Galectin-3 Regulates Indoleamine-2,3-dioxygenase-Dependent Cross-Talk between Colon-Infiltrating Dendritic Cells and T Regulatory Cells and May Represent a Valuable Biomarker for Monitoring the Progression of Ulcerative Colitis


 ,
, 
Abstract
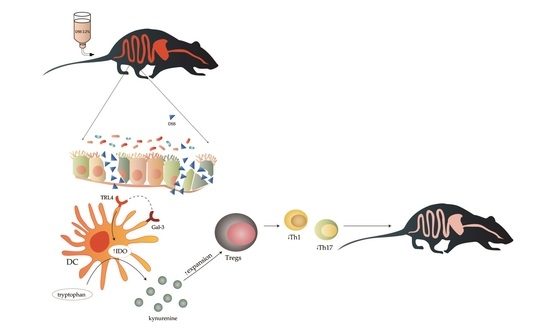
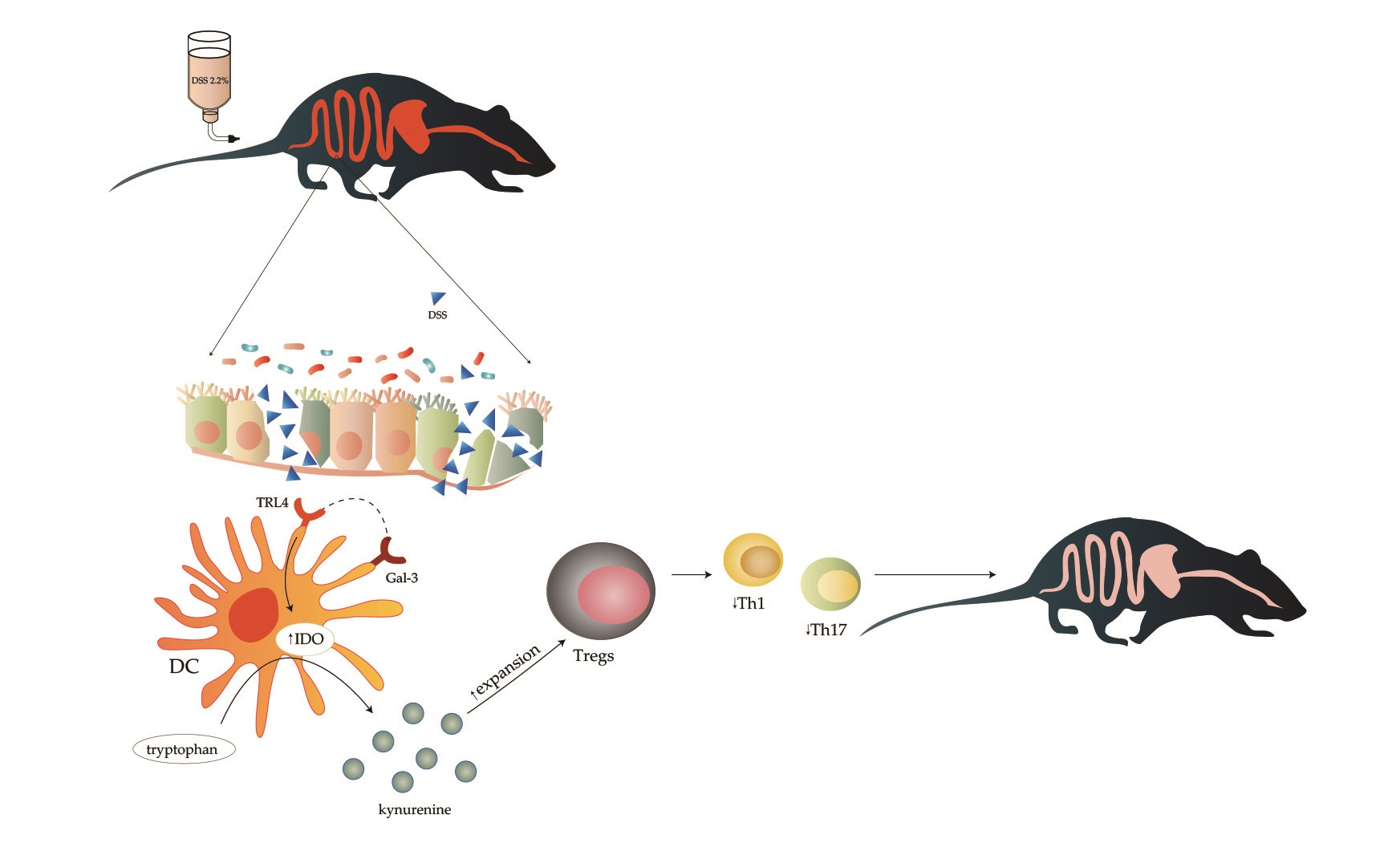
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Measurements of Gal-3, IDO Activity, Concentration of Cytokines, and Fecal Calprotectin in Serum and Fecal Samples of Patients with UC and Healthy Controls
2.3. Isolation of Colon-Infiltrating Tregs from the Colon Samples of UC Patients and Flow Cytometry Analysis of Their Phenotype and Function
2.4. Cytokine Production in T Cells from UC Patients
2.5. Animals
2.6. Induction and Evaluation of Colitis in Mice
2.7. Flow Cytometry Analysis of Colon Infiltrating Immune Cells of DSS-Treated Mice
2.8. Measurements of Cytokines in Serum Samples of DSS-Treated Mice
2.9. Isolation and TLR-4-Priming of DCs
2.10. Pharmacological Inhibition of Gal-3 in DCs
2.11. Determination of IDO1 Activity
2.12. Adoptive Transfer of TLR-4 Primed DCs in DSS-Treated Mice
2.13. Co-culture of TLR-4-Primed DCs and Tregs
2.14. Co-Culture of Tregs and Effector T Cells
2.15. RNA Isolation and Real-Time PCR Analysis
2.16. Statistics
3. Results
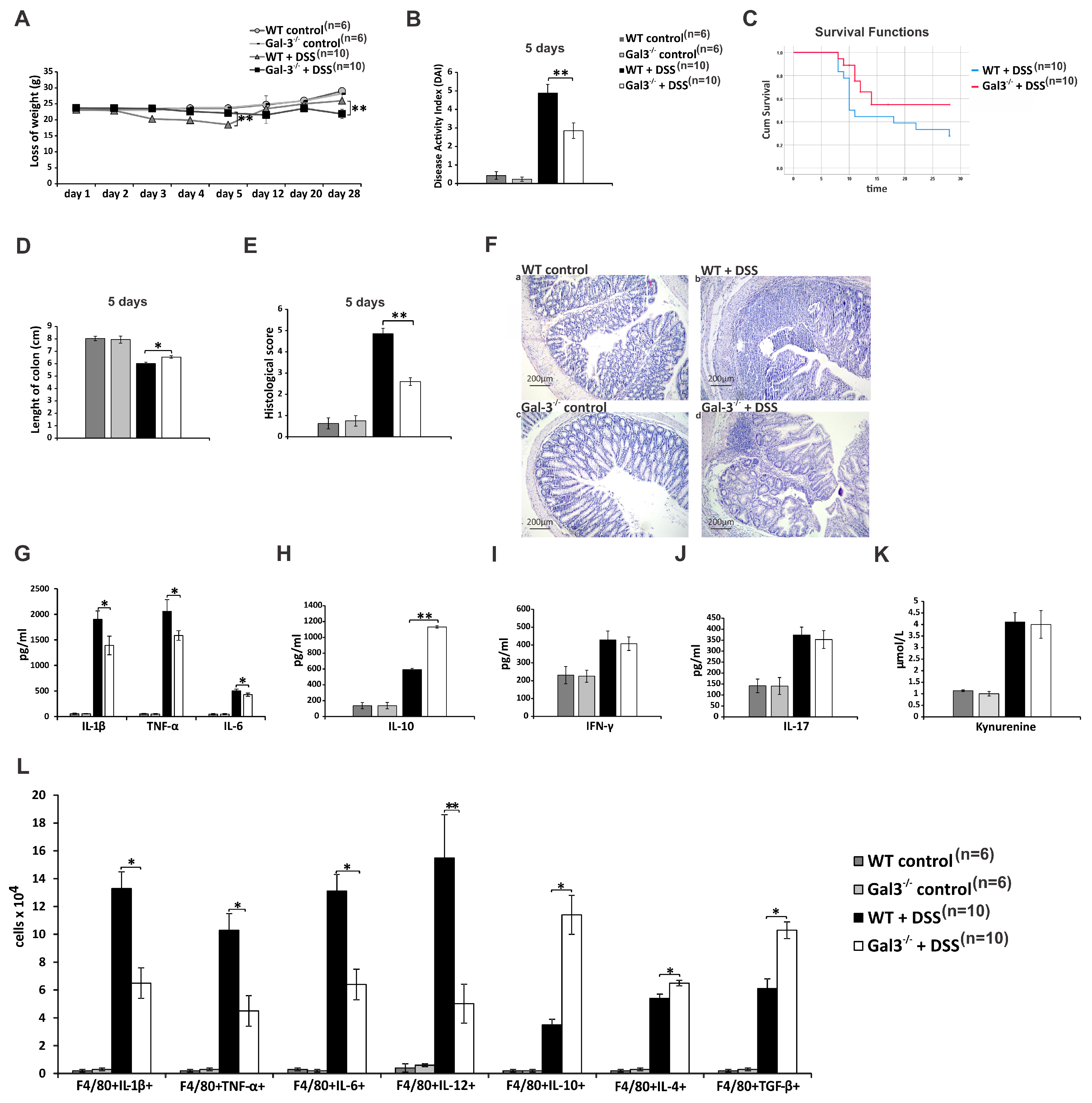
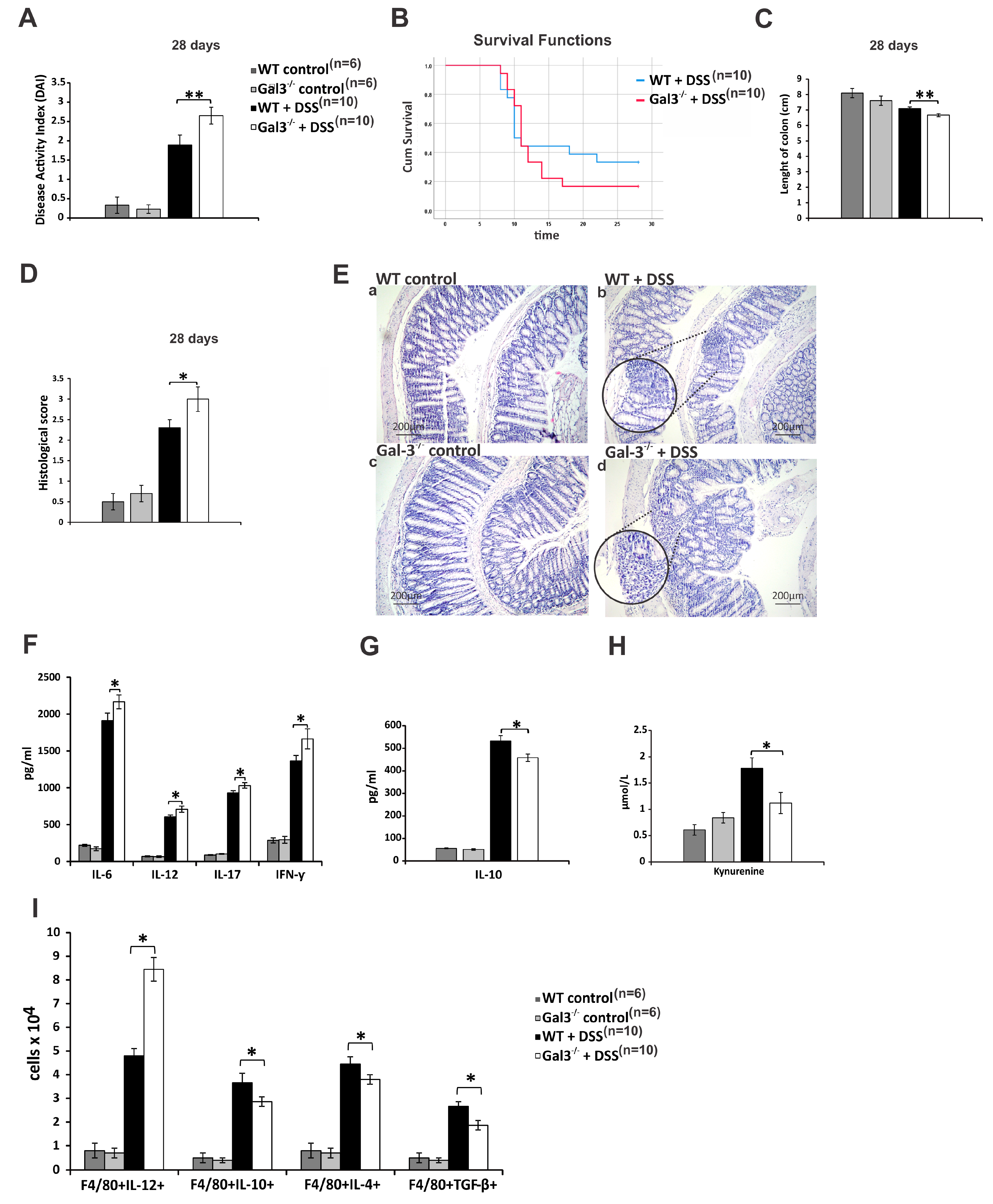
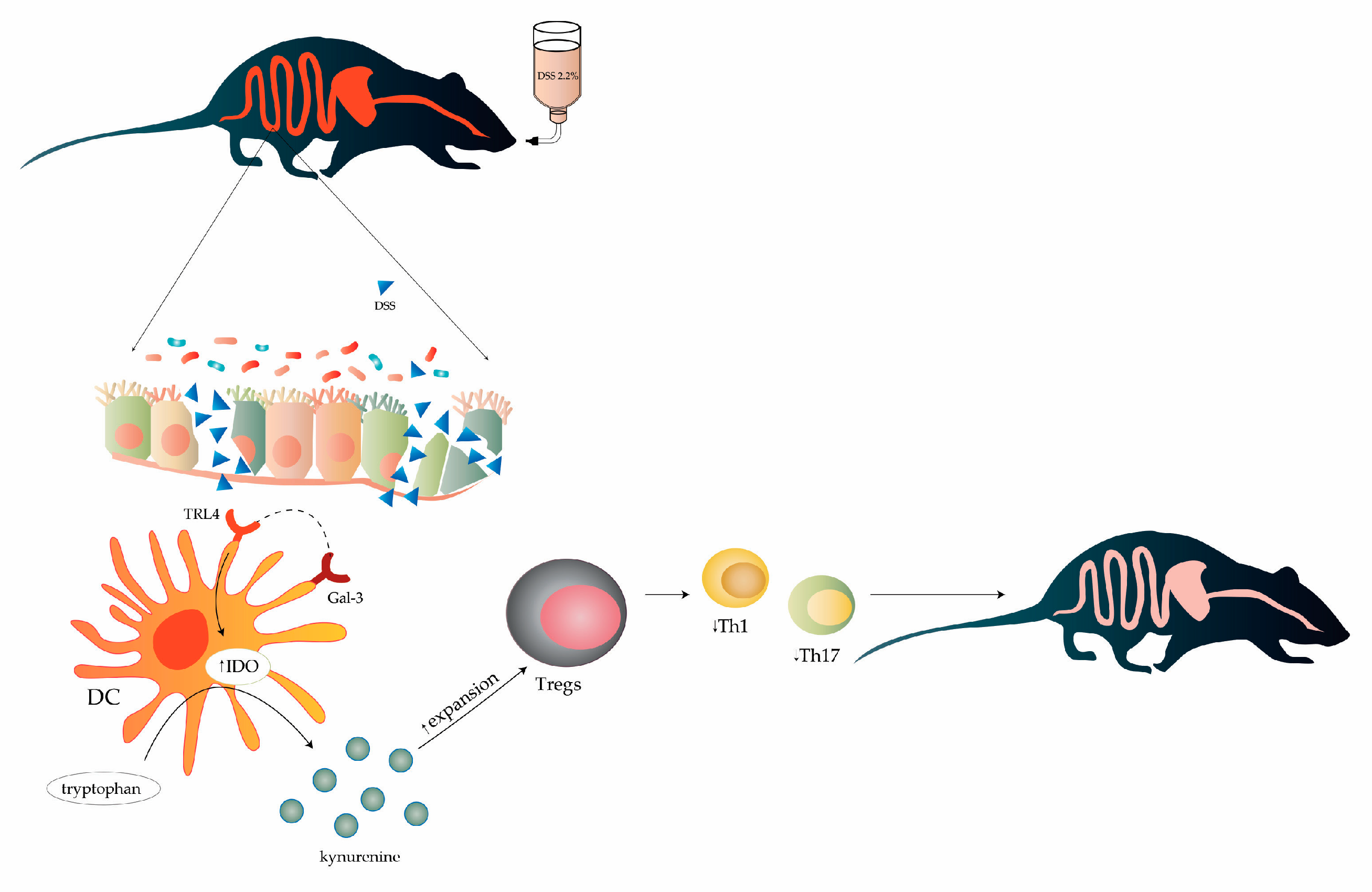
3.1. Gal-3 Had Pro-Inflammatory Role in Induction Phase of Colitis, but Its Deficiency Significantly Impaired Recovery from DSS-Induced Colon Injury
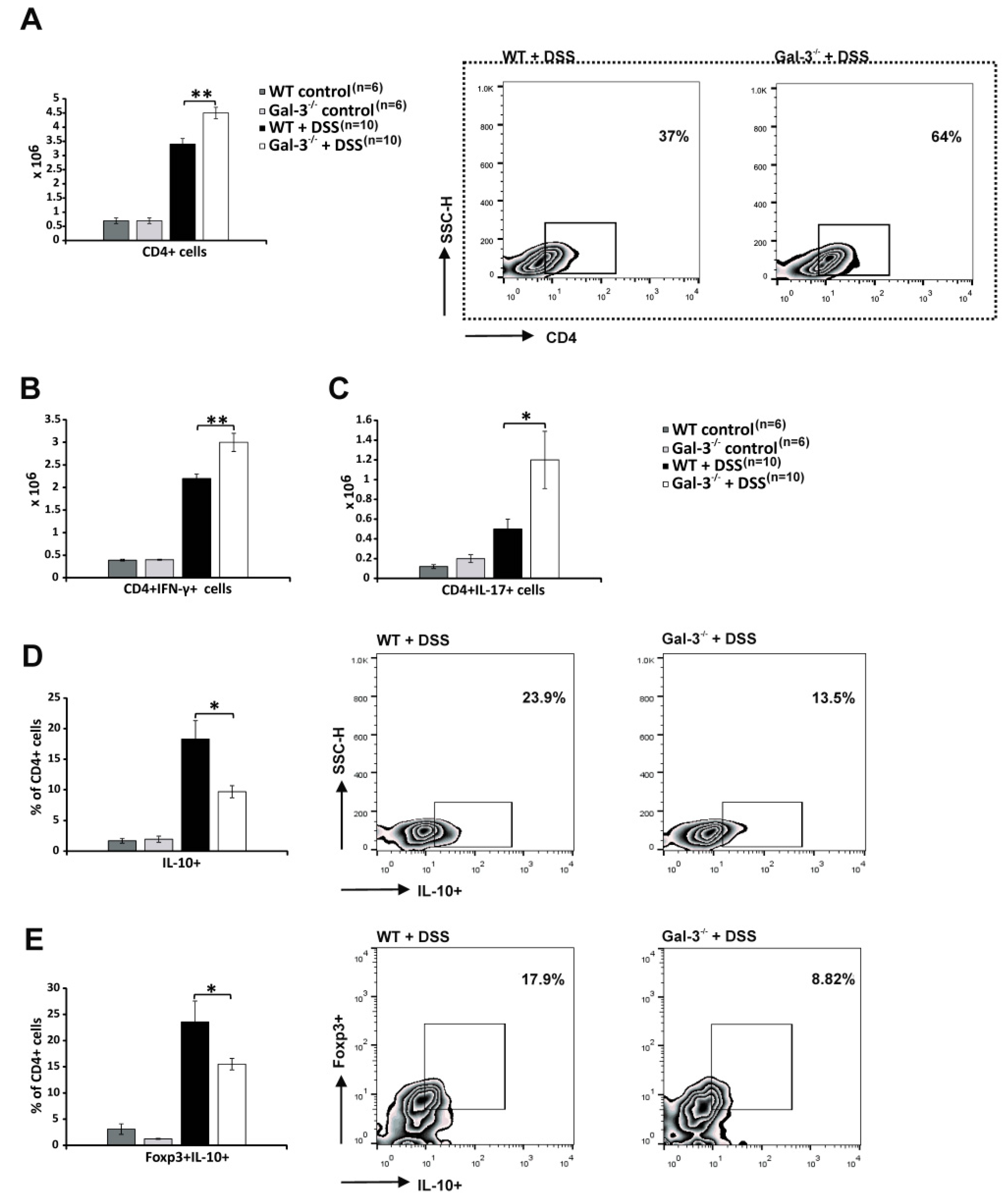
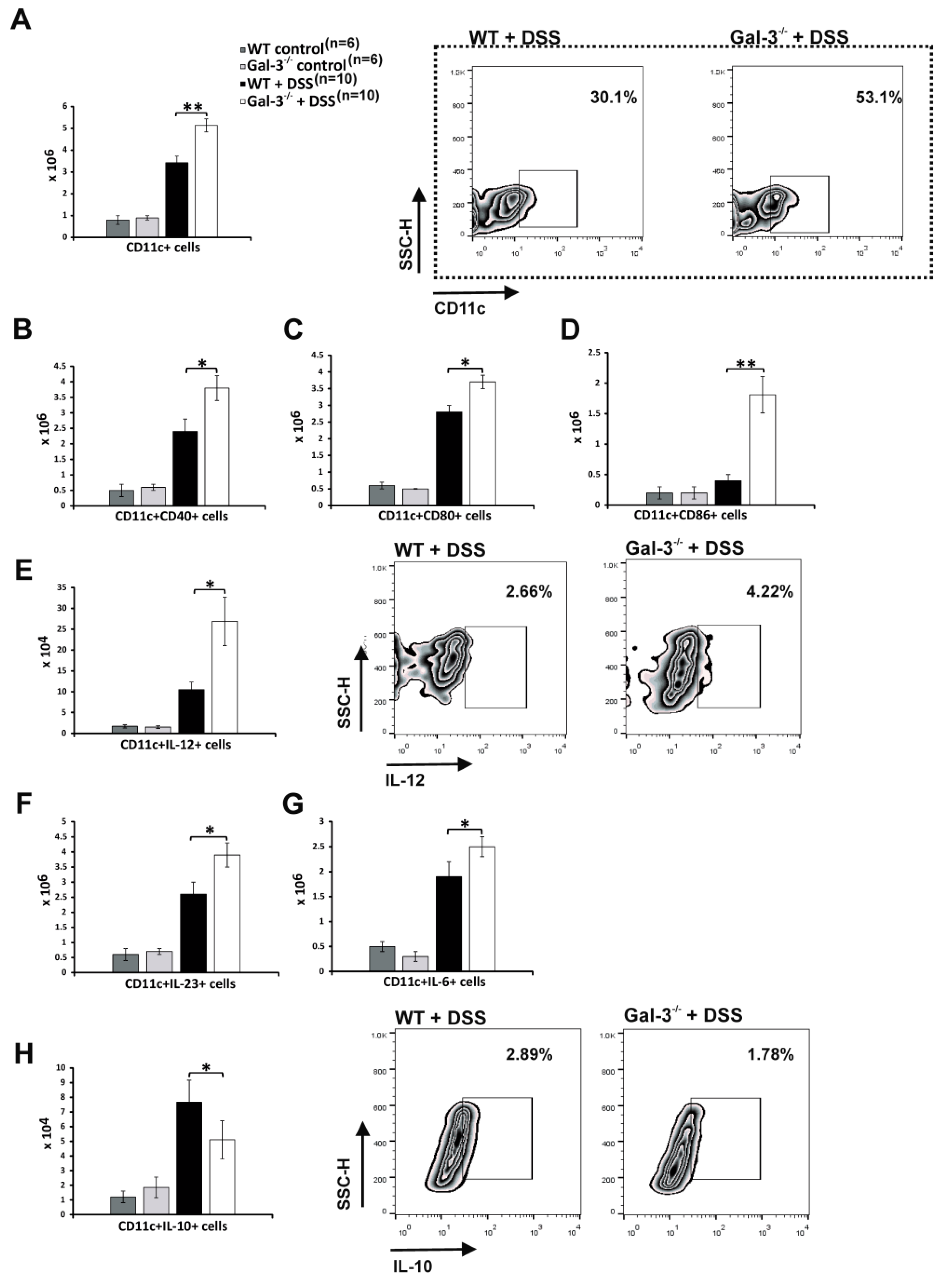
3.2. Gal-3 Deficiency Resulted in Enhanced DC-Dependent Activation of Inflammatory Th1 and Th17 Cells in DSS-Induced Colitis
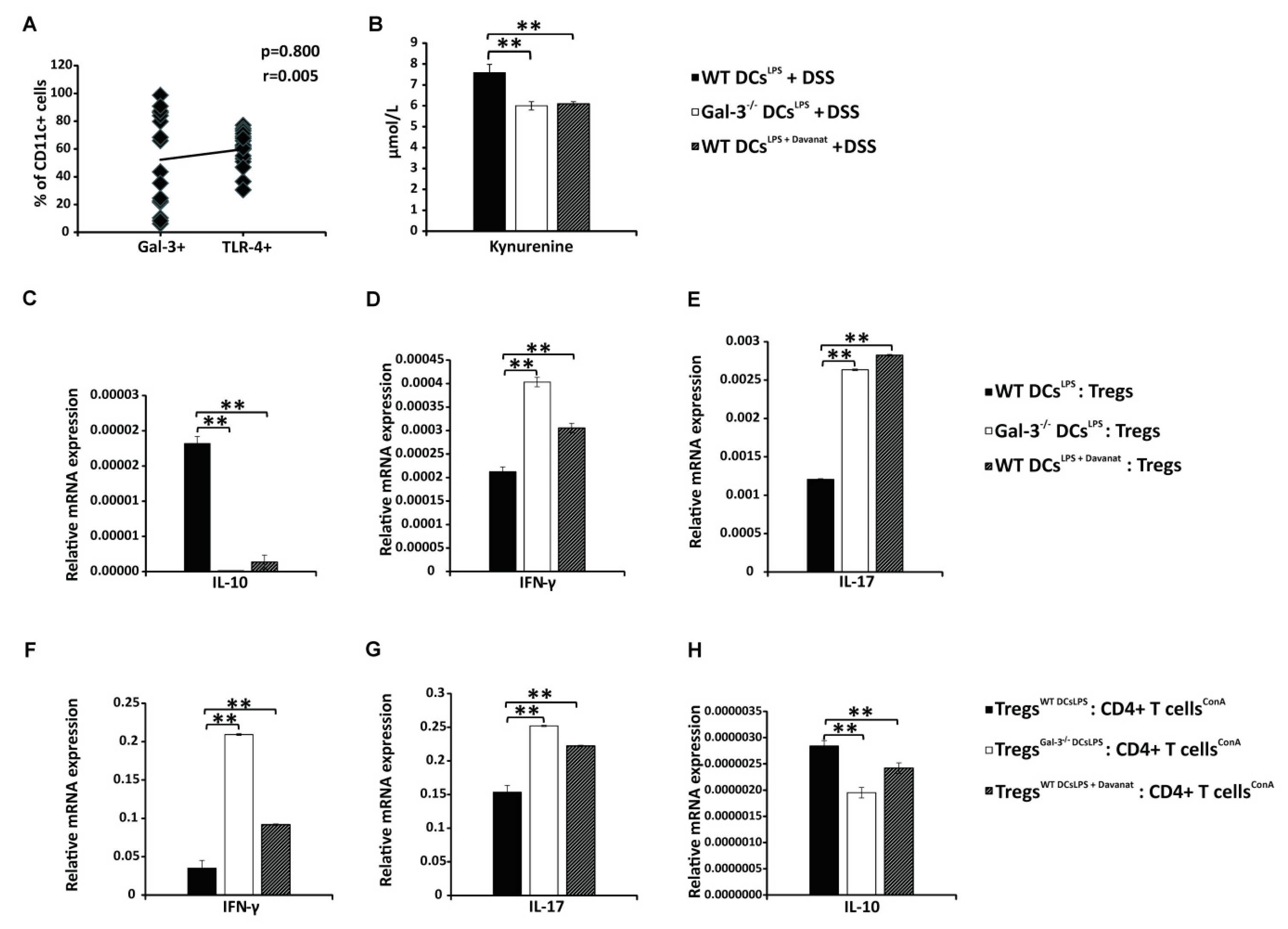
3.3. Gal-3 is Required for Optimal TLR-4-Dependent Production of KYN in DCs
3.4. Gal-3 Deficiency Completely Abrogated Capacity of TLR-4 Primed DCs to Maintain Immunosuppressive Function of Tregs
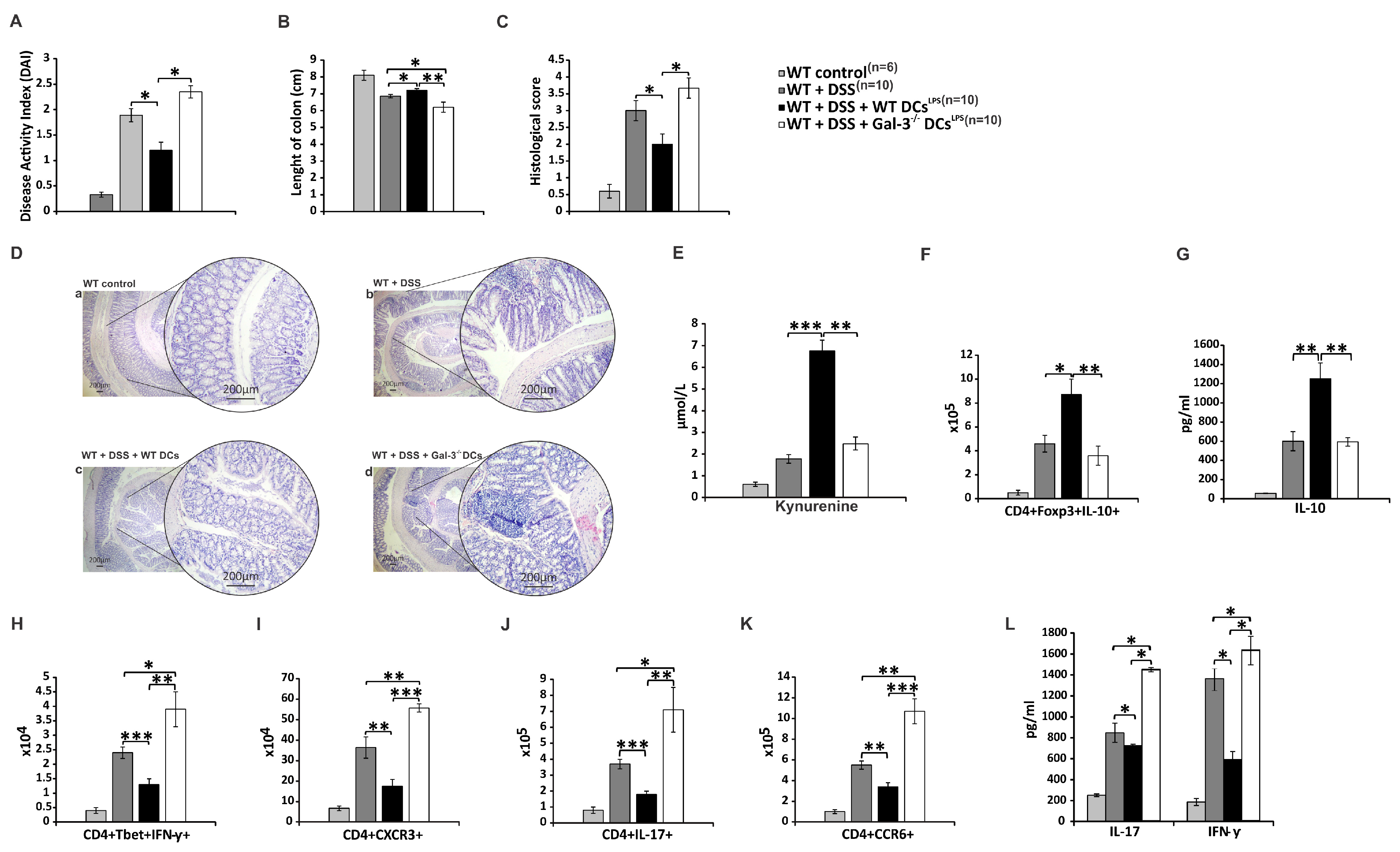
3.5. Genetic Deletion of Gal-3 Diminished Capacity of TLR-4-Primed DCs to Attenuate DSS-Induced Colitis
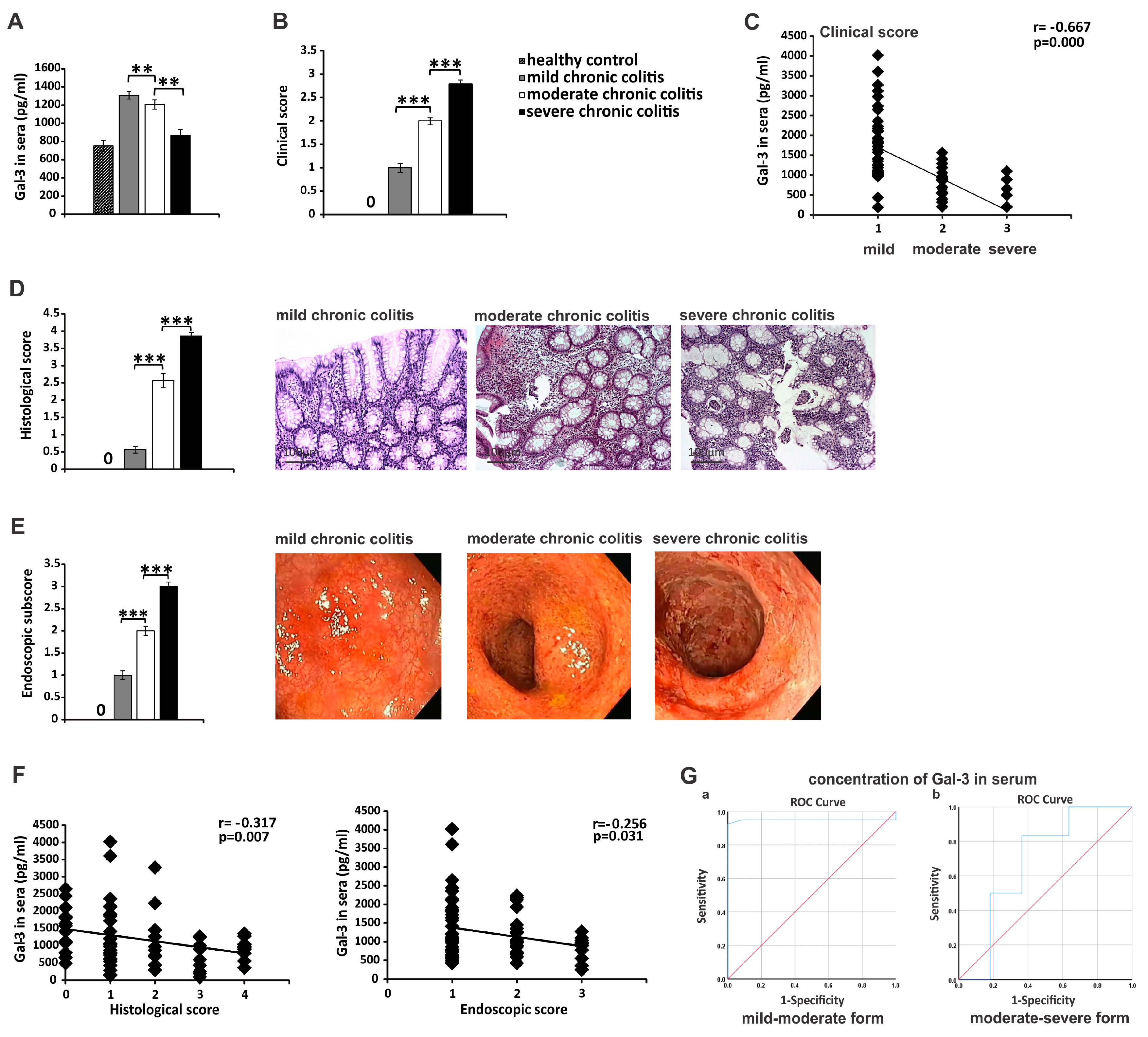
3.6. Serum Gal-3 May Serve as A Valuable Marker for Monitoring the Progression of UC
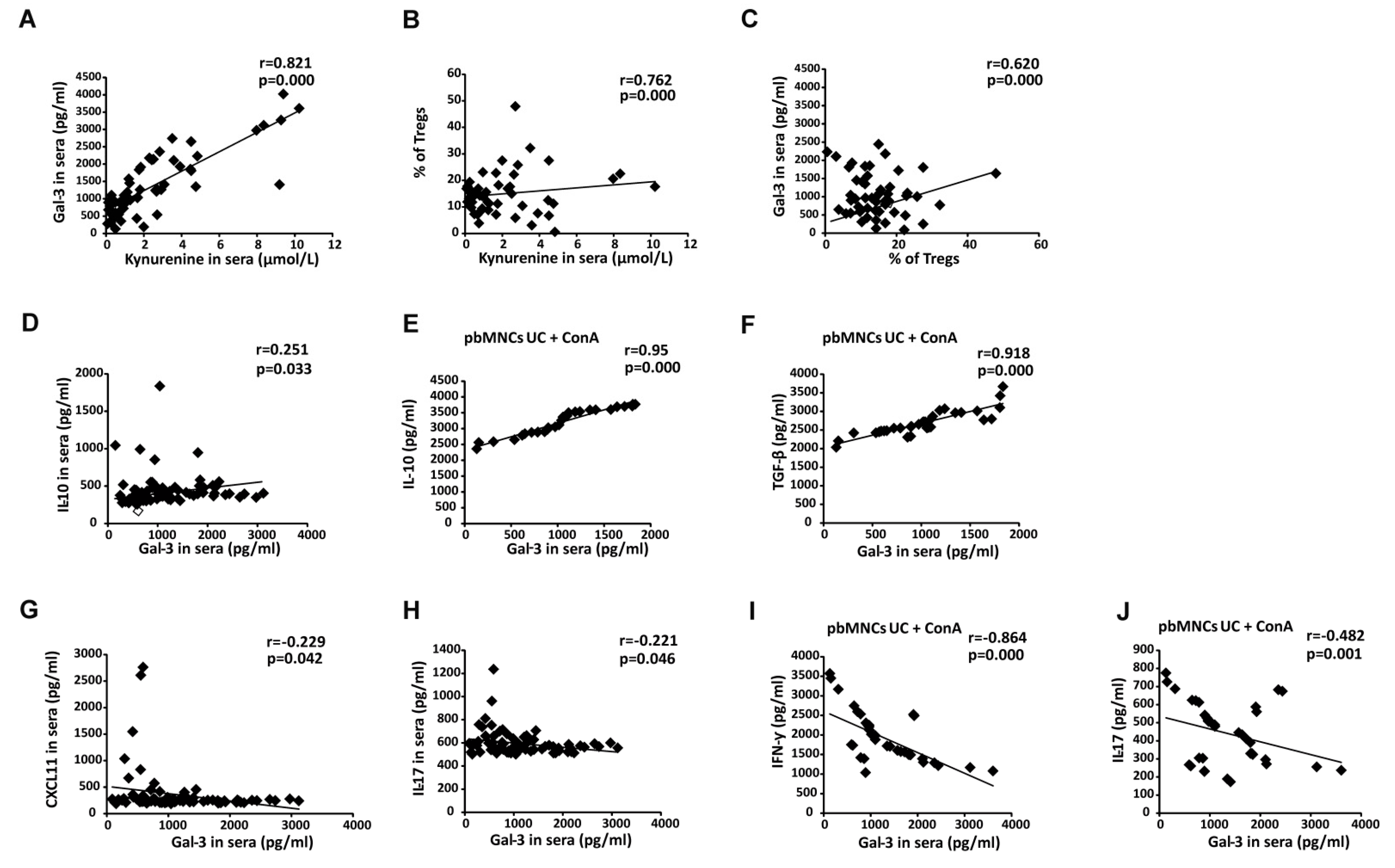
3.7. Increased Serum Levels of Gal-3 Indicates Overproduction of Immunosuppressive Cytokine in UC Patients
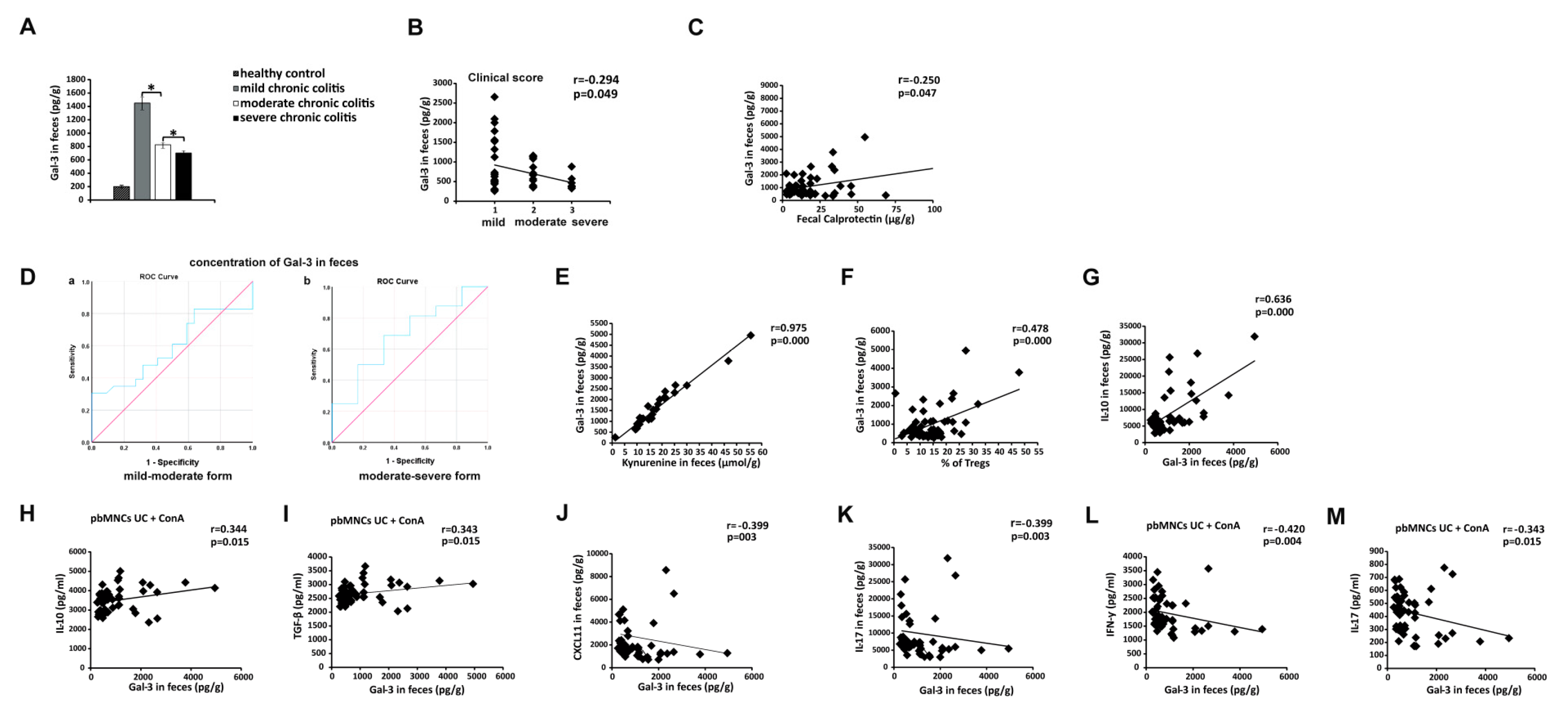
3.8. Measurement of Fecal Gal-3 May Be Used for Assessing Severity of UC
