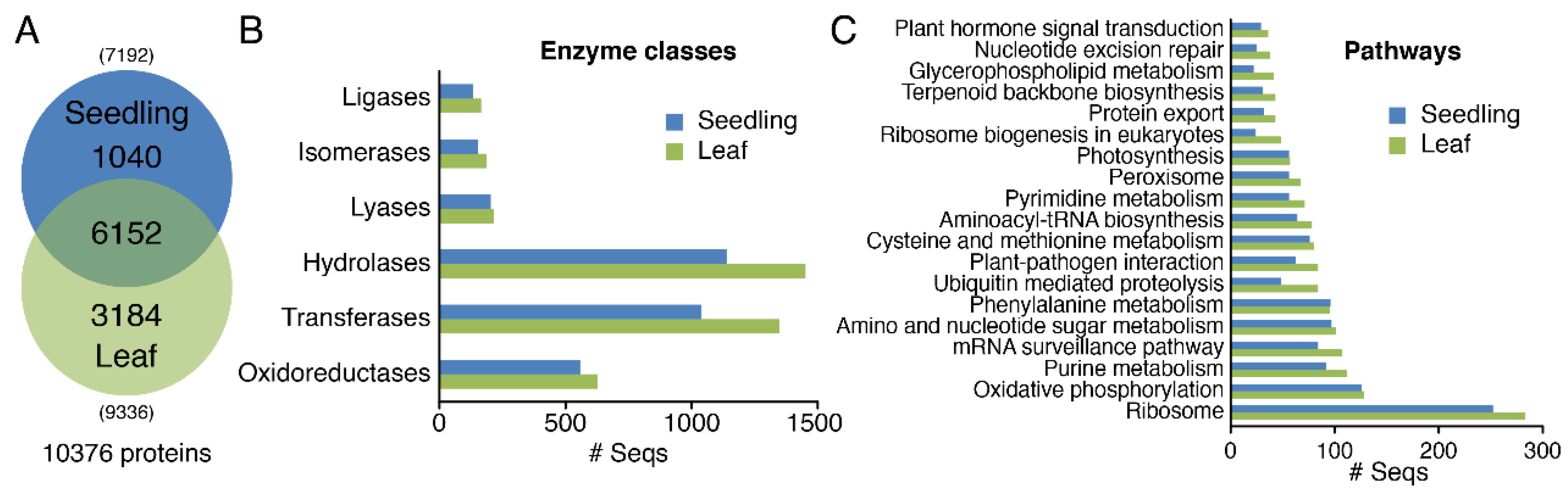
3.2. Analysis of Physiological Process and Metabolic Pathway for All Identified Protein Data
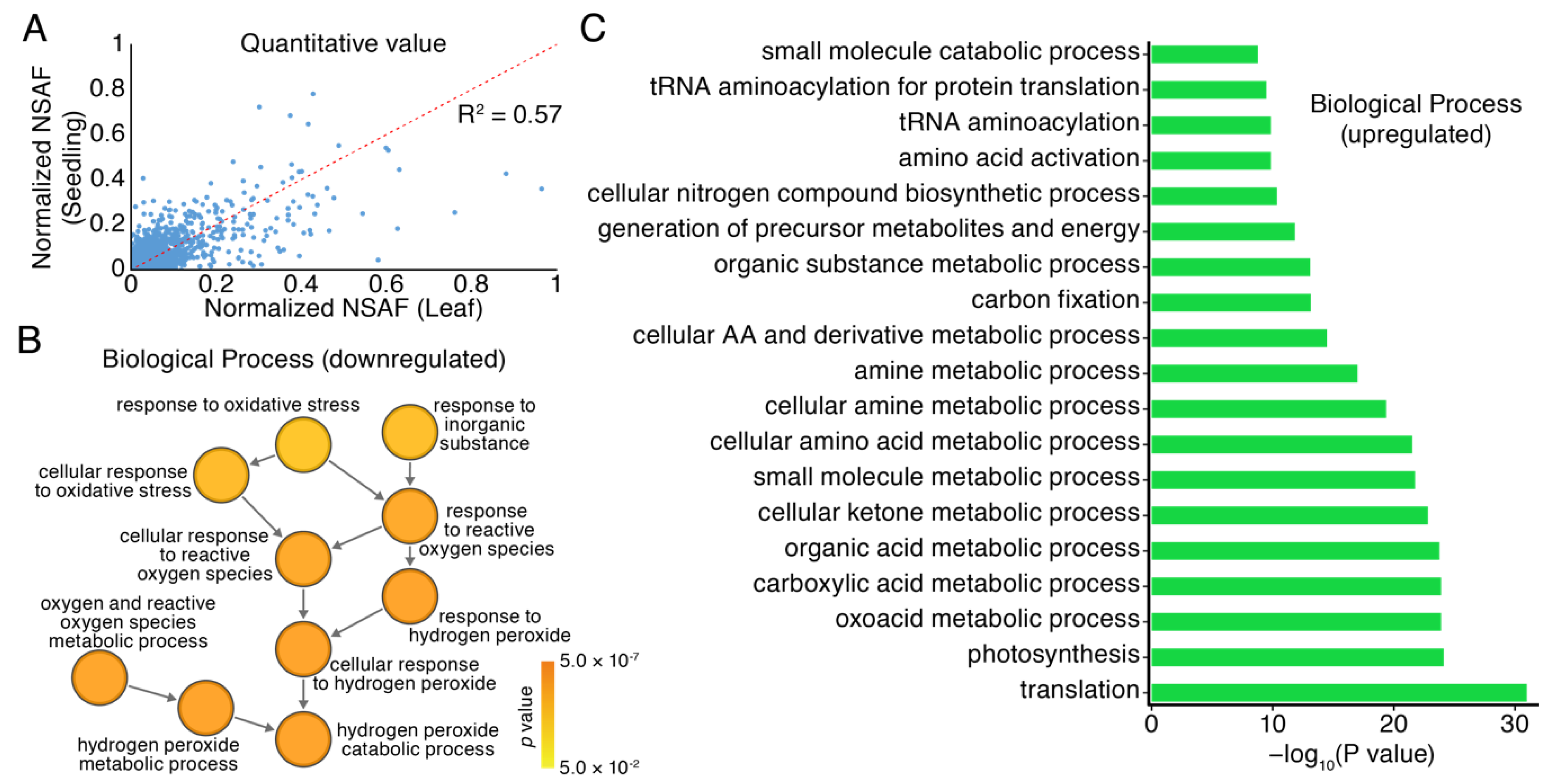
3.3. Differential Expression Profiles of Proteomics in Seedlings and Leaves of Moso Bamboo using High-Throughput Label-Free Quantitative Proteomics
3.4. GO Analysis of Proteomics in Seedlings and Leaves of Moso Bamboo
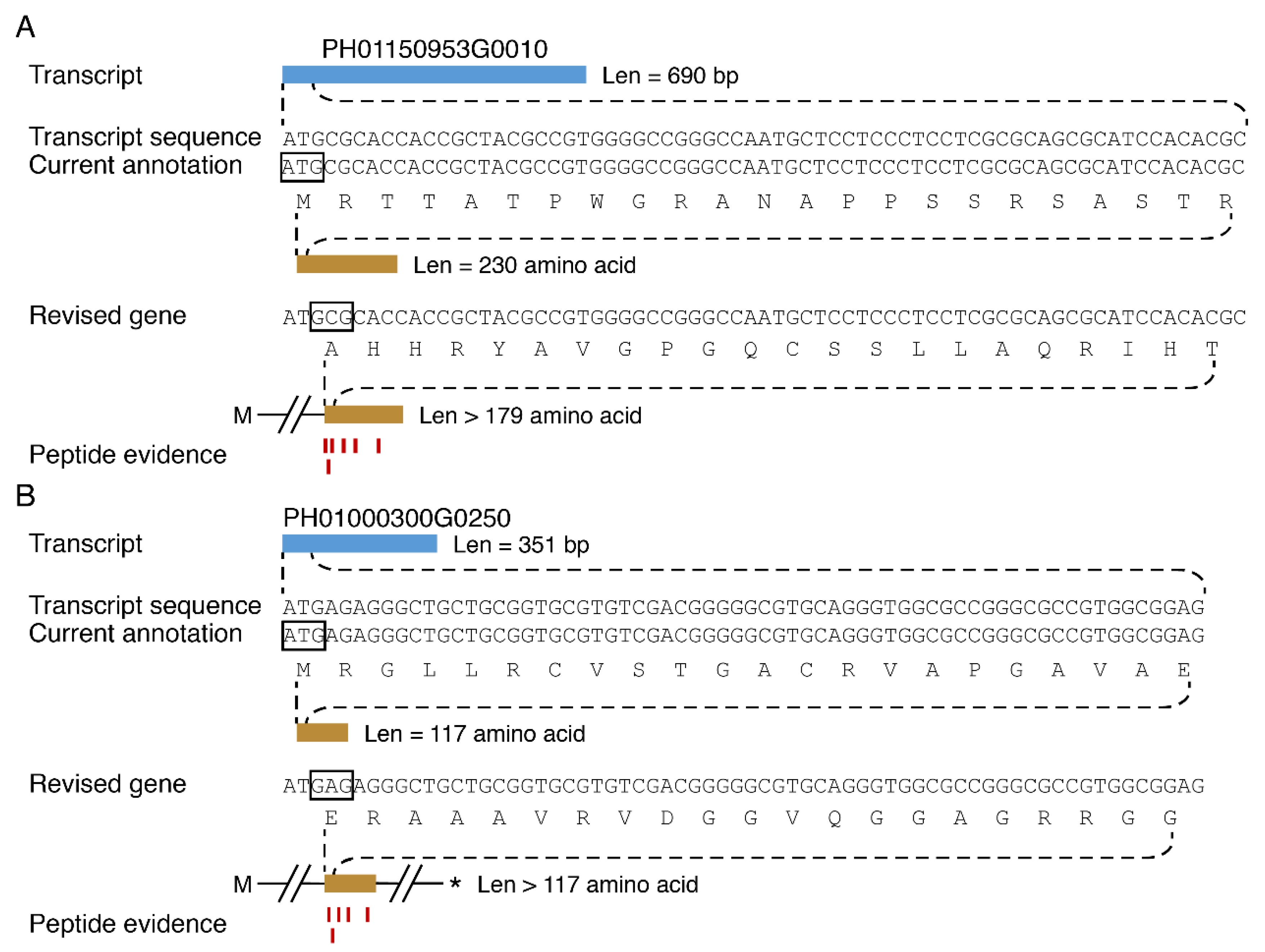
3.5. Discovery of New Protein Coding Genes
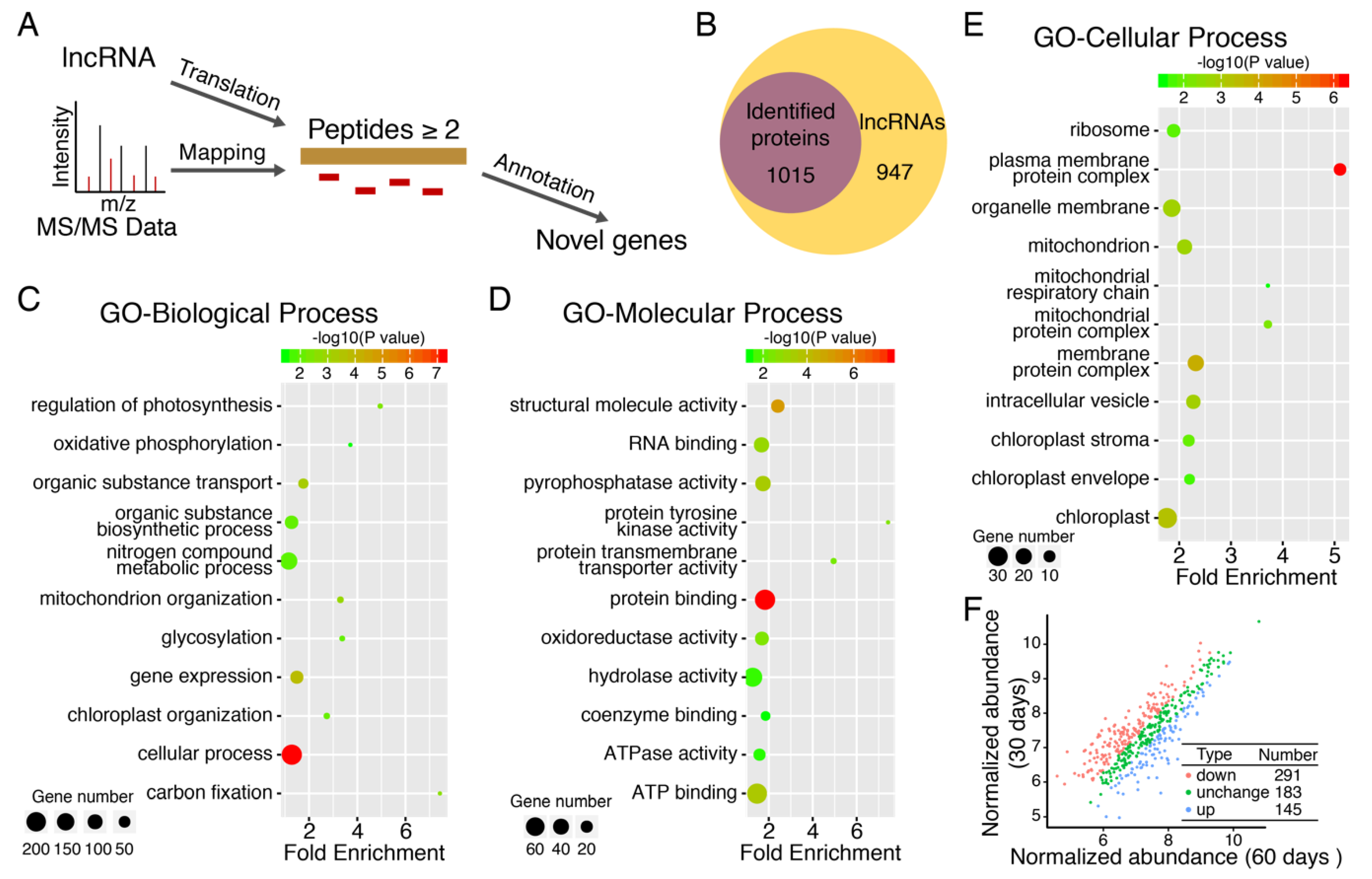
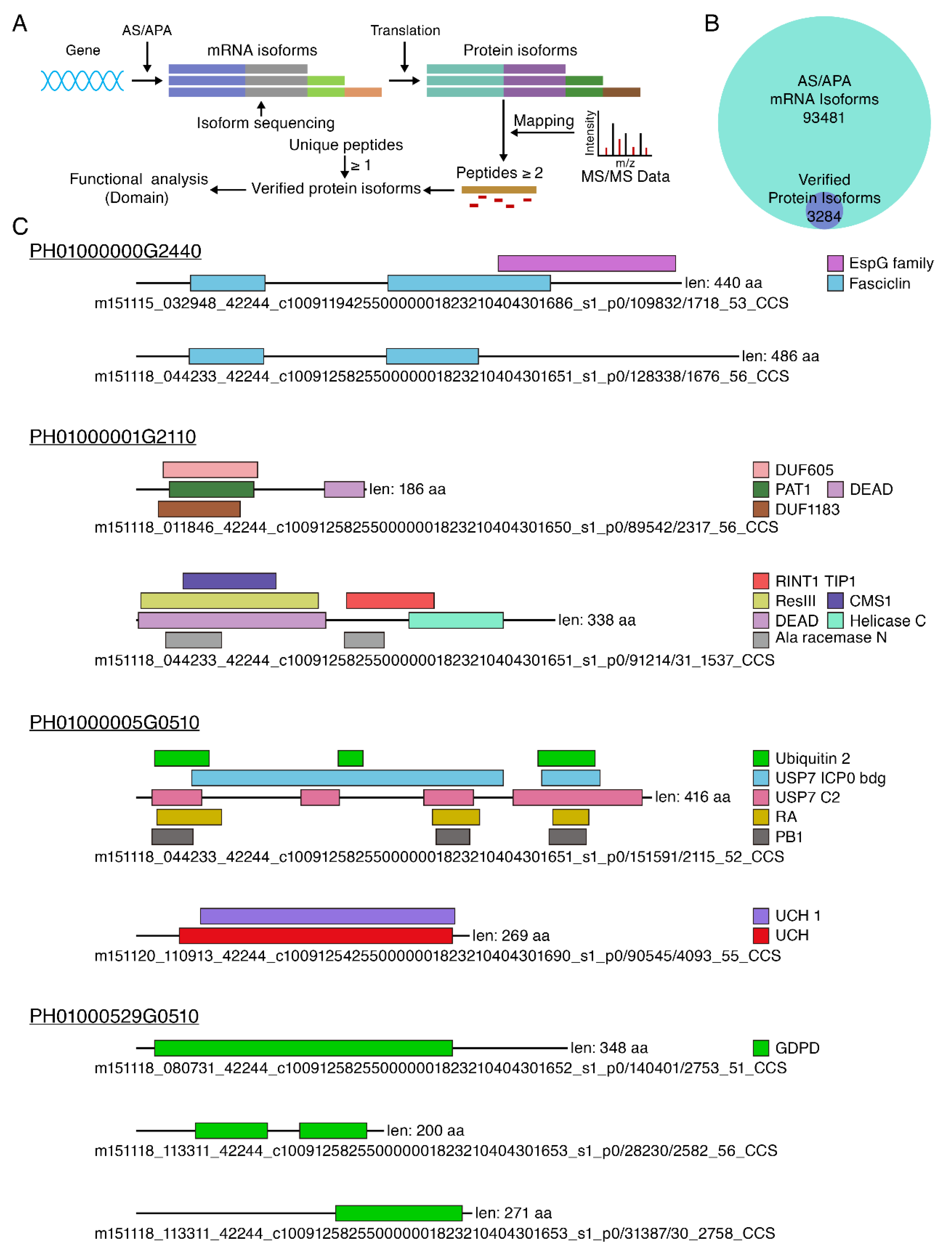
3.6. Large Scale Identification of Protein Isoforms
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lobovikov, M.; Paudel, S.; Piazza, M.; Ren, H.; Wu, J.Q. World Bamboo Resources: A Thematic Study Prepared in the Framework of the Global Forest Resources Assessment 2005; FAO: Rome, Italy, 2007. [Google Scholar]
- Scurlock, J.; Dayton, D.; Hames, B. Bamboo: An overlooked biomass resource? Biomass Bioenergy 2000, 19, 229–244. [Google Scholar] [CrossRef]
- Fu, J. Chinese moso bamboo: Its importance. Bamboo 2001, 22, 5–7. [Google Scholar]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K.; et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wei, W.; Liu, C.; You, W.; Niu, X.; Man, R. Biomass and carbon stock in Moso bamboo forests in subtropical China: Characteristics and implications. J. Trop. For. Sci. 2013, 25, 137–148. [Google Scholar]
- Zhou, R.; Moshgabadi, N.; Adams, K.L. Extensive changes to alternative splicing patterns following allopolyploidy in natural and resynthesized polyploids. Proc. Natl. Acad. Sci. USA 2011, 108, 16122–16127. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhou, G.; Du, H.; Lu, D.; Mo, L.; Xu, X.; Shi, Y.; Zhou, Y. Current and potential carbon stocks in Moso bamboo forests in China. J. Environ. Manag. 