Activation of Cryptic 3′ Splice-Sites by SRSF2 Contributes to Cassette Exon Skipping

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmids Construction
2.2. Cell Culture, Transfection and RT-PCR
2.3. Knockdown of SRSF2 with Lentivirus-Mediated shRNA
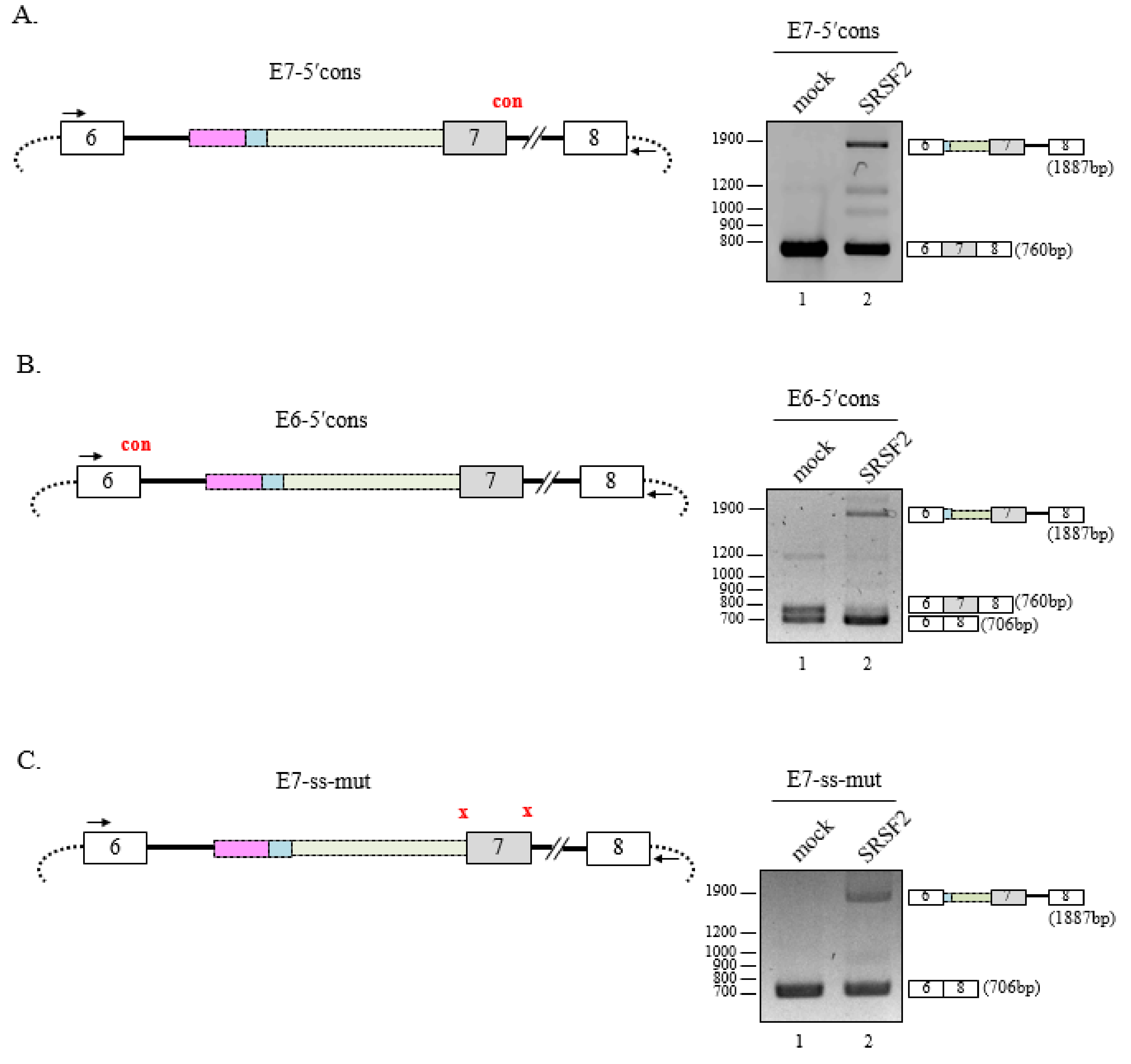
3. Results
3.1. SRSF2 Promotes Cryptic 3′AG′ Activation
3.2. Deletion of 3′AG′ Region Induces SRSF2-Dependent Usage of Alternate 3′AG′
3.3. Multiple SRSF2-Binding Sequences and Clusters Are Necessary for 3′AG′ Activation by SRSF2
3.4. 5′Splice-Site Mutations do not Affect SRSF2-Mediated 3′AG′ Activation
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nelson, K.K.; Green, M.R. Mammalian U2 snRNP has a sequence-specific RNA-binding activity. Genes Dev. 1989, 3, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Berglund, J.A.; Chua, K.; Abovich, N.; Reed, R.; Rosbash, M. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 1997, 89, 781–787. [Google Scholar] [CrossRef]
- Wu, S.; Romfo, C.M.; Nilsen, T.W.; Green, M.R. Functional recognition of the 3’ splice site AG by the splicing factor U2AF35. Nature 1999, 402, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Zamore, P.D.; Patton, J.G.; Green, M.R. Cloning and domain structure of the mammalian splicing factor U2AF. Nature 1992, 355, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Buratti, E.; Chivers, M.; Hwang, G.; Vorechovsky, I. DBASS3 and DBASS5: Databases of aberrant 3’- and 5’-splice sites. Nucleic Acids Res. 2011, 39, D86–D91. [Google Scholar] [CrossRef]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Schwartz, S.H.; Silva, J.; Burstein, D.; Pupko, T.; Eyras, E.; Ast, G. Large-scale comparative analysis of splicing signals and their corresponding splicing factors in eukaryotes. Genome Res. 2008, 18, 88–103. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Graveley, B.R. Sorting out the complexity of SR protein functions. RNA 2000, 6, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Cho, S.; Loh, T.J.; Oh, H.K.; Jang, H.N.; Zhou, J.; Kwon, Y.S.; Liao, D.J.; Jun, Y.; Eom, S.; et al. SRSF2 promotes splicing and transcription of exon 11 included isoform in Ron proto-oncogene. Biochim. Biophys. Acta 2014, 1839, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D. Specific commitment of different pre-mRNAs to splicing by single SR proteins. Nature 1993, 365, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Maniatis, T. Factor required for mammalian spliceosome assembly is localized to discrete regions in the nucleus. Nature 1990, 343, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Maniatis, T. The 35-kDa mammalian splicing factor SC35 mediates specific interactions between U1 and U2 small nuclear ribonucleoprotein particles at the 3’ splice site. Proc. Natl. Acad. Sci. USA 1992, 89, 1725–1729. [Google Scholar] [CrossRef] [PubMed]