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Strober, W.; Fuss, I.; Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Investig. 2007, 117, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Naganuma, M.; Sakuraba, A.; Hibi, T. Ulcerative colitis: Prevention of relapse. Expert. Rev. Gastroenterol. Hepatol. 2013, 7, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Vilela, E.G.; Torres, H.O.; Martins, F.P.; Ferrari, M.L.; Andrade, M.M.; Cunha, A.S. Evaluation of inflammatory activity in Crohn’s disease and ulcerative colitis. World J. Gastroenterol. 2012, 18, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Malter, L.; Hudesman, D. Disease monitoring in inflammatory bowel disease. World J. Gastroenterol. 2015, 21, 11246–11259. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, U.; Rosenfeld, G.; Bressler, B.; Seidman, E. Clinical utility of fecal biomarkers for the diagnosis and management of inflammatory bowel disease. Inflamm. Bowel. Dis. 2014, 20, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Langhorst, J.; Elsenbruch, S.; Koelzer, J.; Rueffer, A.; Michalsen, A.; Dobos, G.J. Noninvasive markers in the assessment of intestinal inflammation in inflammatory bowel diseases: Performance of fecal lactoferrin, calprotectin, and PMN-elastase, CRP, and clinical indices. Am. J. Gastroenterol. 2008, 103, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Ministro, P.; Martins, D. Fecal biomarkers in inflammatory bowel disease: How, when and why? Expert Rev. Gastroenterol. Hepatol. 2017, 11, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Frolová, L.; Smetana, K., Jr.; Borovská, D.; Kitanovicová, A.; Klimesová, K.; Janatková, I.; Malícková, K.; Lukás, M.; Drastich, P.; Benes, Z.; et al. Detection of galectin-3 in patients with inflammatory bowel diseases: New serum marker of active forms of IBD? Inflamm. Res. 2009, 58, 503–512. [Google Scholar] [CrossRef]
- Simovic Markovic, B.; Nikolic, A.; Gazdic, M.; Bojic, S.; Vucicevic, L.; Kosic, M.; Mitrovic, S.; Milosavljevic, M.; Besra, G.; Trajkovic, V.; et al. Galectin-3 Plays an Important Pro-inflammatory Role in the Induction Phase of Acute Colitis by Promoting Activation of NLRP3 Inflammasome and Production of IL-1β in Macrophages. J. Crohn’s Colitis 2016, 10, 593–606. [Google Scholar] [CrossRef]
- Tsai, H.F.; Wu, C.S.; Chen, Y.L.; Liao, H.J.; Chyuan, I.T.; Hsu, P.N. Galectin-3 suppresses mucosal inflammation and reduces disease severity in experimental colitis. J. Mol. Med. 2016, 94, 545–556. [Google Scholar] [CrossRef]