2015, 156, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Gao, Z.; Wang, L.; Wang, J.; Wang, S.; Fei, B.; Chen, C.; Shi, C.; Liu, X.; Zhang, H.; et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). Gigascience 2018, 7. [Google Scholar] [CrossRef]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.; Zhang, H.; Wang, Y.; Lin, C.; Ma, L.; Gu, L. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef]
- Peng, Z.; Zhang, C.; Zhang, Y.; Hu, T.; Mu, S.; Li, X.; Gao, J. Transcriptome sequencing and analysis of the fast growing shoots of moso bamboo (Phyllostachys edulis). PLoS ONE 2013, 8, e78944. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, H.; Zhu, Q.; Gao, Y.; Wang, H.; Zhao, L.; Wang, Y.; Xi, F.; Wang, W.; Yang, Y.; et al. Transcriptome characterization of moso bamboo (Phyllostachys edulis) seedlings in response to exogenous gibberellin applications. BMC Plant Biol. 2018, 18, 125. [Google Scholar] [CrossRef]
- Liu, L.; Cao, X.L.; Bai, R.; Yao, N.; Li, L.B.; He, C.F. Isolation and characterization of the cold-induced Phyllostachys edulis AP2/ERF family transcription factor, peDREB1. Plant Mol. Bio. Rep. 2012, 30, 679–689. [Google Scholar] [CrossRef]
- Cui, X.W.; Zhang, Y.; Qi, F.Y.; Gao, J.; Chen, Y.W.; Zhang, C.L. Overexpression of a moso bamboo (Phyllostachys edulis) transcription factor gene PheWRKY1 enhances disease resistance in transgenic Arabidopsis thaliana. Botany 2013, 91, 486–494. [Google Scholar] [CrossRef]
- Huang, Z.; Zhong, X.J.; He, J.; Jin, S.H.; Guo, H.D.; Yu, X.F.; Zhou, Y.J.; Li, X.; Ma, M.D.; Chen, Q.B.; et al. Genome-wide identification, characterization, and stress-responsive expression profiling of genes encoding LEA (late embryogenesis abundant) proteins in moso bamboo (Phyllostachys edulis). PLoS ONE 2016, 11, e0165953. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT-and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheng, Z.; Ma, Y.; Bai, Q.; Li, X.; Cao, Z.; Wu, Z.; Gao, J. The association of hormone signalling genes, transcription and changes in shoot anatomy during moso bamboo growth. Plant Biotechnol. J. 2018, 16, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S. RNA regulation: A new genetics? Nat. Rev. Genet. 2004, 5, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, J.; Grant, S.G. Proteomics in postgenomic neuroscience: The end of the beginning. Nat. Neurosci. 2004, 7, 440–445. [Google Scholar] [CrossRef]
- Armengaud, J. A perfect genome annotation is within reach with the proteomics and genomics alliance. Curr. Opin. Microbiol. 2009, 12, 292–300. [Google Scholar] [CrossRef]
- Prasad, T.K.; Mohanty, A.K.; Kumar, M.; Sreenivasamurthy, S.K.; Dey, G.; Nirujogi, R.S.; Pinto, S.M.; Madugundu, A.K.; Patil, A.H.; Advani, J.; et al. Integrating transcriptomic and proteomic data for accurate assembly and annotation of genomes. Genome Res. 2017, 27, 133–144. [Google Scholar] [CrossRef]
- Mahesh, H.B.; Subba, P.; Advani, J.; Shirke, M.D.; Loganathan, R.M.; Chandana, S.L.; Shilpa, S.; Chatterjee, O.; Pinto, S.M.; Prasad, T.S.K.; et al. Multi-omics driven assembly and annotation of the sandalwood (Santalum album) genome. Plant Physiol. 2018, 176, 2772–2788. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J. Reannotation of genomes by means of proteomics data. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 585, pp. 201–216. [Google Scholar]
- Bock, T.; Chen, W.-H.; Ori, A.; Malik, N.; Silva-Martin, N.; Huerta-Cepas, J.; Powell, S.T.; Kastritis, P.L.; Smyshlyaev, G.; Vonkova, I.; et al. An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res. 2014, 42, 13525–13533. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Silmon de Monerri, N.C.; Weiss, L.M. Integration of RNA-seq and proteomics data with genomics for improved genome annotation in Apicomplexan parasites. Proteomics 2015, 15, 2557–2559. