Induction of Acquired Resistance towards EGFR Inhibitor Gefitinib in a Patient-Derived Xenograft Model of Non-Small Cell Lung Cancer and Subsequent Molecular Characterization

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. PDX Establishment
2.2. Treatment Experiments In Vivo
2.3. Formalin-Fixed Paraffin-Embedded (FFPE) Samples, Tissue Micro Array (TMA), and Immune-Histochemistry (IHC)
2.4. RNA Isolation
2.5. DNA Isolation
2.6. Reverse Transcription
2.7. qRT-PCR
2.8. Mutational Analysis (Whole Exome Sequencing)
2.9. Reverse Phase Protein Array (RPPA)
2.10. Protein–Protein Association Network Analysis
2.11. Statistical Analysis
3. Results
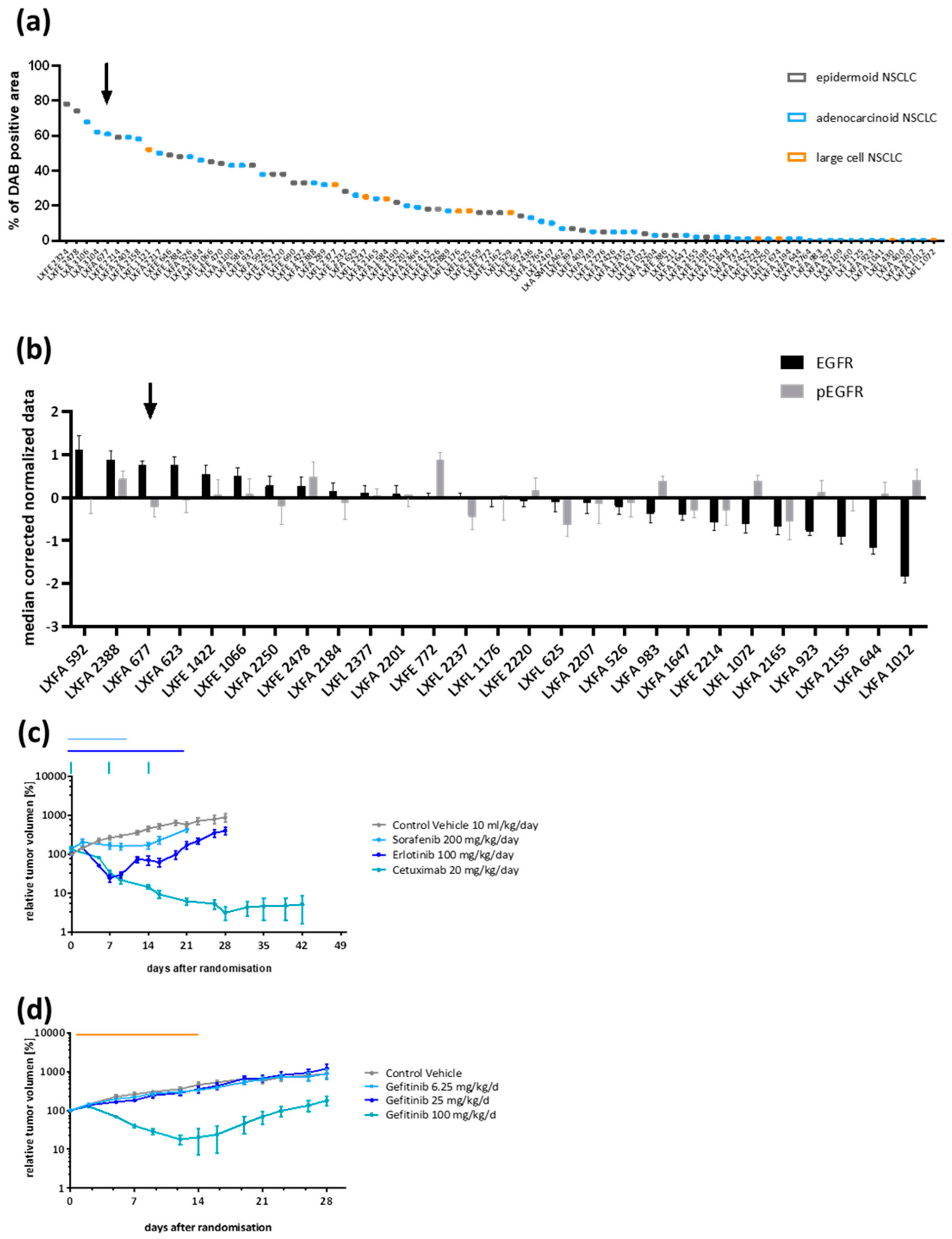
3.1. Patient-Derived NSCLC Xenograft LXFA 677 Showed EGFR Dependency and Dose Dependent Sensitivity towards Gefitinib
3.2. Gefitinib-Resistant Tumors Emerged under Continuous Treatment
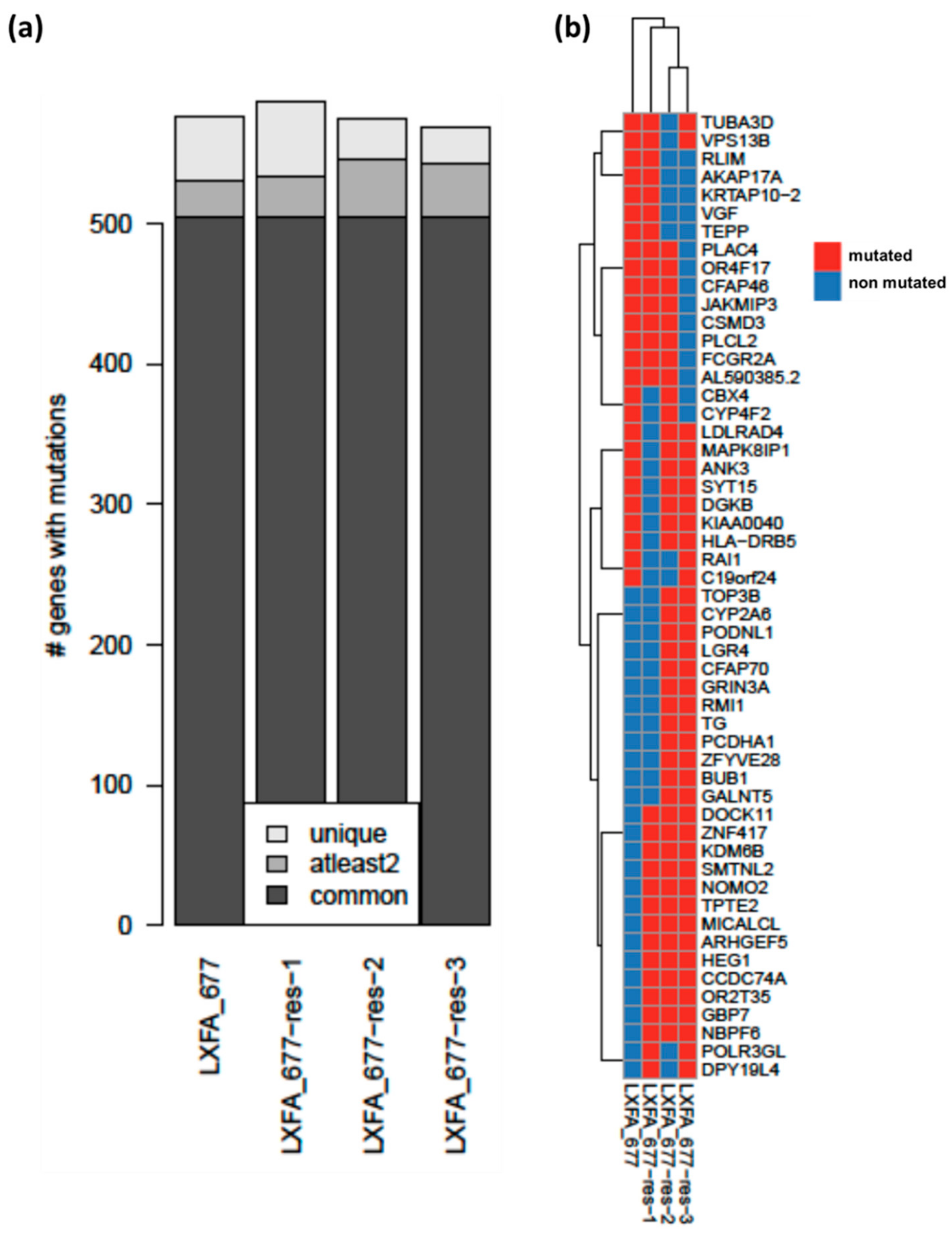
3.3. Whole Exome Sequencing Analysis Revealed High Similarities between the Resistant and the Treatment Naïve Clones of NSCLC PDX LXFA 677
3.4. The NSCLC PDX Lines Emerging under Gefitinib Treatment Displayed Specific Proteomic Profiles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [PubMed]
- Antony, J.; Thiery, J.P.; Huang, R.Y. Epithelial-to-mesenchymal transition: Lessons from development, insights into cancer and the potential of EMT-subtype based therapeutic intervention. Phys. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Siu, M.K.; Abou-Kheir, W.; Yin, J.J.; Chang, Y.S.; Barrett, B.; Suau, F.; Casey, O.; Chen, W.Y.; Fang, L.; Hynes, P.; et al. Loss of EGFR signaling regulated miR-203 promotes prostate cancer bone metastasis and tyrosine kinase inhibitors resistance. Oncotarget 2014, 5, 3770–3784. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Et Biophys. Acta 2019, 1871, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; Zhang, S. Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer. Oncotarget 2017, 8, 90557–90578. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Bao, Y.; Guo, Y.; Yang, W. Proline-Rich Protein Tyrosine Kinase 2 in Inflammation and Cancer. Cancers 2018, 10, 139. [Google Scholar] [CrossRef]