- Lippert, E.; Stieber-Gunckel, M.; Dunger, N.; Falk, W.; Obermeier, F.; Kunst, C. Galectin-3 Modulates Experimental Colitis. Digestion 2015, 92, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Schaffer, T.; Flogerzi, B.; Fleetwood, A.; Weimann, R.; Schoepfer, A.M.; Seibold, F. Galectin-3 modulates T cell activity and is reduced in the inflamed intestinal epithelium in IBD. Inflamm. Bowel. Dis. 2006, 12, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Papa Gobbi, R.; De Francesco, N.; Bondar, C.; Muglia, C.; Chirdo, F.; Rumbo, M.; Rocca, A.; Toscano, M.A.; Sambuelli, A.; Rabinovich, G.A.; et al. A galectin-specific signature in the gut delineates Crohn’s disease and ulcerative colitis from other human inflammatory intestinal disorders. Biofactors 2016, 42, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Prakash, N.; Stumbles, P.; Mansfield, C. Initial validation of cytokine measurement by ELISA in canine feces. Open J. Vet. Med. 2013, 3, 282–288. [Google Scholar] [CrossRef]
- Ling, W.; Zhang, J.; Yuan, Z.; Ren, G.; Zhang, L.; Chen, X.; Rabson, A.B.; Roberts, A.I.; Wang, Y.; Shi, Y. Mesenchymal stem cells use IDO to regulate immunity in tumor microenvironment. Cancer Res. 2014, 74, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Kostas, A.; Siakavellas, S.I.; Kosmidis, C.; Takou, A.; Nikou, J.; Maropoulos, G.; Vlachogiannakos, J.; Papatheodoridis, G.V.; Papaconstantinou, I.; Bamias, G. Fecal calprotectin measurement is a marker of short-term clinical outcome and presence of mucosal healing in patients with inflammatory bowel disease. World J. Gastroenterol. 2017, 23, 7387–7396. [Google Scholar] [CrossRef]
- Acovic, A.; Simovic Markovic, B.; Gazdic, M.; Arsenijevic, A.; Jovicic, N.; Gajovic, N.; Jovanovic, M.; Zdravkovic, N.; Kanjevac, T.; Harrell, C.R.; et al. Indoleamine 2,3-dioxygenase-dependent expansion of T-regulatory cells maintains mucosal healing in ulcerative colitis. Therap. Adv. Gastroenterol. 2018, 11, 1756284818793558. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Milovanovic, M.; Ljujic, B.; Pejnovic, N.; Arsenijevic, N.; Nilsson, U.; Leffler, H.; Lukic, M.L. Galectin-3 Deficiency Prevents Concanavalin A-Induced Hepatitis in Mice. Hepatology 2012, 55, 1954–1964. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Fahlgren, A.; Sundstedt, C.; Hammarström, S.; Danielsson, A.; Hammarström, M.L. Chronic colitis induces expression of β-defensins in murine intestinal epithelial cells. Clin. Exp. Immunol. 2011, 163, 123–130. [Google Scholar] [CrossRef]