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J. Next-generation proteomics faces new challenges in environmental biotechnology. Curr. Opin. Biotechnol. 2016, 38, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, H.; Zhang, B.; Fei, B.; Burgert, I. Cell wall structure and formation of maturing fibres of moso bamboo (Phyllostachys pubescens) increase buckling resistance. J. R. Soc. Interface 2011, 9, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; He, C.Y.; Zhang, J.G.; Duan, A.G.; Zeng, Y.F. Temporal and spatial profiling of internode elongation-associated protein expression in rapidly growing culms of bamboo. J. Proteome Res. 2012, 11, 2492–2507. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.; Gu, L.; Reddy, A.S. Analysis of transcriptome and epitranscriptome in plants using PacBio Iso-Seq and Nanopore-based direct RNA sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Xu, Z.; Peters, R.J.; Weirather, J.; Luo, H.; Liao, B.; Zhang, X.; Zhu, Y.; Ji, A.; Zhang, B.; Hu, S.; et al. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 2015, 82, 951–961. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef]
- Li, L.; Hu, T.; Li, X.; Mu, S.; Cheng, Z.; Ge, W.; Gao, J. Genome-wide analysis of shoot growth-associated alternative splicing in moso bamboo. Mol. Genet. Genom. 2016, 291, 1695–1714. [Google Scholar] [CrossRef]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Drechsel, G.; Kahles, A.; Kesarwani, A.K.; Stauffer, E.; Behr, J.; Drewe, P.; Rätsch, G.; Wachter, A. Nonsense-mediated decay of alternative precursor mRNA splicing variants is a major determinant of the Arabidopsis steady state transcriptome. Plant Cell 2013, 25, 3726–3742. [Google Scholar] [CrossRef]
- Kalyna, M.; Simpson, C.G.; Syed, N.H.; Lewandowska, D.; Marquez, Y.; Kusenda, B.; Marshall, J.; Fuller, J.; Cardle, L.; McNicol, J.; et al. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2011, 40, 2454–2469. [Google Scholar] [CrossRef]
