Convolutional Neural Network and Bidirectional Long Short-Term Memory-Based Method for Predicting Drug–Disease Associations

Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset
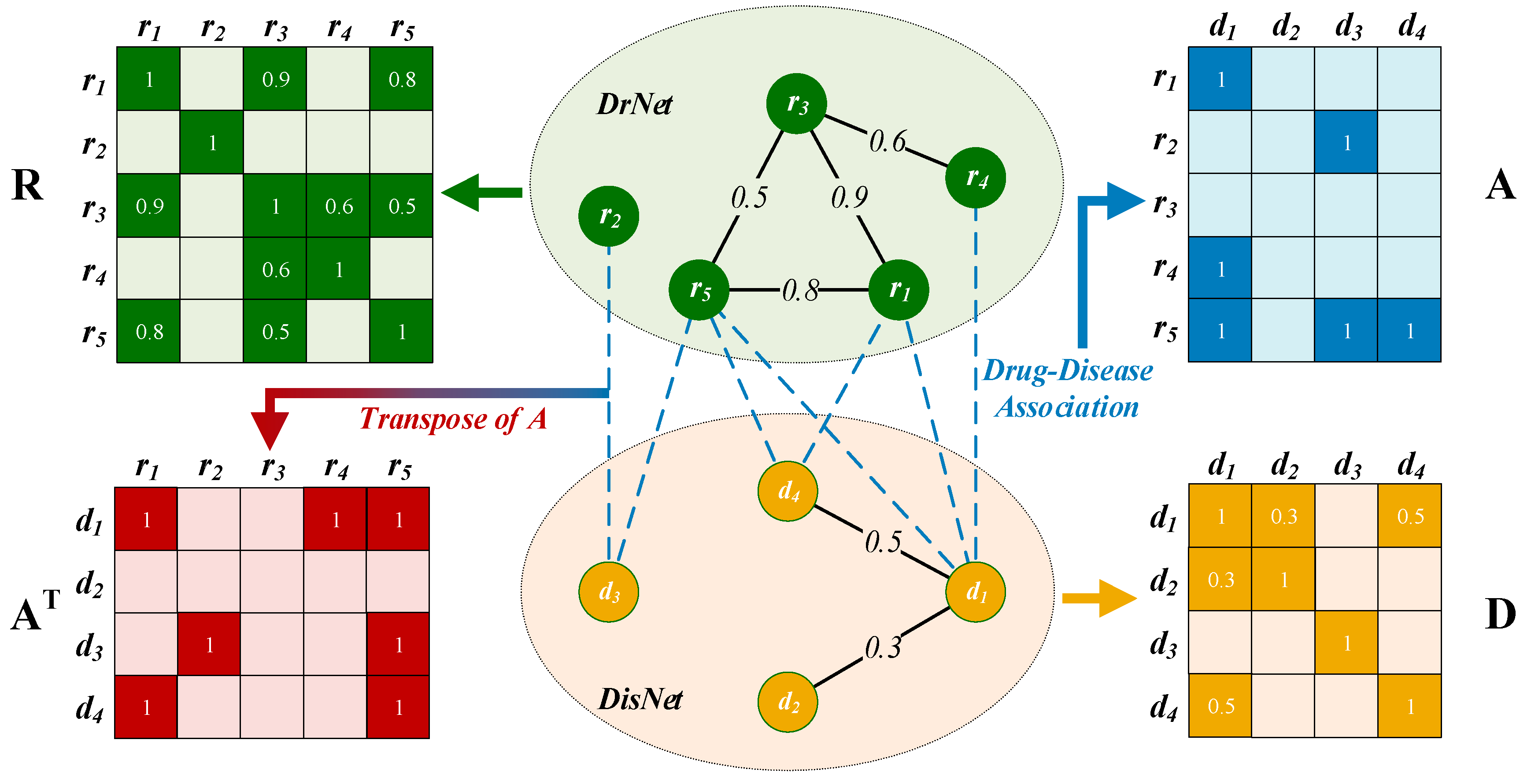
2.2. Construction of a Drug–Disease Network
2.2.1. Drug Network Construction
2.2.2. Disease Network Construction
2.2.3. Edges between DrNet and DisNet
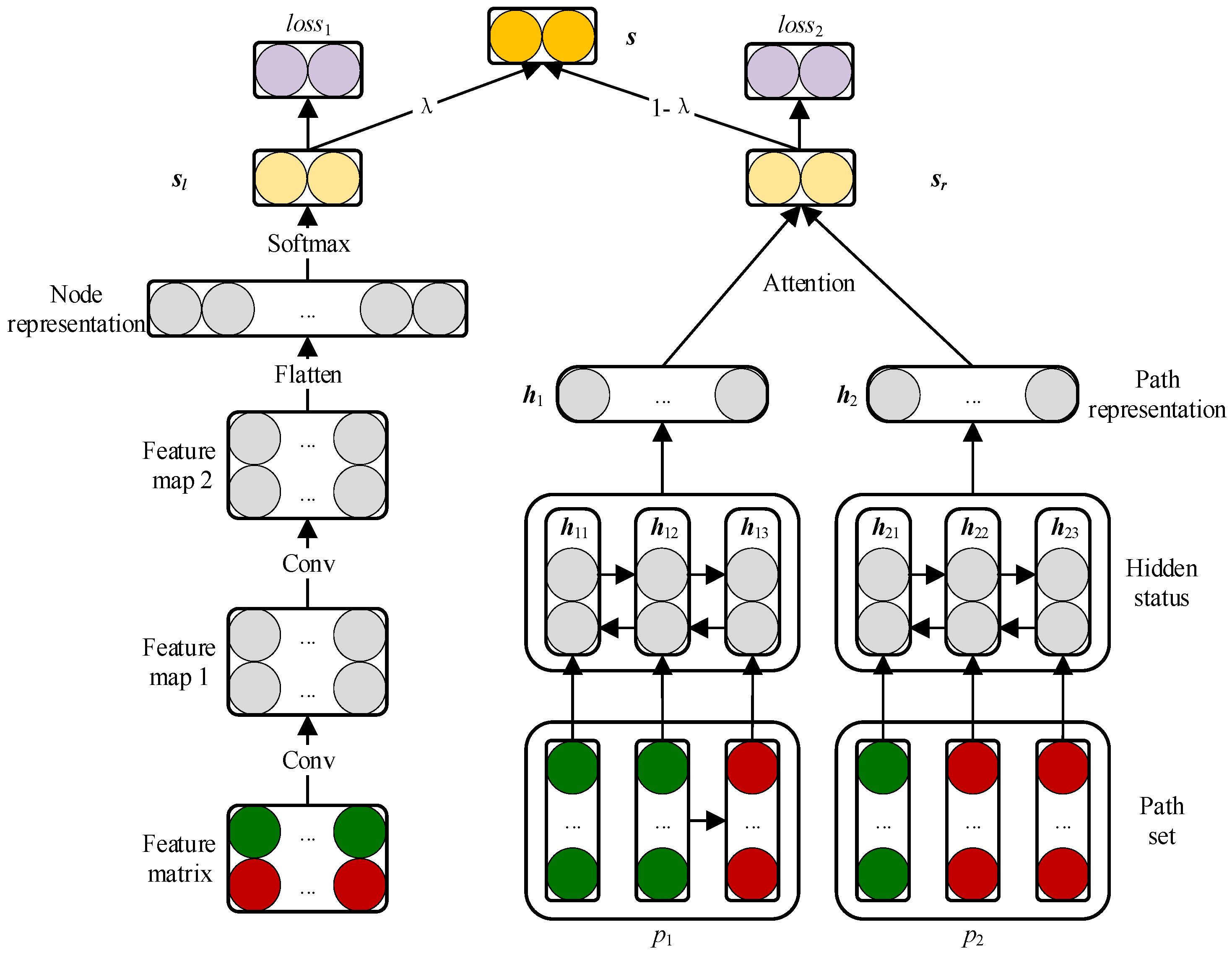
2.3. Prediction Model Based on CNN and BiLSTM Module
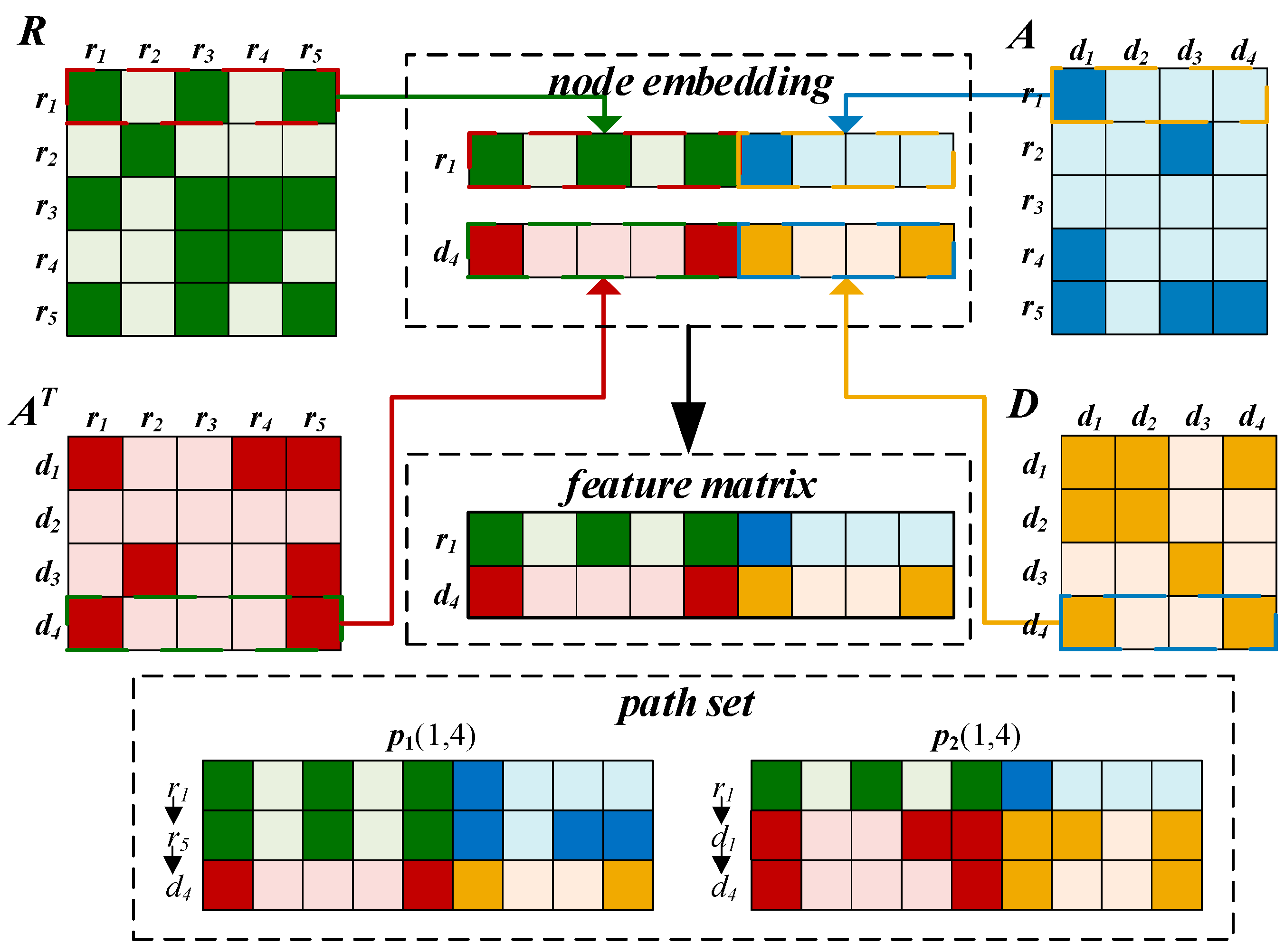
2.3.1. Embedding Layer
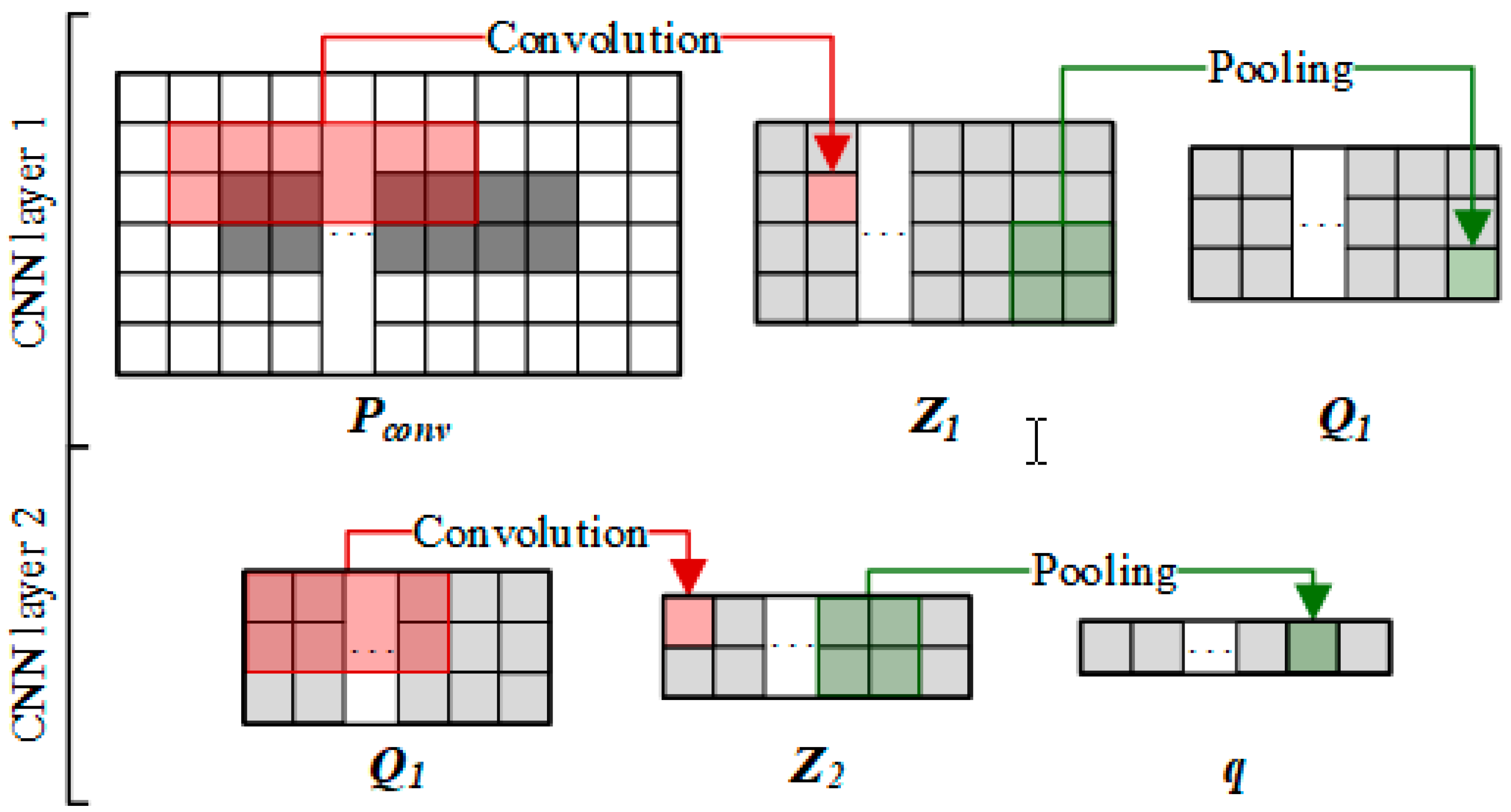
2.3.2. Convolutional Module on the Left
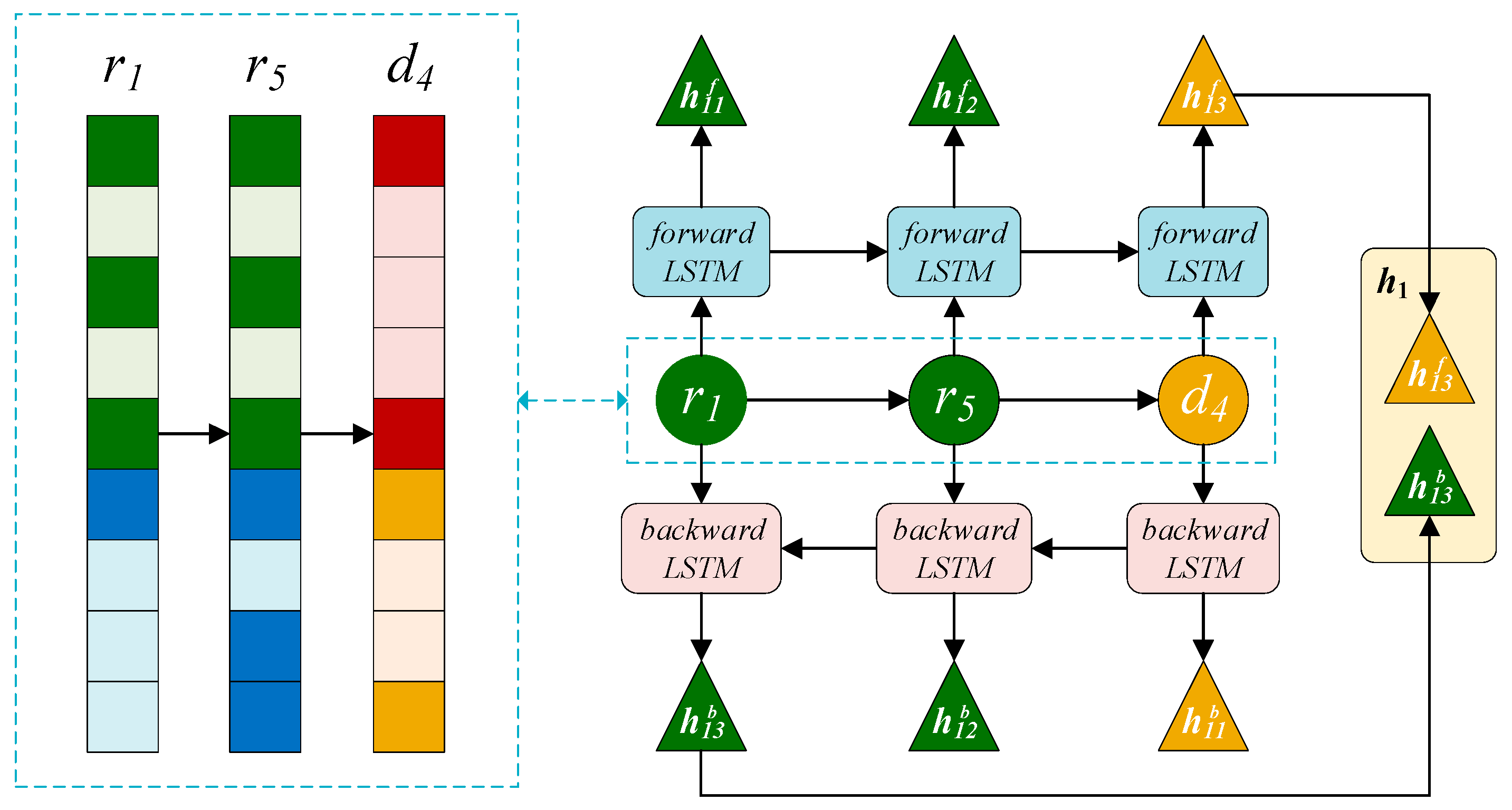
2.3.3. BiLSTM Module on the Right
2.3.4. Attention Mechanism at Path Level
2.3.5. Combined Strategy
3. Experimental Evaluation and Discussion
3.1. Evaluation Metrics
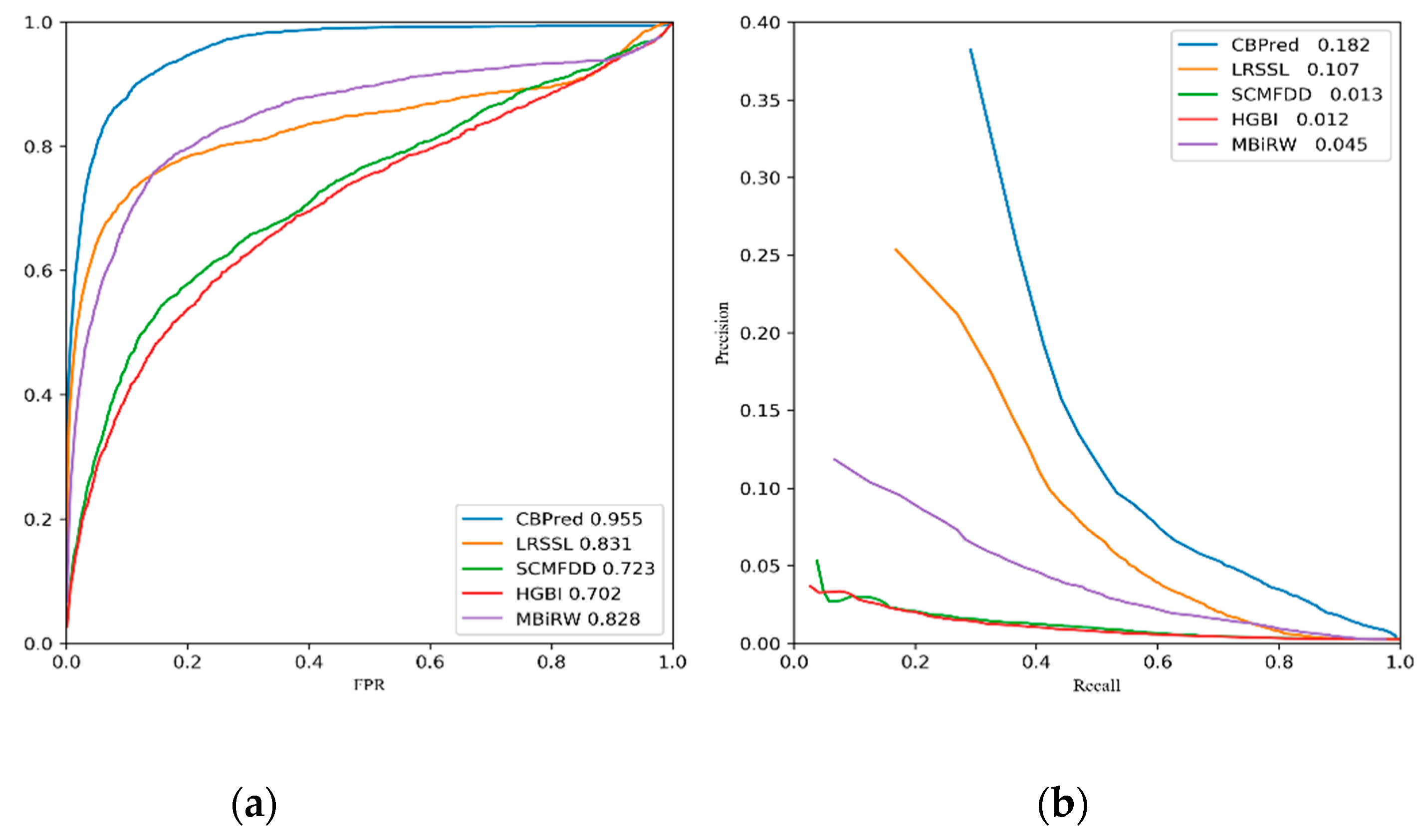
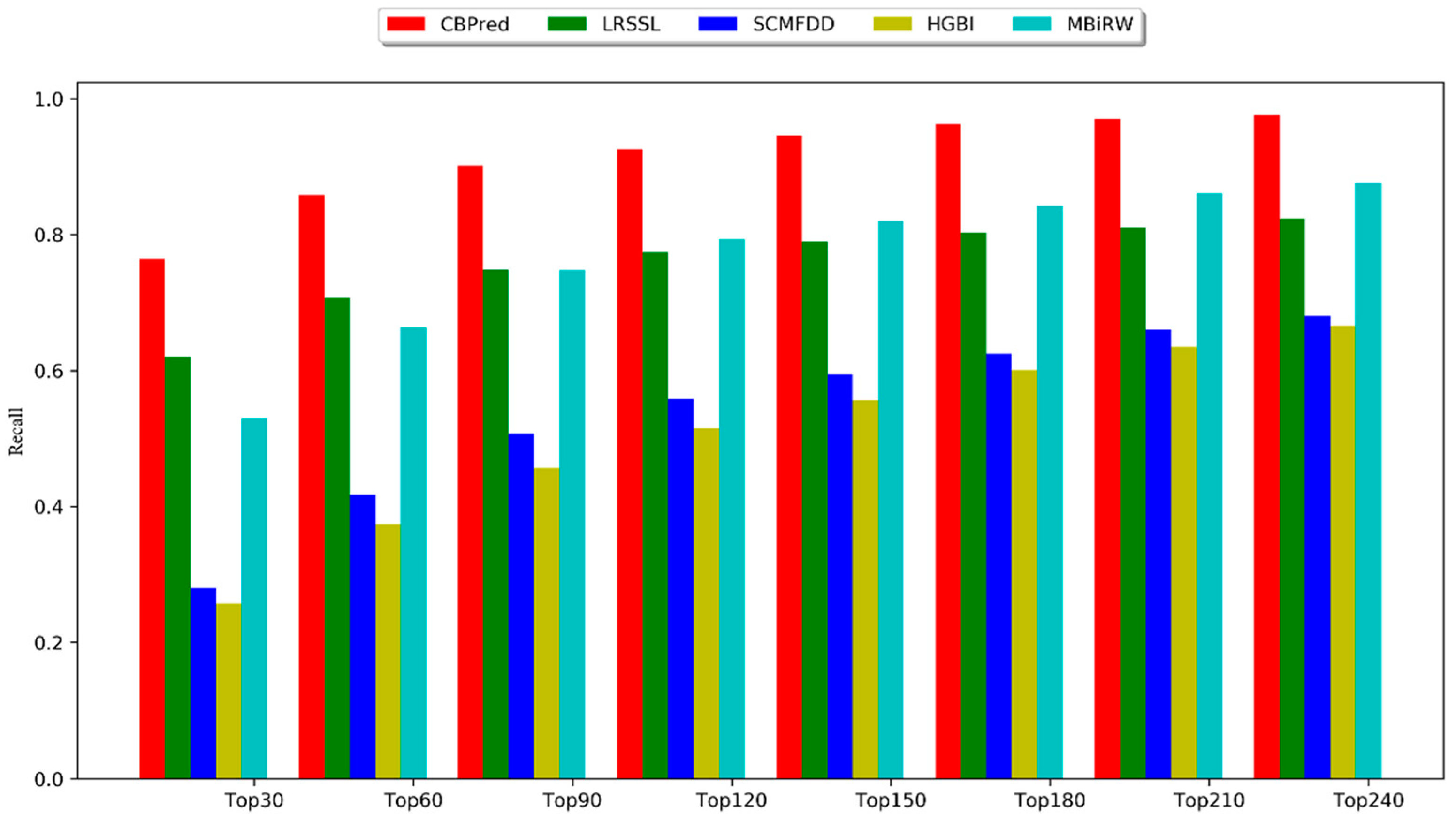
3.2. Comparison with Other Methods
3.3. Case Studies of Five Drugs
3.4. Prediction of Novel Drug–Disease Associations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liang, X.; Zhang, P.; Yan, L.; Fu, Y.; Peng, F.; Qu, L.; Shao, M.; Chen, Y.; Chen, Z. LRSSL: Predict and interpret drug–disease associations based on data integration using sparse subspace learning. Bioinformatics 2017, 33, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, A.; Oraiopoulos, N.; Drakeman, D.L. Renovation as innovation: Is repurposing the future of drug discovery research? Drug Discov. Today 2019, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Vohora, D. Drug Discovery and Development: An Overview. In Pharmaceutical Medicine and Translational Clinical Research; Vohora, D., Singh, G., Eds.; Elsevier: Dutch, The Netherlands, 2018; pp. 19–32. [Google Scholar]