- Murthy, S.N.; Cooper, H.S.; Shim, H.; Shah, R.S.; Ibrahim, S.A.; Sedergran, D.J. Treatment of dextran sulfate sodium-induced murine colitis by intracolonic cyclosporine. Dig. Dis. Sci. 1993, 38, 1722–1734. [Google Scholar] [CrossRef]
- Whittem, C.G.; Williams, A.D.; Williams, C.S. Murine Colitis Modeling using Dextran Sulfate Sodium. J. Vis. Exp. 2010, 35, 1652. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, F.; Kojouharoff, G.; Hans, W.; Schölmerich, J.; Gross, V.; Falk, W. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin. Exp. Immunol. 1999, 116, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, Y.; Onoé, K. Enhanced IL-10 production by TLR4- and TLR2-primed dendritic cells upon TLR restimulation. J. Immunol. 2007, 178, 6173–6180. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Sasaki, Y.; Segawa, R.; Asakawa, S.; Hiratsuka, M.; Hirasawa, N. LPS priming in early life decreases antigen uptake of dendritic cells via NO production. Immunobiology 2018, 223, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Demotte, N.; Bigirimana, R.; Wieërs, G.; Stroobant, V.; Squifflet, J.L.; Carrasco, J.; Thielemans, K.; Baurain, J.F.; Van Der Smissen, P.; Courtoy, P.J.; et al. A short treatment with galactomannan GM-CT-01 corrects the functions of freshly isolated human tumor-infiltrating lymphocytes. Clin. Cancer Res. 2014, 20, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Nguyen, K.P.; Fine, S.D.; Mo, J.H.; Shen, C.; Shenouda, S.; Corr, M.; Jung, S.; Lee, J.; Eckmann, L.; et al. Conventional dendritic cells regulate the outcome of colonic inflammation independently of T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 17022–17027. [Google Scholar] [CrossRef]
- Kourepini, E.; Aggelakopoulou, M.; Alissafi, T.; Paschalidis, N.; Simoes, D.C.; Panoutsakopoulou, V. Osteopontin expression by CD103- dendritic cells drives intestinal inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, E856–E865. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Kondo, Y.; Yokosawa, M.; Tsuboi, H.; Takahashi, S.; Shibayama, S.; Matsumoto, I.; Sumida, T. T-bet regulates differentiation of forkhead box protein 3+ regulatory T cells in programmed cell death-1-deficient mice. Clin. Exp. Immunol. 2015, 179, 197–209. [Google Scholar] [CrossRef]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. Proc. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef]
- Saksida, T.; Nikolic, I.; Vujicic, M.; Nilsson, U.J.; Leffler, H.; Lukic, M.L.; Stojanovic, I.; Stosic-Grujicic, S. Galectin-3 deficiency protects pancreatic islet cells from cytokine-triggered apoptosis In Vitro. J. Cell. Physiol. 2013, 7, 1568–1576. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.P.; Elson, C.O. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 1994, 107, 1643–1652. [Google Scholar] [CrossRef]