- Belostotsky, D.A.; Sieburth, L.E. Kill the messenger: mRNA decay and plant development. Curr. Opin. Plant Biol. 2009, 12, 96–102. [Google Scholar] [CrossRef]
- Yang, X.; Coulombe-Huntington, J.; Kang, S.; Sheynkman, G.M.; Hao, T.; Richardson, A.; Sun, S.; Yang, F.; Shen, Y.A.; Murray, R.R.; et al. Widespread expansion of protein interaction capabilities by alternative splicing. Cell 2016, 164, 805–817. [Google Scholar] [CrossRef]
- Ezkurdia, I.; Rodriguez, J.M.; Carrillo-de Santa Pau, E.; Vázquez, J.; Valencia, A.; Tress, M.L. Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res. 2015, 14, 1880–1887. [Google Scholar] [CrossRef]
- Liu, Y.; Gonzàlez-Porta, M.; Santos, S.; Brazma, A.; Marioni, J.C.; Aebersold, R.; Venkitaraman, A.R.; Wickramasinghe, V.O. Impact of alternative splicing on the human proteome. Cell Rep. 2017, 20, 1229–1241. [Google Scholar] [CrossRef]
- Reixachs-Sole, M.; Ruiz-Orera, J.; Alba, M.; Eyras, E. Ribosome profiling at isoform level reveals an evolutionary conserved impact of differential splicing on the proteome. BioRxiv 2019. [Google Scholar] [CrossRef]
- Chaudhary, S.; Jabre, I.; Reddy, A.S.; Staiger, D.; Syed, N.H. Perspective on alternative splicing and proteome complexity in plants. Trends Plant Sci. 2019, 24, 496–506. [Google Scholar] [CrossRef]
- Brosch, M.; Saunders, G.I.; Frankish, A.; Collins, M.O.; Yu, L.; Wright, J.; Verstraten, R.; Adams, D.J.; Harrow, J.; Choudhary, J.S.; et al. Shotgun proteomics aids discovery of novel protein-coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome. Genome Res. 2011, 21, 756–767. [Google Scholar] [CrossRef]
- Tress, M.L.; Martelli, P.L.; Frankish, A.; Reeves, G.A.; Wesselink, J.J.; Yeats, C.; ĺsólfur Ólason, P.; Albrecht, M.; Hegyi, H.; Giorgetti, A.; et al. The implications of alternative splicing in the ENCODE protein complement. Proc. Natl. Acad. Sci. USA 2007, 104, 5495–5500. [Google Scholar] [CrossRef]