- Xuan, P.; Cao, Y.; Zhang, T.; Wang, X.; Pan, S.; Shen, T. Drug repositioning through integration of prior knowledge and projections of drugs and diseases. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.P. Parexel’s Pharmaceutical R&D Statistical Sourcebook; PAREXEL International Corporation: Waltham, MA, USA, 2007. [Google Scholar]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- von Richter, O.; Lemke, L.; Haliduola, H.; Fuhr, R.; Koernicke, T.; Schuck, E.; Velinova, M.; Skerjanec, A.; Poetzl, J.; Jauch-Lembach, J. GP2017, an adalimumab biosimilar: Pharmacokinetic similarity to its reference medicine and pharmacokinetics comparison of different administration methods. Expert Opin. Biol. Ther. 2019. [Google Scholar] [CrossRef]
- Xu, C.; Ai, D.; Suo, S.; Chen, X.; Yan, Y.; Cao, Y.; Sun, N.; Chen, W.; McDermott, J.; Zhang, S. Accurate Drug Repositioning through Non-tissue-Specific Core Signatures from Cancer Transcriptomes. Cell Rep. 2018, 25, 523–535. [Google Scholar] [CrossRef]
- Xu, Y.; Guo, M.; Liu, X.; Wang, C.; Liu, Y.; Liu, G. Identify bilayer modules via pseudo-3D clustering: Applications to miRNA-gene bilayer networks. Nucleic Acids Res. 2016, 44, e152. [Google Scholar] [CrossRef]
- Xu, Y.; Guo, M.; Liu, X.; Wang, C.; Liu, Y. Inferring the soybean (Glycine max) microRNA functional network based on target gene network. Bioinformatics 2013, 30, 94–103. [Google Scholar] [CrossRef]
- Karaman, B.; Sippl, W. Computational Drug Repurposing: Current Trends. In Current Medicinal Chemistry; Bentham Science Publishers: Sharjah, UAE, 2019. [Google Scholar]