- Daubner, G.M.; Clery, A.; Jayne, S.; Stevenin, J.; Allain, F.H. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J. 2012, 31, 162–174. [Google Scholar] [CrossRef]
- Tacke, R.; Manley, J.L. The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J. 1995, 14, 3540–3551. [Google Scholar] [CrossRef]
- Moon, H.; Cho, S.; Loh, T.J.; Jang, H.N.; Liu, Y.; Choi, N.; Oh, J.; Ha, J.; Zhou, J.; Cho, S.; et al. SRSF2 directly inhibits intron splicing to suppresses cassette exon inclusion. BMB Rep. 2017, 50, 423–428. [Google Scholar] [CrossRef]
- Pandit, S.; Zhou, Y.; Shiue, L.; Coutinho-Mansfield, G.; Li, H.; Qiu, J.; Huang, J.; Yeo, G.W.; Ares, M., Jr.; Fu, X.D. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol. Cell 2013, 50, 223–235. [Google Scholar] [CrossRef]
- Lin, S.; Coutinho-Mansfield, G.; Wang, D.; Pandit, S.; Fu, X.D. The splicing factor SC35 has an active role in transcriptional elongation. Nat. Struct. Mol. Biol. 2008, 15, 819–826. [Google Scholar] [CrossRef]
- Das, R.; Yu, J.; Zhang, Z.; Gygi, M.P.; Krainer, A.R.; Gygi, S.P.; Reed, R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 2007, 26, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Crawford, T.O.; Pardo, C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Harahap, N.I.; Hamamura, Y.; Ar Rochmah, M.; Shima, A.; Morisada, N.; Shinohara, M.; Saito, T.; Saito, K.; Lai, P.S.; et al. Alternative splicing of a cryptic exon embedded in intron 6 of SMN1 and SMN2. Hum. Genome Var. 2016, 3, 16040. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Singh, N.N.; Ottesen, E.W.; Lee, B.M.; Singh, R.N. A novel human-specific splice isoform alters the critical C-terminus of Survival Motor Neuron protein. Sci. Rep. 2016, 6, 30778. [Google Scholar] [CrossRef] [PubMed]
- Kashima, T.; Manley, J.L. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet. 2003, 34, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef]
- Cho, S.; Moon, H.; Loh, T.J.; Jang, H.N.; Liu, Y.; Zhou, J.; Ohn, T.; Zheng, X.; Shen, H. Splicing inhibition of U2AF65 leads to alternative exon skipping. Proc. Natl. Acad. Sci. USA 2015, 112, 9926–9931. [Google Scholar] [CrossRef]
- Bose, J.K.; Wang, I.F.; Hung, L.; Tarn, W.Y.; Shen, C.K. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J. Biol. Chem. 2008, 283, 28852–28859. [Google Scholar] [CrossRef]