- Takeda, M.; Nakagawa, K. First- and Second-Generation EGFR-TKIs Are All Replaced to Osimertinib in Chemo-Naive EGFR Mutation-Positive Non-Small Cell Lung Cancer? Int. J. Mol. Sci. 2019, 20, 146. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Ohashi, K.; Maruvka, Y.E.; Michor, F.; Pao, W. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor–Resistant Disease. J. Clin. Oncol. 2013, 31, 1070–1080. [Google Scholar] [CrossRef]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, ra26–ra75. [Google Scholar] [CrossRef]
- Lee, C.K.; Kim, S.; Lee, J.S.; Lee, J.E.; Kim, S.M.; Yang, I.S.; Kim, H.R.; Lee, J.H.; Kim, S.; Cho, B.C. Next-generation sequencing reveals novel resistance mechanisms and molecular heterogeneity in EGFR-mutant non-small cell lung cancer with acquired resistance to EGFR-TKIs. Lung Cancer 2017, 113, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Herter-Sprie, G.S.; Kung, A.L.; Wong, K.K. New cast for a new era: Preclinical cancer drug development revisited. J. Clin. Investig. 2013, 123, 3639–3645. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A. Tumor models for efficacy determination. Mol. Cancer Ther. 2006, 5, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Reviews. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Conway, T.; Wazny, J.; Bromage, A.; Tymms, M.; Sooraj, D.; Williams, E.D.; Beresford-Smith, B. Xenome—a tool for classifying reads from xenograft samples. Bioinformatics 2012, 28, i172–i178. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- Dayem Ullah, A.Z.; Oscanoa, J.; Wang, J.; Nagano, A.; Lemoine, N.R.; Chelala, C. SNPnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018, 46, W109–W113. [Google Scholar] [CrossRef]
- Meseure, D.; Vacher, S.; Lallemand, F.; Alsibai, K.D.; Hatem, R.; Chemlali, W.; Nicolas, A.; De Koning, L.; Pasmant, E.; Callens, C.; et al. Prognostic value of a newly identified MALAT1 alternatively spliced transcript in breast cancer. Br. J. Cancer 2016, 114, 1395–1404. [Google Scholar] [CrossRef]
- Troncale, S.; Barbet, A.; Coulibaly, L.; Henry, E.; He, B.; Barillot, E.; Dubois, T.; Hupe, P.; de Koning, L. NormaCurve: A SuperCurve-based method that simultaneously quantifies and normalizes reverse phase protein array data. PLoS ONE 2012, 7, e38686. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef]
- Sebens, S.; Schafer, H. The tumor stroma as mediator of drug resistance—A potential target to improve cancer therapy? Curr. Pharm. Biotechnol. 2012, 13, 2259–2272. [Google Scholar] [CrossRef]