- Shameer, K.; Readhead, B.; Dudley, J.T. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr. Top. Med. Chem. 2015, 15, 5–20. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Guan, J.; Luo, L.; Zhuang, Z. Inferring new indications for approved drugs via random walk on drug-disease heterogenous networks. BMC bioinformatics 2016, 17, 539. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.-X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, S.; Zhang, X.; Li, J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014, 30, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Berger, B.; Peng, J. Diffusion component analysis: Unraveling functional topology in biological networks. In Proceedings of the International Conference on Research in Computational Molecular Biology, Warsaw, Poland, 12–15 April 2015; pp. 62–64. [Google Scholar]
- Zhang, W.; Yue, X.; Lin, W.; Wu, W.; Liu, R.; Huang, F.; Liu, F. Predicting drug-disease associations by using similarity constrained matrix factorization. BMC bioinformatics 2018, 19, 233. [Google Scholar] [CrossRef] [PubMed]
- Bengio, Y.; LeCun, Y. Scaling learning algorithms towards AI. Large-scale Kernel Mach. 2007, 34, 1–41. [Google Scholar]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Cheminformatics 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, Y.; Luo, J.; Zhao, W.; Zhou, X. Deep learning of the splicing (epi) genetic code reveals a novel candidate mechanism linking histone modifications to ESC fate decision. Nucleic Acids Res. 2017, 45, 12100–12112. [Google Scholar] [CrossRef]
- Zou, Q.; Mrozek, D.; Ma, Q.; Xu, Y. Scalable data mining algorithms in computational biology and biomedicine. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, P.; Cao, N.; Hu, J.; Sorrentino, R. Exploring the associations between drug side-effects and therapeutic indications. J. Biomed. Inform. 2014, 51, 15–23. [Google Scholar] [CrossRef]