- Cho, S.; Moon, H.; Loh, T.J.; Oh, H.K.; Cho, S.; Choy, H.E.; Song, W.K.; Chun, J.S.; Zheng, X.; Shen, H. hnRNP M facilitates exon 7 inclusion of SMN2 pre-mRNA in spinal muscular atrophy by targeting an enhancer on exon 7. Biochim. Biophys. Acta 2014, 1839, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Moon, H.; Loh, T.J.; Oh, H.K.; Williams, D.R.; Liao, D.J.; Zhou, J.; Green, M.R.; Zheng, X.; Shen, H. PSF contacts exon 7 of SMN2 pre-mRNA to promote exon 7 inclusion. Biochim. Biophys. Acta 2014, 1839, 517–525. [Google Scholar] [CrossRef][Green Version]
- Hofmann, Y.; Wirth, B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum. Mol. Genet. 2002, 11, 2037–2049. [Google Scholar] [CrossRef] [PubMed]
- Young, P.J.; DiDonato, C.J.; Hu, D.; Kothary, R.; Androphy, E.J.; Lorson, C.L. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2 beta 1. Hum. Mol. Genet. 2002, 11, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Stuani, C.; De Prato, G.; Baralle, F.E. SR protein-mediated inhibition of CFTR exon 9 inclusion: Molecular characterization of the intronic splicing silencer. Nucleic Acids Res. 2007, 35, 4359–4368. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.B.; Patton, J.G. Activation of alpha-tropomyosin exon 2 is regulated by the SR protein 9G8 and heterogeneous nuclear ribonucleoproteins H and F. Mol. Cell. Biol. 2006, 26, 8791–8802. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.W.; Maniatis, T. Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev. 1996, 10, 2089–2101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 2015, 112, E4726–E4734. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, H.; Jang, H.N.; Liu, Y.; Choi, N.; Oh, J.; Ha, J.; Zheng, X.; Shen, H. Activation of Cryptic 3′ Splice-Sites by SRSF2 Contributes to Cassette Exon Skipping. Cells 2019, 8, 696. https://doi.org/10.3390/cells8070696
Moon H, Jang HN, Liu Y, Choi N, Oh J, Ha J, Zheng X, Shen H. Activation of Cryptic 3′ Splice-Sites by SRSF2 Contributes to Cassette Exon Skipping. Cells. 2019; 8(7):696. https://doi.org/10.3390/cells8070696
Chicago/Turabian StyleMoon, Heegyum, Ha Na Jang, Yongchao Liu, Namjeong Choi, Jagyeong Oh, Jiyeon Ha, Xuexiu Zheng, and Haihong Shen. 2019. "Activation of Cryptic 3′ Splice-Sites by SRSF2 Contributes to Cassette Exon Skipping" Cells 8, no. 7: 696. https://doi.org/10.3390/cells8070696
APA StyleMoon, H., Jang, H. N., Liu, Y., Choi, N., Oh, J., Ha, J., Zheng, X., & Shen, H. (2019). Activation of Cryptic 3′ Splice-Sites by SRSF2 Contributes to Cassette Exon Skipping. Cells, 8(7), 696. https://doi.org/10.3390/cells8070696