- Tress, M.L.; Bodenmiller, B.; Aebersold, R.; Valencia, A. Proteomics studies confirm the presence of alternative protein isoforms on a large scale. Genome Biol. 2008, 9, R162. [Google Scholar] [CrossRef]
- Abascal, F.; Ezkurdia, I.; Rodriguez-Rivas, J.; Rodriguez, J.M.; del Pozo, A.; Vázquez, J.; Valencia, A.; Tress, M.L. Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput. Biol. 2015, 11, e1004325. [Google Scholar] [CrossRef]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci. 2017, 42, 98–110. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 2010, 10, 1265–1269. [Google Scholar] [CrossRef]
- Zybailov, B.; Mosley, A.L.; Sardiu, M.E.; Coleman, M.K.; Florens, L.; Washburn, M.P. Statistical analysis of membrane proteome expression changes in Saccharomyces c erevisiae. J. Proteome Res. 2006, 5, 2339–2347. [Google Scholar] [CrossRef]
- Zybailov, B.L.; Florens, L.; Washburn, M.P. Quantitative shotgun proteomics using a protease with broad specificity and normalized spectral abundance factors. Mol. Biosyst. 2007, 3, 354–360. [Google Scholar] [CrossRef]
- Florens, L.; Carozza, M.J.; Swanson, S.K.; Fournier, M.; Coleman, M.K.; Workman, J.L.; Washburn, M.P. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 2006, 40, 303–311. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Stone, J.M.; Walker, J.C. Plant protein kinase families and signal transduction. Plant Physiol. 1995, 108, 451–457. [Google Scholar] [CrossRef]
- Filichkin, S.A.; Mockler, T.C. Unproductive alternative splicing and nonsense mRNAs: A widespread phenomenon among plant circadian clock genes. Biol. Direct 2012, 7, 20. [Google Scholar] [CrossRef]