- Campos-Parra, A.D.; Zuloaga, C.; Manriquez, M.E.; Aviles, A.; Borbolla-Escoboza, J.; Cardona, A.; Meneses, A.; Arrieta, O. KRAS mutation as the biomarker of response to chemotherapy and EGFR-TKIs in patients with advanced non-small cell lung cancer: Clues for its potential use in second-line therapy decision making. Am. J. Clin. Oncol. 2015, 38, 33–40. [Google Scholar] [CrossRef]
- Zhang, J.; Babic, A. Regulation of the MET oncogene: Molecular mechanisms. Carcinogenesis 2016, 37, 345–355. [Google Scholar] [CrossRef]
- Yun, H.S.; Baek, J.H.; Yim, J.H.; Um, H.D.; Park, J.K.; Song, J.Y.; Park, I.C.; Kim, J.S.; Lee, S.J.; Lee, C.W.; et al. Radiotherapy diagnostic biomarkers in radioresistant human H460 lung cancer stem-like cells. Cancer Biol. Ther. 2016, 17, 208–218. [Google Scholar] [CrossRef]
- Haffner, C.; Frauli, M.; Topp, S.; Irmler, M.; Hofmann, K.; Regula, J.T.; Bally-Cuif, L.; Haass, C. Nicalin and its binding partner Nomo are novel Nodal signaling antagonists. EMBO J. 2004, 23, 3041–3050. [Google Scholar] [CrossRef]
- Kalyan, A.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Chae, Y.K.; Matsangou, M.; Hendrix, M.J.C.; Giles, F. Nodal Signaling as a Developmental Therapeutics Target in Oncology. Mol. Cancer Ther. 2017, 16, 787–792. [Google Scholar] [CrossRef]
- He, P.; Wu, W.; Wang, H.; Liao, K.; Zhang, W.; Xiong, G.; Wu, F.; Meng, G.; Yang, K. Co-expression of Rho guanine nucleotide exchange factor 5 and Src associates with poor prognosis of patients with resected non-small cell lung cancer. Oncol. Rep. 2013, 30, 2864–2870. [Google Scholar] [CrossRef]
- Komiya, Y.; Onodera, Y.; Kuroiwa, M.; Nomimura, S.; Kubo, Y.; Nam, J.M.; Kajiwara, K.; Nada, S.; Oneyama, C.; Sabe, H.; et al. The Rho guanine nucleotide exchange factor ARHGEF5 promotes tumor malignancy via epithelial-mesenchymal transition. Oncogenesis 2016, 5, e258. [Google Scholar] [CrossRef]
- DiBardino, D.M.; Rawson, D.W.; Saqi, A.; Heymann, J.J.; Pagan, C.A.; Bulman, W.A. Next-generation sequencing of non-small cell lung cancer using a customized, targeted sequencing panel: Emphasis on small biopsy and cytology. CytoJournal 2017, 14, 7. [Google Scholar] [CrossRef]
- Gadea, G.; Blangy, A. Dock-family exchange factors in cell migration and disease. Eur. J. Cell Biol. 2014, 93, 466–477. [Google Scholar] [CrossRef]
- Gordon, E.A.; Whisenant, T.C.; Zeller, M.; Kaake, R.M.; Gordon, W.M.; Krotee, P.; Patel, V.; Huang, L.; Baldi, P.; Bardwell, L. Combining docking site and phosphosite predictions to find new substrates: Identification of smoothelin-like-2 (SMTNL2) as a c-Jun N-terminal kinase (JNK) substrate. Cell. Signal. 2013, 25, 2518–2529. [Google Scholar] [CrossRef]