- Melgar, S.; Karlsson, A.; Michaëlsson, E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: Correlation between symptoms and inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Seo, J.N.; Suh, Y.H.; Park, H.J.; Kim, J.H.; Kim, J.Y.; Oh, K.I. Involvement of lymphocytes in dextran sulfate sodium-induced experimental colitis. World J. Gastroenterol. 2006, 12, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Salazar, F.; Awuah, D.; Negm, O.H.; Shakib, F.; Ghaemmaghami, A.M. The role of indoleamine 2,3-dioxygenase-aryl hydrocarbon receptor pathway in the TLR4-induced tolerogenic phenotype in human DCs. Sci. Rep. 2017, 7, 43337. [Google Scholar] [CrossRef] [PubMed]
- Matteoli, G.; Mazzini, E.; Iliev, I.D.; Mileti, E.; Fallarino, F.; Puccetti, P.; Chieppa, M.; Rescigno, M. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut 2010, 59, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.P.; Kane, S. Faecal markers: Calprotectin and lactoferrin. Gastroenterol. Clin. N. Am. 2012, 41, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, L.; Xie, C.; Zou, K.; Tu, L.; Yan, W.; Hou, X. Comparison of non-invasive biomarkers faecal BAFF, calprotectin and FOBT in discriminating IBS from IBD and evaluation of intestinal inflammation. Sci. Rep. 2017, 7, 2669. [Google Scholar] [CrossRef] [PubMed]
- Mak, W.Y.; Buisson, A.; Andersen, M.J., Jr.; Lei, D.; Pekow, J.; Cohen, R.D.; Kahn, S.A.; Pereira, B.; Rubin, D.T. Fecal Calprotectin in Assessing Endoscopic and Histological Remission in Patients with Ulcerative Colitis. Dig. Dis. Sci. 2018, 63, 1294–1301. [Google Scholar] [CrossRef]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G. Signaling for inflammation and repair in inflammatory bowel disease. Rom. J. Gastroenterol. 2004, 13, 309–316. [Google Scholar] [PubMed]
- Trivedi, P.J.; Adams, D.H. Chemokines and Chemokine Receptors as Therapeutic Targets in Inflammatory Bowel Disease; Pitfalls and Promise. J. Crohns Colitis 2018, 12, S641–S652. [Google Scholar] [CrossRef] [PubMed]
- McAlindon, M.E.; Hawkey, C.J.; Mahida, Y.R. Expression of interleukin 1 beta and interleukin 1 beta converting enzyme by intestinal macrophages in health and inflammatory bowel disease. Gut 1998, 42, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Isidro, R.A.; Appleyard, C.B. Colonic macrophage polarization in homeostasis, inflammation, and cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G59–G73. [Google Scholar] [CrossRef] [PubMed]