- Bodenreider, O. The unified medical language system (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004, 32, D267–D270. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Lu, M.; Song, F.; Cui, Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics 2010, 26, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Gers, F.A.; Schmidhuber, J.; Cummins, F. Learning to forget: Continual prediction with LSTM. In Proceedings of the 9th International Conference on Artificial Neural Networks: ICANN ’99, Edinburgh, UK, 7–10 September 1999; pp. 812–815. [Google Scholar]
- Ghaeini, R.; Hasan, S.A.; Datla, V.; Liu, J.; Lee, K.; Qadir, A.; Ling, Y.; Prakash, A.; Fern, X.Z.; Farri, O. Dr-bilstm: Dependent reading bidirectional lstm for natural language inference. arXiv 2018, arXiv:1802.05577. [Google Scholar]
- Firat, O.; Cho, K.; Bengio, Y. Multi-way, multilingual neural machine translation with a shared attention mechanism. arXiv 2016, arXiv:1601.01073. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Zhang, P. Model selection via multifold cross validation. Ann. Stat. 1993, 299–313. [Google Scholar] [CrossRef]
- Xuan, P.; Sun, C.; Zhang, T.; Ye, Y.; Shen, T.; Dong, Y. A Gradient Boosting Decision Tree-based Method for Predicting Interactions between Target Genes and Drugs. Front. Genet. 2019, 10, 459. [Google Scholar] [CrossRef] [PubMed]
- Glas, A.S.; Lijmer, J.G.; Prins, M.H.; Bonsel, G.J.; Bossuyt, P.M. The diagnostic odds ratio: A single indicator of test performance. J. Clin. Epidemiol. 2003, 56, 1129–1135. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef]
- Bradley, A.P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997, 30, 1145–1159. [Google Scholar] [CrossRef]