- Kim, B.H.; Shenoy, A.R.; Kumar, P.; Das, R.; Tiwari, S.; MacMicking, J.D. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 2011, 332, 717–721. [Google Scholar] [CrossRef]
- Miura, K. ERK2-binding domain is required for phosphorylation of EBITEIN1, a potential downstream interactor of ERK2. Biochem. Biophys. Res. Commun. 2008, 375, 367–371. [Google Scholar] [CrossRef]
- Silva, M.P.; Barros-Silva, J.D.; Vieira, J.; Lisboa, S.; Torres, L.; Correia, C.; Vieira-Coimbra, M.; Martins, A.T.; Jeronimo, C.; Henrique, R.; et al. NCOA2 is a candidate target gene of 8q gain associated with clinically aggressive prostate cancer. Geneschromosomes Cancer 2016, 55, 365–374. [Google Scholar] [CrossRef]
- Xiao, Z.G.; Shen, J.; Zhang, L.; Li, L.F.; Li, M.X.; Hu, W.; Li, Z.J.; Cho, C.H. The Roles of Histone Demethylase UTX and JMJD3 (KDM6B) in Cancers: Current Progress and Future Perspectives. Curr. Med. Chem. 2016, 23, 3687–3696. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, M.; Zhu, H.; Xu, J. A 15-gene signature for prediction of colon cancer recurrence and prognosis based on SVM. Gene 2017, 604, 33–40. [Google Scholar] [CrossRef]
- Ali, G.; Bruno, R.; Poma, A.M.; Affinito, O.; Monticelli, A.; Piaggi, P.; Ricciardi, S.; Lucchi, M.; Melfi, F.; Chella, A.; et al. Whole transcriptome targeted gene quantification provides new insights on pulmonary sarcomatoid carcinomas. Sci. Rep. 2019, 9, 3536. [Google Scholar] [CrossRef]
- Cathcart-Rake, E.; Lopez-Chavez, A. Young male with fanconi anemia and EGFR-mutant non-small-cell lung cancer. J. Thorac. Oncol. 2014, 9, e83–e85. [Google Scholar] [CrossRef]
- Pfaffle, H.N.; Wang, M.; Gheorghiu, L.; Ferraiolo, N.; Greninger, P.; Borgmann, K.; Settleman, J.; Benes, C.H.; Sequist, L.V.; Zou, L.; et al. EGFR-activating mutations correlate with a Fanconi anemia-like cellular phenotype that includes PARP inhibitor sensitivity. Cancer Res. 2013, 73, 6254–6263. [Google Scholar] [CrossRef]
- Ye, H.; Zhang, X.; Chen, Y.; Liu, Q.; Wei, J. Ranking novel cancer driving synthetic lethal gene pairs using TCGA data. Oncotarget 2016, 7, 55352–55367. [Google Scholar] [CrossRef]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’Andrea, A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Li, L.; Gu, X.; Yue, J.; Zhao, Q.; Lv, D.; Chen, H.; Xu, L. Acquisition of EGFR TKI resistance and EMT phenotype is linked with activation of IGF1R/NF-kappaB pathway in EGFR-mutant NSCLC. Oncotarget 2017, 8, 92240–92253. [Google Scholar]