- Kojouharoff, G.; Hans, W.; Obermeier, F.; Männel, D.N.; Andus, T.; Schölmerich, J.; Gross, V.; Falk, W. Neutralization of tumour necrosis factor (TNF) but not of IL-1 reduces inflammation in chronic dextran sulphate sodium-induced colitis in mice. Clin. Exp. Immunol. 1997, 107, 353–358. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, A.C.; Farnworth, S.L.; Hodkinson, P.S.; Henderson, N.C.; Atkinson, K.M.; Leffler, H.; Nilsson, U.J.; Haslett, C.; Forbes, S.J.; Sethi, T. Regulation of alternative macrophage activation by galectin-3. J. Immunol. 2008, 180, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Matricon, J.; Barnich, N.; Ardid, D. Immunopathogenesis of inflammatory bowel disease. Self Nonself 2010, 1, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of regulatory T cell in the pathogenesis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 2195–2205. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chen, F.; Liu, Z.; Cong, Y. Microbiota-specific Th17 Cells: Yin and Yang in Regulation of Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2016, 22, 1473–1482. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Häsler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516. [Google Scholar] [CrossRef]
- Etienne-Mesmin, L.; Chassaing, B.; Gewirtz, A.T. Tryptophan: A gut microbiota-derived metabolites regulating inflammation. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 7–9. [Google Scholar] [CrossRef]
- Islam, J.; Sato, S.; Watanabe, K.; Watanabe, T.; Hirahara, K.A.; Aoyama, Y.; Tomita, S.; Aso, H.; Komai, M.; Shirakawa, H. Dietary tryptophan alleviates dextran sodium sulfate-induced colitis through aryl hydrocarbon receptor in mice. J. Nutr. Biochem. 2017, 42, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Shizuma, T.; Mori, H.; Fukuyama, N. Protective effect of tryptophan against dextran sulfate sodium- induced experimental colitis. Turk. J. Gastroenterol. 2013, 24, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Boasso, A.; Vaccari, M.; Hryniewicz, A.; Fuchs, D.; Nacsa, J.; Cecchinato, V.; Andersson, J.; Franchini, G.; Shearer, G.M.; Chougnet, C. Regulatory T-cell markers, indoleamine 2,3-dioxygenase, and virus levels in spleen and gut during progressive simian immunodeficiency virus infection. J. Virol. 2007, 81, 11593–11603. [Google Scholar] [CrossRef] [PubMed]