- Marquez, Y.; Brown, J.W.; Simpson, C.; Barta, A.; Kalyna, M. Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 2012, 22, 1184–1195. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Wilkinson, M.F. Nonsense-mediated RNA decay in the brain: Emerging modulator of neural development and disease. Nat. Rev. Neurosci. 2018, 19, 715–728. [Google Scholar] [CrossRef]
- Rödiger, A.; Agne, B.; Baerenfaller, K.; Baginsky, S. Arabidopsis proteomics: A simple and standardizable workflow for quantitative proteome characterization. Methods Mol. Biol. 2014, 1072, 275–288. [Google Scholar]
- Castellana, N.E.; Payne, S.H.; Shen, Z.; Stanke, M.; Bafna, V.; Briggs, S.P. Discovery and revision of Arabidopsis genes by proteogenomics. Proc. Natl. Acad. Sci. USA 2008, 105, 21034–21038. [Google Scholar] [CrossRef]
- Castellana, N.E.; Shen, Z.X.; He, Y.P.; Walley, J.W.; Cassidy, C.J.; Briggs, S.P.; Bafna, V. An automated proteogenomic method uses mass spectrometry to reveal novel genes in Zea mays. Mol. Cell. Proteomics 2014, 13, 157–167. [Google Scholar] [CrossRef]
- Helmy, M.; Tomita, M.; Ishihama, Y. OryzaPG-DB: Rice proteome database based on shotgun proteogenomics. BMC Plant Biol 2011, 11, 63. [Google Scholar] [CrossRef]
- Watanabe, K.A.; Homayouni, A.; Tufano, T.; Lopez, J.; Ringler, P.; Rushton, P.; Shen, Q.J. Tiling Assembly: A new tool for reference annotation-independent transcript assembly and novel gene identification by RNA-sequencing. DNA Res. 2015, 22, 319–329. [Google Scholar] [CrossRef]
- Kay, R.; Barton, C.; Ratcliffe, L.; Matharoo-Ball, B.; Brown, P.; Roberts, J.; Teale, P.; Creaser, C. Enrichment of low molecular weight serum proteins using acetonitrile precipitation for mass spectrometry based proteomic analysis. Rapid Commun. Mass Spectrom. 2008, 22, 3255–3260. [Google Scholar] [CrossRef]
- Sedgwick, G.W.; Fenton, T.F.; Thompson, J.R. Effect of protein precipitating agents on the recovery of plasma free aminoacids. Can. J. Anim. Sci. 1991, 71, 953–957. [Google Scholar] [CrossRef]