- Matusan-Ilijas, K.; Damante, G.; Fabbro, D.; Dordevic, G.; Hadzisejdic, I.; Grahovac, M.; Avirovic, M.; Grahovac, B.; Jonjic, N.; Lucin, K. EGFR expression is linked to osteopontin and Nf-kappaB signaling in clear cell renal cell carcinoma. Clin. Transl. Oncol. 2013, 15, 65–71. [Google Scholar] [CrossRef]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef]
- Baud, V.; Collares, D. Post-Translational Modifications of RelB NF-kappaB Subunit and Associated Functions. Cells 2016, 5, 22. [Google Scholar]
- Lin, C.; Lu, W.; Ren, Z.; Tang, Y.; Zhang, C.; Yang, R.; Chen, Y.; Cao, W.; Wang, L.; Wang, X.; et al. Elevated RET expression enhances EGFR activation and mediates EGFR inhibitor resistance in head and neck squamous cell carcinoma. Cancer Lett. 2016, 377, 1–10. [Google Scholar] [CrossRef]
- Wang, S.E.; Narasanna, A.; Perez-Torres, M.; Xiang, B.; Wu, F.Y.; Yang, S.; Carpenter, G.; Gazdar, A.F.; Muthuswamy, S.K.; Arteaga, C.L. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006, 10, 25–38. [Google Scholar] [CrossRef]
- Guo, G.; Gong, K.; Wohlfeld, B.; Hatanpaa, K.J.; Zhao, D.; Habib, A.A. Ligand-Independent EGFR Signaling. Cancer Res. 2015, 75, 3436–3441. [Google Scholar] [CrossRef]
- Han, W.; Carpenter, R.L.; Cao, X.; Lo, H.W. STAT1 gene expression is enhanced by nuclear EGFR and HER2 via cooperation with STAT3. Mol. Carcinog. 2013, 52, 959–969. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef]
- Guo, G.; Gong, K.; Ali, S.; Ali, N.; Shallwani, S.; Hatanpaa, K.J.; Pan, E.; Mickey, B.; Burma, S.; Wang, D.H.; et al. A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma. Nat. Neurosci. 2017, 20, 1074–1084. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) Increases Resistance to EGFR-TKI and Activation of AKT and ERK1/2 in Non-Small Cell Lung Cancer Cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef]
- Qu, Y.; Wu, X.; Yin, Y.; Yang, Y.; Ma, D.; Li, H. Antitumor activity of selective MEK1/2 inhibitor AZD6244 in combination with PI3K/mTOR inhibitor BEZ235 in gefitinib-resistant NSCLC xenograft models. J. Exp. Clin. Cancer Res. 2014, 33, 52. [Google Scholar] [CrossRef]