- Pencina, M.J.; D’Agostino, R.B.; Vasan, R.S. Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond. Stat. Med. 2008, 27, 157–172. [Google Scholar] [CrossRef]
- Davis, J.; Goadrich, M. The relationship between Precision-Recall and ROC curves. In Proceedings of the 23rd international conference on Machine learning, Pittsburgh, PA, USA, 25–29 June 2006; pp. 233–240. [Google Scholar]
- Flach, P.; Kull, M. Precision-recall-gain curves: PR analysis done right. In Proceedings of the Advances in Neural Information Processing Systems 28 (NIPS 2015), Montreal, QC, Canada, 7–12 December 2015; pp. 838–846. [Google Scholar]
- van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Gehan, E.A. A generalized Wilcoxon test for comparing arbitrarily singly-censored samples. Biometrika 1965, 52, 203–224. [Google Scholar] [CrossRef] [PubMed]
- Fix, E.; Hodges, J., Jr. Significance probabilities of the Wilcoxon test. Annals Math. Statistics 1955, 26, 301–312. [Google Scholar] [CrossRef]
- Vexler, A.; Yu, J.; Zhao, Y.; Hutson, A.D.; Gurevich, G. Expected p-values in light of an ROC curve analysis applied to optimal multiple testing procedures. Stat. Methods Med. Res. 2018, 27, 3560–3576. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Li, J.; Ju, P.; Peng, J.; Wang, Y. SemFunSim: A new method for measuring disease similarity by integrating semantic and gene functional association. PLoS ONE 2014, 9, e99415. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Name | AUC | ||||
|---|---|---|---|---|---|
| CBPred | LRSSL | SCMFDD | HGBI | MBiRW | |
| Ave AUC on 763 drugs | 0.955 | 0.831 | 0.723 | 0.702 | 0.828 |
| ampicillin | 0.909 | 0.885 | 0.861 | 0.786 | 0.906 |
| cefepime | 0.953 | 0.932 | 0.898 | 0.910 | 0.872 |
| cefotaxime | 0.906 | 0.902 | 0.911 | 0.870 | 0.967 |
