The Role of Adipose Tissue in the Pathogenesis and Therapeutic Outcomes of Inflammatory Bowel Disease

 ,
,  , and
, and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
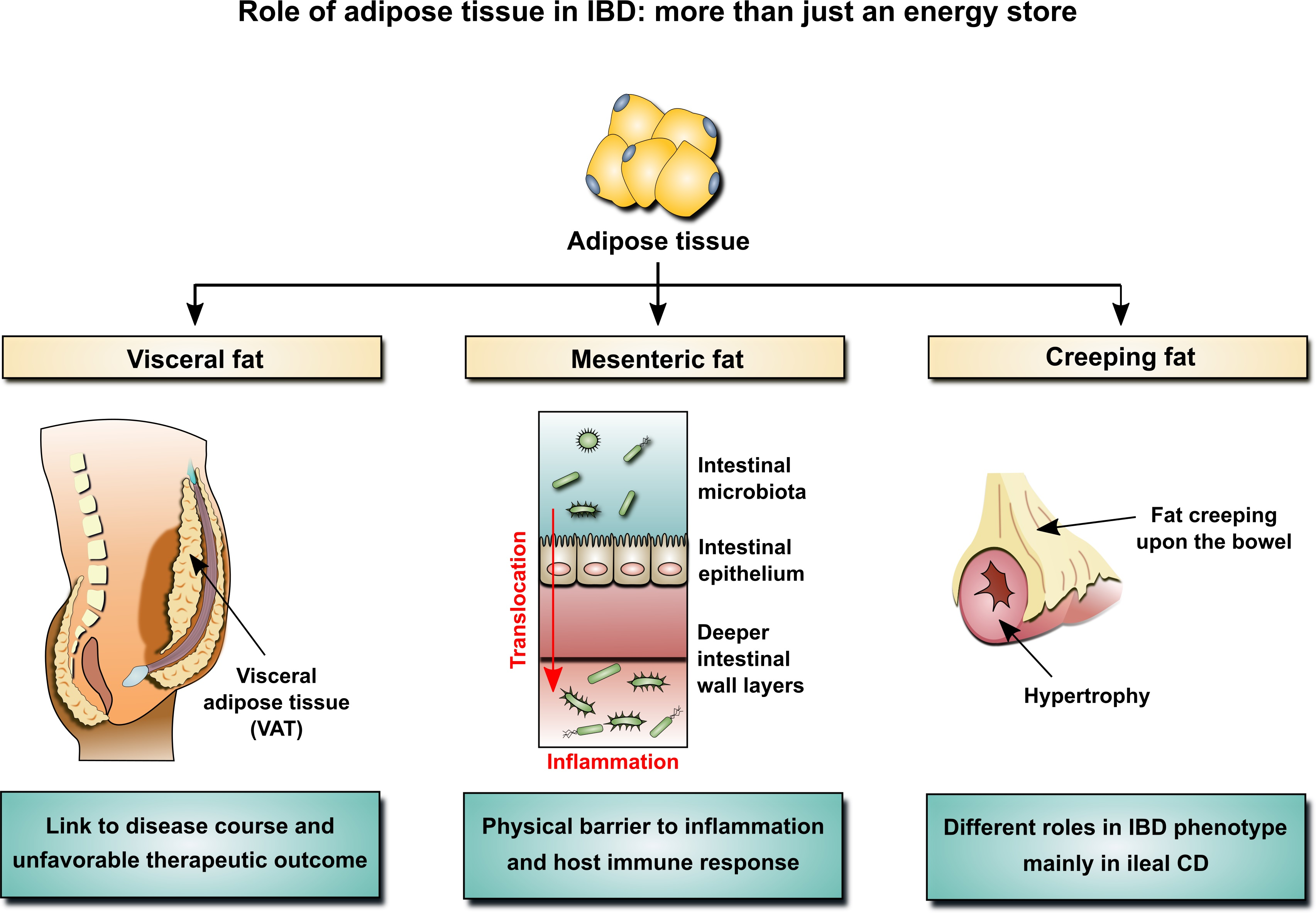
2. Adipose Tissue and the Pathogenesis of IBD
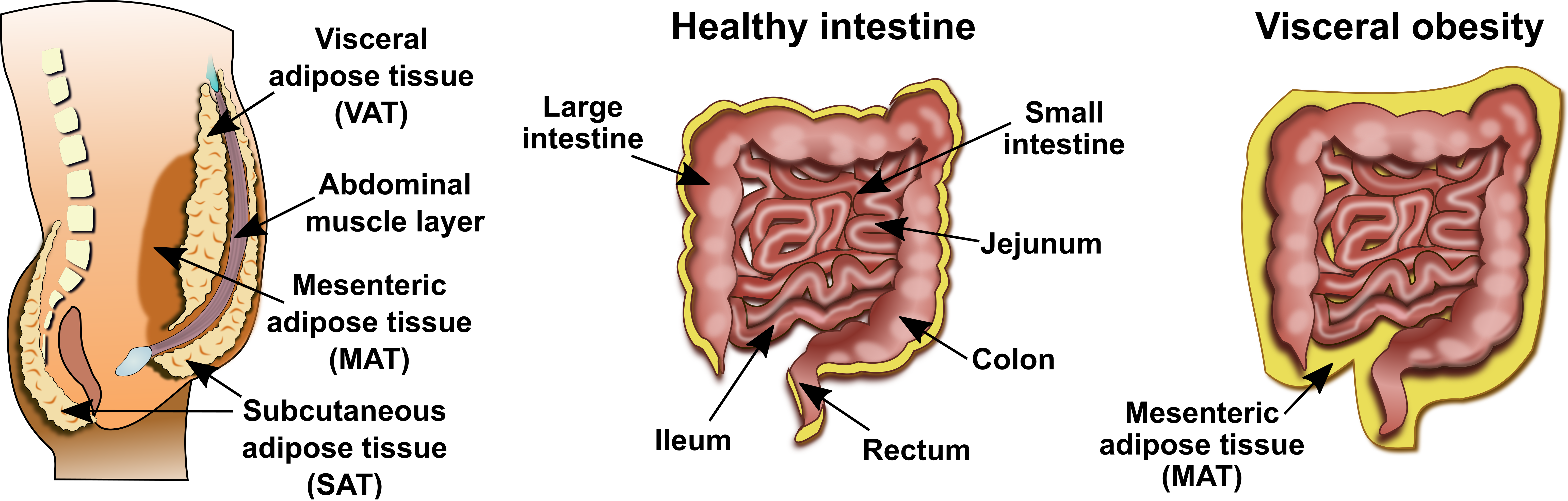
2.1. IBD and Adiposity-Shortcomings of BMI
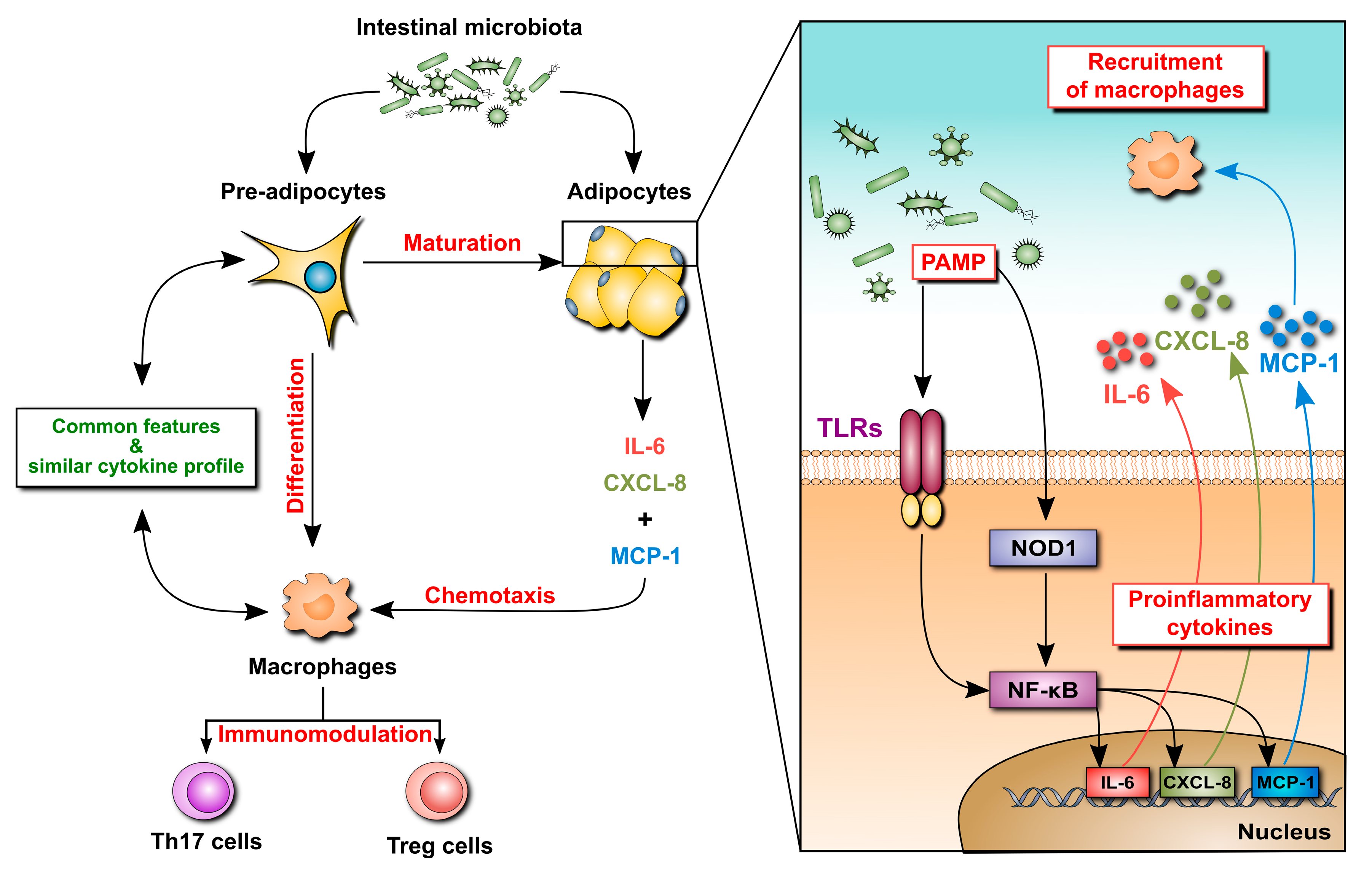
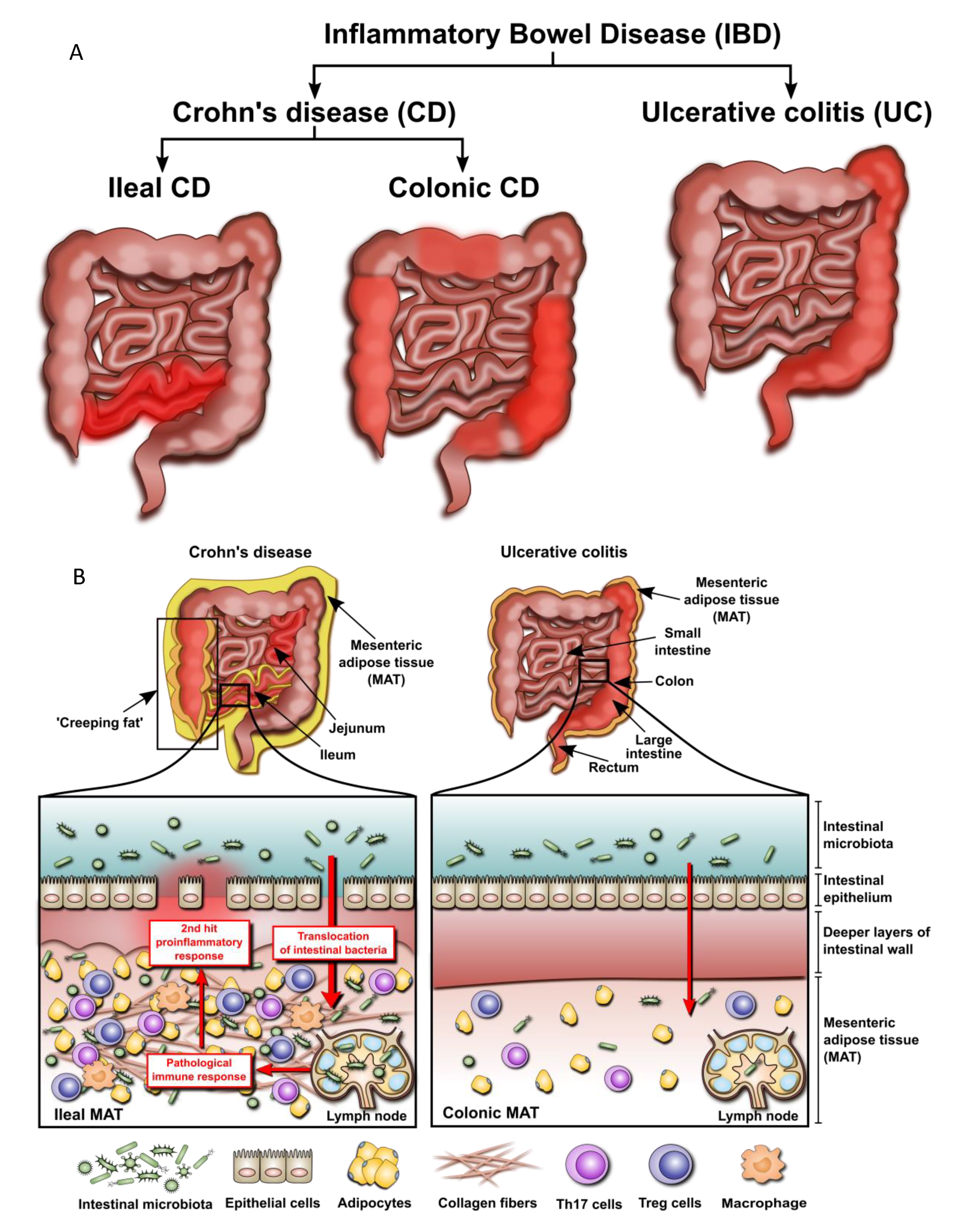
2.2. Mesenteric Adipose Tissue in IBD: A Unique Environment and A Double-Edged Sword
2.3. Mesenteric Adipose Tissue: Dissimilarities between IBD Phenotypes
2.4. Adipocytokines
2.4.1. Leptin
2.4.2. Adiponectin
2.4.3. Resistin
2.4.4. Visfatin
2.4.5. Chemerin
2.4.6. Ghrelin
2.4.7. Other Notable Mentions
3. The Impact of Adipose Tissue on Clinical Course and Therapeutic Outcomes in IBD
3.1. Adipose Tissue and Clinical Course of IBD