- Buonato, J.M.; Lazzara, M.J. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014, 74, 309–319. [Google Scholar] [CrossRef]
- Kachroo, P.; Lee, M.H.; Zhang, L.; Baratelli, F.; Lee, G.; Srivastava, M.K.; Wang, G.; Walser, T.C.; Krysan, K.; Sharma, S.; et al. IL-27 inhibits epithelial-mesenchymal transition and angiogenic factor production in a STAT1-dominant pathway in human non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2013, 32, 97. [Google Scholar] [CrossRef]
- Kaowinn, S.; Kaewpiboon, C.; Koh, S.S.; Kramer, O.H.; Chung, Y.H. STAT1HDAC4 signaling induces epithelialmesenchymal transition and sphere formation of cancer cells overexpressing the oncogene, CUG2. Oncol. Rep. 2018, 40, 2619–2627. [Google Scholar]
- Li, B.; Huang, C. Regulation of EMT by STAT3 in gastrointestinal cancer (Review). Int. J. Oncol. 2017, 50, 753–767. [Google Scholar] [CrossRef]
- Schmitt, N.C.; Trivedi, S.; Ferris, R.L. STAT1 Activation Is Enhanced by Cisplatin and Variably Affected by EGFR Inhibition in HNSCC Cells. Mol. Cancer Ther. 2015, 14, 2103–2111. [Google Scholar] [CrossRef]
- Cheng, C.C.; Lin, H.C.; Tsai, K.J.; Chiang, Y.W.; Lim, K.H.; Chen, C.G.; Su, Y.W.; Peng, C.L.; Ho, A.S.; Huang, L.; et al. Epidermal growth factor induces STAT1 expression to exacerbate the IFNr-mediated PD-L1 axis in epidermal growth factor receptor-positive cancers. Mol. Carcinog. 2018, 57, 1588–1598. [Google Scholar] [CrossRef]
- Phuchareon, J.; McCormick, F.; Eisele, D.W.; Tetsu, O. EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function. Proc. Natl. Acad. Sci. USA 2015, 112, E3855–E3863. [Google Scholar] [CrossRef]
- Tetsu, O.; Eisele, D.W.; McCormick, F. Resistance to EGFR-targeted therapy by Ets-1 inactivation. Cell Cycle 2015, 14, 3211–3212. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Biopsy | Pre-Treatment | Stage | Pts Age | Pts Gender | Ethnicity | HLA-Type |
|---|---|---|---|---|---|---|---|
| undifferentiated adenocarcinoma of the lung | primary tumor | Cisplatin | T1N1Mx | 62 years | male | Caucasian | A*31:01 B*56:01 DQB1*05:01 DRB1*10:01 DQB1*05:01 DRB1*01:01 A*11:01 B*37:01 C*06:02 no C*01:02 no |
| Compound | Supplier | Daily Dose [mg/kg/d] | Route | Schedule |
|---|---|---|---|---|
| Sorafenib | # S8502, LC Laboratories, USA | 200 | per os | 0–11 |
| Lapatinib | #L-4899, LC Laboratories, USA | 150 | per os | 0–21 |
| Osimertinib | #O-7200, LC Laboratories, USA | 5 | per os | 0–21 |