| cefotetan | 0.889 | 0.892 | 0.897 | 0.908 | 0.866 |
| cefoxitin | 0.913 | 0.911 | 0.899 | 0.909 | 0.907 |
| ceftazidime | 0.940 | 0.925 | 0.939 | 0.924 | 0.916 |
| ceftizoxime | 0.902 | 0.894 | 0.841 | 0.823 | 0.854 |
| ceftriaxone | 0.863 | 0.925 | 0.808 | 0.779 | 0.851 |
| ciprofloxacin | 0.917 | 0.893 | 0.810 | 0.790 | 0.844 |
| doxorubicin | 0.921 | 0.749 | 0.361 | 0.486 | 0.918 |
| erythromycin | 0.859 | 0.817 | 0.769 | 0.734 | 0.857 |
| itraconazole | 0.942 | 0.543 | 0.701 | 0.560 | 0.897 |
| levofloxacin | 0.910 | 0.852 | 0.824 | 0.819 | 0.867 |
| moxifloxacin | 0.909 | 0.792 | 0.841 | 0.849 | 0.826 |
| ofloxacin | 0.899 | 0.884 | 0.851 | 0.845 | 0.896 |
| Disease Name | AUPR | ||||
|---|---|---|---|---|---|
| CBPred | LRSSL | SCMFDD | HGBI | MBiRW | |
| Ave AUPR on 763 drugs | 0.182 | 0.107 | 0.013 | 0.012 | 0.045 |
| ampicillin | 0.249 | 0.220 | 0.059 | 0.089 | 0.058 |
| cefepime | 0.258 | 0.562 | 0.101 | 0.137 | 0.279 |
| cefotaxime | 0.276 | 0.273 | 0.072 | 0.098 | 0.266 |
| cefotetan | 0.177 | 0.724 | 0.093 | 0.131 | 0.152 |
| cefoxitin | 0.227 | 0.136 | 0.051 | 0.081 | 0.186 |
| ceftazidime | 0.201 | 0.187 | 0.132 | 0.164 | 0.119 |
| ceftizoxime | 0.328 | 0.168 | 0.125 | 0.174 | 0.153 |
| ceftriaxone | 0.269 | 0.138 | 0.081 | 0.101 | 0.123 |
| ciprofloxacin | 0.471 | 0.256 | 0.061 | 0.074 | 0.071 |
| doxorubicin | 0.164 | 0.159 | 0.006 | 0.007 | 0.075 |
| erythromycin | 0.194 | 0.034 | 0.013 | 0.013 | 0.052 |
| itraconazole | 0.334 | 0.057 | 0.008 | 0.006 | 0.097 |
| levofloxacin | 0.263 | 0.512 | 0.086 | 0.111 | 0.177 |
| moxifloxacin | 0.301 | 0.158 | 0.095 | 0.126 | 0.098 |
| ofloxacin | 0.221 | 0.214 | 0.114 | 0.158 | 0.095 |
| p-Value between CBPred and Another Method | LRSSL | SCMFDD | HGBI | MBiRW |