- Acovic, A.; Gazdic, M.; Jovicic, N.; Harrell, C.R.; Fellabaum, C.; Arsenijevic, N.; Volarevic, V. Role of indoleamine 2,3-dioxygenase in pathology of the gastrointestinal tract. Therap. Adv. Gastroenterol. 2018, 11, 1756284818815334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Yuan, Z.W.; Qu, C.; Yu, X.T.; Huang, T.; Chen, P.V.; Su, Z.R.; Dou, Y.X.; Wu, J.Z.; Zeng, H.F.; et al. Palmatine ameliorated murine colitis by suppressing tryptophan metabolism and regulating gut microbiota. Pharmacol. Res. 2018, 137, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Ferdinande, L.; Demetter, P.; Perez-Novo, C.; Waeytens, A.; Taildeman, J.; Rottiers, I.; Rottiers, P.; De Vos, M.; Cuvelier, C.A. Inflamed intestinal mucosa features a specific epithelial expression pattern of indoleamine 2,3-dioxygenase. Int. J. Immunopathol. Pharmacol. 2008, 21, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Furuzawa-Carballeda, J.; Fonseca-Camarillo, G.; Lima, G.; Yamamoto-Furusho, J.K. Indoleamine 2,3-dioxygenase: Expressing cells in inflammatory bowel disease-a cross-sectional study. Clin. Dev. Immunol. 2013, 2013, 278035. [Google Scholar] [CrossRef] [PubMed]
- Fougeray, S.; Mami, I.; Bertho, G.; Beaune, P.; Thervet, E.; Pallet, N. Tryptophan depletion and the kinase GCN2 mediate IFN-γ-induced autophagy. J. Immunol. 2012, 189, 2954–2964. [Google Scholar] [CrossRef] [PubMed]
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012, 1, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Deierborg, T.; Burguillos, M.A. A new “sweet” ligand for Toll-like receptor 4. Oncotarget 2015, 6, 19928–19929. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling networks: An integration of adaptor molecules, kinases, and cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Block, M.; Mölne, J.; Leffler, H.; Börjesson, L.; Breimer, M.E. Immunohistochemical Studies on Galectin Expression in Colectomised Patients with Ulcerative Colitis. Biomed. Res. Int. 2016, 2016, 5989128. [Google Scholar] [CrossRef] [PubMed]
- Cibor, D.; Szczeklik, K.; Brzozowski, B.; Mach, T.; Owczarek, D. Serum galectin 3, galectin 9 and galectin 3-binding proteins in patients with active and inactive inflammatory bowel disease. J. Physiol. Pharmacol. 2019, 70, 95–104. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Mild Chronic Colitis | Moderate Chronic Colitis | Severe Chronic Colitis |
|---|---|---|---|
| Number of patients | 39 | 18 | 8 |
| Age (range) | 26–74 | 29–82 | 30–73 |
| Disease location Proctitis/left sided-distal UC/pancolitis | 10/23/6 | 1/10/7 | 0/4/4 |
| Hb, median (IQR) | 134 (8) | 109 (18.25) | 110.5 (13) |
| Fe, median (IQR) | 14.5 (6.10) | 8.15 (6.93) | 6.5 (4.4) |
| Ferritin, median (IQR) | 239 (109) | 53.5 (102) | 20.5 (65.75) |