| Afatinib | #A-8644, LC Laboratories, USA | 12.5 | per os | 0–21 |
| Necitumumab | #LY3012211, local pharmacy | 20 | intraperitoneal | twice weekly for 3 weeks |
| Erlotinib | # E-4007-25G, LC Laboratories, USA | 100 | per os | 0–21 |
| Cetuximab | PZN 00493528, Merck Serono, USA | 20 | intravenous | 0, 7, 14 |
| Sunitinib | #S-8803, LC Laboratories, USA | 30 | per os | 0–21 |
| Gefitinib | #G-4408-1G, LC Laboratories, USA | up to 50 | per os | 0—end of experiment |
| Target | Forward Primer | Reverse Primer | Used for |
|---|---|---|---|
| GAPDH | CAA′ATT′CCA′TGG′CAC′CGT′C | GCC′ACA′CCA′TCC′TAG′TTG′C | Gene copy number (housekeeping gene) |
| ERBB2 | CTGAACTGGTGTATGCAGATTGC | TTCCGAGCGGCCAAGTC | Gene copy number |
| cMET | GACATTTCCAGTCCTGCAGTCA | CTCCGATCGCACACATTTGT | Gene copy number/mRNA expression |
| ERBB3 | AATAAAAGGGCTATGAGGCGATACT | AGCTTCCTTAGCTCTGTCTCTTTGA | mRNA expression |
| PTEN | CAGTAAGCGTTTTTTTTCTTTGAAGA | TGTGTAAGGTCAAAAGGGTGGAA | mRNA expression |
| HGF | GAATACTGCAGACCAATGTGCT | TTGCAAGTGAATGGAAGTCCT | mRNA expression |
| 18S | CTACCACATCCAAGGAAGGCA | TTTTTCGTCACTACCTCCCCG | mRNA expression (housekeeping gene) |
| Index | Designation | Source | Mono/Polyclonal |
|---|---|---|---|
| 1 | Phospho-Akt (Ser473) (193H12) | Rabbit | Monoclonal |
| 2 | Phospho-PTEN (ser380/Thr382/383) | Rabbit | Polyclonal |
| 3 | p44/42 MAPK | Rabbit | Polyclonal |
| 4 | Stat3 | Rabbit | Polyclonal |
| 5 | Phospho-p38 MAPK (Thr180/Tyr182) | Rabbit | Monoclonal |
| 6 | Phospho-Stat3 (Ser727) | Rabbit | Polyclonal |
| 7 | Stat1 | Rabbit | Polyclonal |
| 8 | Phospho-MEK1/2 (Ser217/221) | Rabbit | Monoclonal |
| 9 | Phospho-p44/42 MAPK (Thr202/Tyr204) | Rabbit | Monoclonal |
| 10 | Phospho-c-jun (Ser73) (D47G9) | Rabbit | Monoclonal |
| 11 | NF-kB p65 | Rabbit | Monoclonal |
| 12 | Phospho-NF-kB p65 (Ser536) | Rabbit | Monoclonal |
| 13 | Phospho-Met (Tyr1234/1235) | Rabbit | Monoclonal |
| 14 | p38 MAPK | Rabbit | Monoclonal |
| 15 | phospho-mTOR (Ser2448) | Rabbit | Monoclonal |
| 16 | Phospho-Her3/Erbb3 (tyr1289) | Rabbit | Monoclonal |
| 17 | HER3/ErbB3 (c-17) | Rabbit | Polyclonal |
| 18 | mTOR | Rabbit | Polyclonal |
| 19 | c-Jun (N-term) | Rabbit | Monoclonal |
| 20 | Phospho-eIF4B (Ser422) | Rabbit | Polyclonal |
| 21 | eIF4B | Rabbit | Monoclonal |
| 22 | MEK1/2 | Rabbit | Polyclonal |
| 23 | Akt (pan) (C67E7) | Rabbit | Monoclonal |