|---|---|---|---|---|
| p-value of ROC curve | 3.577 × 10−13 | 1.218 × 10−75 | 1.460 × 10−80 | 3.724 × 10−32 |
| p-value of P–R curve | 2.591 × 10−15 | 1.122 × 10−76 | 6.075 × 10−80 | 4.577 × 10−38 |
| Rank | Disease Name | Description | Rank | Disease Name | Description | |
|---|---|---|---|---|---|---|
| Ciprofloxacin | 1 | Conjunctivitis, Bacterial | ClinicalTrials | 6 | Campylobacter Infections | Drugbank |
| 2 | Chlamydia Infections | CTD | 7 | Neurocysticercosis | Drugbank | |
| 3 | Thrombocytopenic, Idiopathic | Drugbank | 8 | Respiration Disorders | ClinicalTrials | |
| 4 | Acanthamoeba Keratitis | Drugbank | 9 | Anthrax | CTD | |
| 5 | Scalp Dermatoses | PubChem | 10 | Skin Diseases | CTD | |
| Ceftriaxone | 1 | Panic Disorder | Drugbank | 6 | Bacteroides Infections | PubChem |
| 2 | Respiration Disorders | ClinicalTrials | 7 | Bone Diseases, Infectious | ClinicalTrials | |
| 3 | Respiratory Distress Syndrome, Adult | ClinicalTrials | 8 | Multiple Myeloma | Drugbank | |
| 4 | Rickettsia Infections | PubChem | 9 | Rectal Neoplasms | inferred candidate by 2 literature | |
| 5 | Respiratory Distress Syndrome, Newborn | ClinicalTrials | 10 | Maxillary Sinusitis | Drugbank | |
| Ofloxacin | 1 | Trichuriasis | inferred candidate by 1 study | 6 | Pulmonary Valve Stenosis | PubChem |
| 2 | Corneal Ulcer | PubChem | 7 | Schizophrenia | CTD | |
| 3 | Nausea | CTD | 8 | Peritonitis | CTD | |
| 4 | Rectal Neoplasms | ClinicalTrials | 9 | Mouth Diseases | CTD | |
| 5 | Epididymitis | Drugbank | 10 | Proteus Infections | CTD | |
| Ampicillin | 1 | Keratosis | inferred candidate by 1 literature | 6 | Pneumonia, Bacterial | CTD, ClinicalTrials |
| 2 | Bacterial Infections | CTD | 7 | Toothache | ClinicalTrials | |