| Platelets, median (IQR) | 392 (110) | 457.5 (124.25) | 491.5 (138.25) |
| UIBC, median (IQR) | 44 (13) | 32.5 (16.25) | 32 (21.5) |
| TIBC, median (IQR) | 52 (12) | 56.5 (12.5) | 53 (23) |
| CEA, median (IQR) | 1.8 (1.95) | 3.75 (2.32) | 4.1 (9.59) |
| CA 19-9, median (IQR) | 2.9 (2.4) | 10.58 (12.62) | 17.82 (21.1) |
| Disease duration (months) (median; range) | 4.4 (1.5–16.5) | 6,2 (2.7–10.9) | 16.5 (11.4–17.9) |
| Current therapy AS/CS/IS/IS+BT/BT | 38/0/1/0/0 | 4/0/10/2/2 | 0/0/4/2/2 |
| Former therapies AS/CS/IS/IS+BT/BT | 34/5/0/0/0 | 2/14/0/1/1 | 0/6/0/0/2 |
| Per oral steroids at diagnosis Yes/No | 5/34 | 14/4 | 6/2 |
| Other diseases (Glaucoma/Gonarthrosis) | 1/0/ | 1/1 | 1/0 |
| Other medications (PG Eye drops/HA) | 1/0 | 1/1 | 1/0 |
| Smoking status Yes/No | 3/36 | 2/16 | 0/8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volarevic, V.; Zdravkovic, N.; Harrell, C.R.; Arsenijevic, N.; Fellabaum, C.; Djonov, V.; Lukic, M.L.; Simovic Markovic, B. Galectin-3 Regulates Indoleamine-2,3-dioxygenase-Dependent Cross-Talk between Colon-Infiltrating Dendritic Cells and T Regulatory Cells and May Represent a Valuable Biomarker for Monitoring the Progression of Ulcerative Colitis. Cells 2019, 8, 709. https://doi.org/10.3390/cells8070709
Volarevic V, Zdravkovic N, Harrell CR, Arsenijevic N, Fellabaum C, Djonov V, Lukic ML, Simovic Markovic B. Galectin-3 Regulates Indoleamine-2,3-dioxygenase-Dependent Cross-Talk between Colon-Infiltrating Dendritic Cells and T Regulatory Cells and May Represent a Valuable Biomarker for Monitoring the Progression of Ulcerative Colitis. Cells. 2019; 8(7):709. https://doi.org/10.3390/cells8070709
Chicago/Turabian StyleVolarevic, Vladislav, Natasa Zdravkovic, Carl Randall Harrell, Nebojsa Arsenijevic, Crissy Fellabaum, Valentin Djonov, Miodrag L. Lukic, and Bojana Simovic Markovic. 2019. "Galectin-3 Regulates Indoleamine-2,3-dioxygenase-Dependent Cross-Talk between Colon-Infiltrating Dendritic Cells and T Regulatory Cells and May Represent a Valuable Biomarker for Monitoring the Progression of Ulcerative Colitis" Cells 8, no. 7: 709. https://doi.org/10.3390/cells8070709
APA StyleVolarevic, V., Zdravkovic, N., Harrell, C. R., Arsenijevic, N., Fellabaum, C., Djonov, V., Lukic, M. L., & Simovic Markovic, B. (2019). Galectin-3 Regulates Indoleamine-2,3-dioxygenase-Dependent Cross-Talk between Colon-Infiltrating Dendritic Cells and T Regulatory Cells and May Represent a Valuable Biomarker for Monitoring the Progression of Ulcerative Colitis. Cells, 8(7), 709. https://doi.org/10.3390/cells8070709