- Chang, Y.-F.; Imam, J.S.; Wilkinson, M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef]
- Schwerk, C.; Schulze-Osthoff, K. Regulation of apoptosis by alternative pre-mRNA splicing. Mol. Cell 2005, 19, 1–13. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenol Extraction | ||||
|---|---|---|---|---|
| Acetonitrile | Tca/Acetone | |||
| Seedling | Leaf | Seedling | Leaf | |
| Spectra | 255470 | 295637 | 243561 | 226212 |
| Peptide | 19470 | 30881 | 3155 | 1694 |
| Protein | 7192 | 9336 | 1937 | 1365 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Wang, Y.; Kohnen, M.V.; Piao, M.; Tu, M.; Gao, Y.; Lin, C.; Zuo, Z.; Gu, L. Large Scale Profiling of Protein Isoforms Using Label-Free Quantitative Proteomics Revealed the Regulation of Nonsense-Mediated Decay in Moso Bamboo (Phyllostachys edulis). Cells 2019, 8, 744. https://doi.org/10.3390/cells8070744
Yu X, Wang Y, Kohnen MV, Piao M, Tu M, Gao Y, Lin C, Zuo Z, Gu L. Large Scale Profiling of Protein Isoforms Using Label-Free Quantitative Proteomics Revealed the Regulation of Nonsense-Mediated Decay in Moso Bamboo (Phyllostachys edulis). Cells. 2019; 8(7):744. https://doi.org/10.3390/cells8070744
Chicago/Turabian StyleYu, Xiaolan, Yongsheng Wang, Markus V. Kohnen, Mingxin Piao, Min Tu, Yubang Gao, Chentao Lin, Zecheng Zuo, and Lianfeng Gu. 2019. "Large Scale Profiling of Protein Isoforms Using Label-Free Quantitative Proteomics Revealed the Regulation of Nonsense-Mediated Decay in Moso Bamboo (Phyllostachys edulis)" Cells 8, no. 7: 744. https://doi.org/10.3390/cells8070744
APA StyleYu, X., Wang, Y., Kohnen, M. V., Piao, M., Tu, M., Gao, Y., Lin, C., Zuo, Z., & Gu, L. (2019). Large Scale Profiling of Protein Isoforms Using Label-Free Quantitative Proteomics Revealed the Regulation of Nonsense-Mediated Decay in Moso Bamboo (Phyllostachys edulis). Cells, 8(7), 744. https://doi.org/10.3390/cells8070744