| 3 | Respiratory Syncytial Virus Infections | inferred candidate by 1 study | 8 | Respiratory Tract Fistula | PubChem | |
| 4 | Respiratory Tract Diseases | ClinicalTrials | 9 | Mouth Diseases | ClinicalTrials | |
| 5 | Burns | CTD | 10 | Sarcoma, Ewings | PubChem | |
| Levofloxacin | 1 | Pneumonia, Mycoplasma | ClinicalTrials | 6 | Respiratory Syncytial Virus Infections | CTD |
| 2 | Rhinitis | PubChem | 7 | Soft Tissue Infections | Drugbank | |
| 3 | Bacteroides Infections | PubChem | 8 | Respiratory Tract Fistula | PubChem | |
| 4 | Tuberculosis, Pulmonary | ClinicalTrials | 9 | Listeriosis | PubChem | |
| 5 | Respiratory Tract Diseases | ClinicalTrials | 10 | Mouth Diseases | ClinicalTrials |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, P.; Ye, Y.; Zhang, T.; Zhao, L.; Sun, C. Convolutional Neural Network and Bidirectional Long Short-Term Memory-Based Method for Predicting Drug–Disease Associations. Cells 2019, 8, 705. https://doi.org/10.3390/cells8070705
Xuan P, Ye Y, Zhang T, Zhao L, Sun C. Convolutional Neural Network and Bidirectional Long Short-Term Memory-Based Method for Predicting Drug–Disease Associations. Cells. 2019; 8(7):705. https://doi.org/10.3390/cells8070705
Chicago/Turabian StyleXuan, Ping, Yilin Ye, Tiangang Zhang, Lianfeng Zhao, and Chang Sun. 2019. "Convolutional Neural Network and Bidirectional Long Short-Term Memory-Based Method for Predicting Drug–Disease Associations" Cells 8, no. 7: 705. https://doi.org/10.3390/cells8070705
APA StyleXuan, P., Ye, Y., Zhang, T., Zhao, L., & Sun, C. (2019). Convolutional Neural Network and Bidirectional Long Short-Term Memory-Based Method for Predicting Drug–Disease Associations. Cells, 8(7), 705. https://doi.org/10.3390/cells8070705