| 24 | PTEN (D4.3) XP | Rabbit | Monoclonal |
| 25 | Phospho-Stat1 (Y701) | Rabbit | Monoclonal |
| 26 | EGFR (D38B1) | Rabbit | Monoclonal |
| 27 | RelB | Rabbit | Monoclonal |
| 28 | Met (D1C2) XP | Rabbit | Monoclonal |
| 29 | Axl (C2B12) | Rabbit | Monoclonal |
| 30 | IkB alpha (44D4) | Rabbit | Monoclonal |
| 31 | HER2/ErbB2 | Mouse | Monoclonal |
| 32 | IGF-I receptor B | Rabbit | Monoclonal |
| 33 | Phospho-EGFR (Tyr1068) (D7A5) | Rabbit | Monoclonal |
| LXFA 677 Untreated | LXFA 677 Treated | LXFA 677res1 Untreated | LXFA 677res1 Treated | LXFA 677res2 Untreated | LXFA 677res2 Treated | LXFA 677res3 Untreated | LXFA 677res3 Treated | |
|---|---|---|---|---|---|---|---|---|
| Mean [d] | 11.85 | 22.98 | 7.54 | 5.37 | 15.03 | 14.43 | 13.68 | 14.38 |
| Std. Deviation [d] | 0.07 | 7.62 | 0.86 | 0.23 | 2.36 | 3.78 | 0.00 | 2.31 |
| Nr. of values [n] | 2 | 2 | 2 | 2 | 2 | 2 | 1 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schueler, J.; Tschuch, C.; Klingner, K.; Bug, D.; Peille, A.-L.; de Koning, L.; Oswald, E.; Klett, H.; Sommergruber, W. Induction of Acquired Resistance towards EGFR Inhibitor Gefitinib in a Patient-Derived Xenograft Model of Non-Small Cell Lung Cancer and Subsequent Molecular Characterization. Cells 2019, 8, 740. https://doi.org/10.3390/cells8070740
Schueler J, Tschuch C, Klingner K, Bug D, Peille A-L, de Koning L, Oswald E, Klett H, Sommergruber W. Induction of Acquired Resistance towards EGFR Inhibitor Gefitinib in a Patient-Derived Xenograft Model of Non-Small Cell Lung Cancer and Subsequent Molecular Characterization. Cells. 2019; 8(7):740. https://doi.org/10.3390/cells8070740
Chicago/Turabian StyleSchueler, Julia, Cordula Tschuch, Kerstin Klingner, Daniel Bug, Anne-Lise Peille, Leanne de Koning, Eva Oswald, Hagen Klett, and Wolfgang Sommergruber. 2019. "Induction of Acquired Resistance towards EGFR Inhibitor Gefitinib in a Patient-Derived Xenograft Model of Non-Small Cell Lung Cancer and Subsequent Molecular Characterization" Cells 8, no. 7: 740. https://doi.org/10.3390/cells8070740
APA StyleSchueler, J., Tschuch, C., Klingner, K., Bug, D., Peille, A.-L., de Koning, L., Oswald, E., Klett, H., & Sommergruber, W. (2019). Induction of Acquired Resistance towards EGFR Inhibitor Gefitinib in a Patient-Derived Xenograft Model of Non-Small Cell Lung Cancer and Subsequent Molecular Characterization. Cells, 8(7), 740. https://doi.org/10.3390/cells8070740