3.2. Adipose Tissue and IBD Therapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abraham, C.; Cho, J.H. Inflammatory Bowel Disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.; Panaccione, R.; Ghosh, S.; Barkema, H.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.; Panaccione, R.; Ghosh, S.; Wu, J.C.; Chan, F.K.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Moran, G.W.; Dubeau, M.F.; Kaplan, G.G.; Panaccione, R.; Ghosh, S. The increasing weight of Crohn’s disease subjects in clinical trials: A hypothesis-generatings time-trend analysis. Inflamm. Bowel Dis. 2013, 19, 2949–2956. [Google Scholar] [CrossRef]
- Nic Suibhne, T.; Raftery, T.C.; McMahon, O.; Walsh, C.; O’Morain, C.; O’Sullivan, M. High prevalence of overweight and obesity in adults with Crohn’s disease: Associations with disease and lifestyle factors. J. Crohn’s Colitis 2013, 7, e241–e248. [Google Scholar] [CrossRef]
- Fink, C.; Karagiannides, I.; Bakirtzi, K.; Pothoulakis, C. Adipose tissue and inflammatory bowel disease pathogenesis. Inflamm. Bowel Dis. 2012, 18, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.S.; Luben, R.; Olsen, A.; Tjonneland, A.; Kaaks, R.; Teucher, B.; Lindgren, S.; Grip, O.; Key, T.; Crowe, F.L.; et al. Body mass index and the risk for Crohn’s disease and ulcerative colitis: Data from a European Prospective Cohort Study (The IBD in EPIC Study). Am. J. Gastroenterol. 2013, 108, 575–582. [Google Scholar] [CrossRef]
- Khalili, H.; Ananthakrishnan, A.N.; Konijeti, G.G.; Konijeti, G.G.; Higuchi, L.M.; Fuchs, C.S.; Richter, J.M.; Chan, A.T. Measures of obesity and risk of Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2015, 21, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Steed, H.; Walsh, S.; Reynolds, N. A brief report of the epidemiology of obesity in the inflammatory bowel disease population of Tayside, Scotland. Obes. Facts 2009, 2, 370–372. [Google Scholar] [CrossRef]
- Flores, A.; Burstein, E.; Cipher, D.J.; Feagins, L.A. Obesity in Inflammatory Bowel Disease: A Marker of Less Severe Disease. Dig. Dis Sci 2015, 60, 2436–2445. [Google Scholar] [CrossRef]
- Hu, Q.; Ren, J.; Li, G.; Wu, X.; Li, J. The Impact of Obesity on the Clinical Course of Inflammatory Bowel Disease: A Meta-Analysis. Med. Sci. Monit. 2017, 23, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Rothman, K.J. BMI-related errors in the measurement of obesity. Int. J. Obes. 2008, 32 (Suppl. 3), S56–S59. [Google Scholar] [CrossRef]
- Tomiyama, A.J.; Hunger, J.M.; Nguyen-Cuu, J.; Wells, C. Misclassification of cardiometabolic health when using body mass index categories in NHANES 2005–2012. Int. J. Obes. 2016, 40, 883–886. [Google Scholar] [CrossRef]
- Versini, M.; Jeandel, P.Y.; Rosenthal, E.; Shoenfeld, Y. Obesity in autoimmune diseases: Not a passive bystander. Autoimmun. Rev. 2014, 13, 981–1000. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Zisman, T.L. Interaction of obesity and inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 7868–7881. [Google Scholar] [CrossRef]
- Crohn, B.B.; Ginzburg, L.; Oppenheimer, G.D. Regional ileitis: A pathological and clinical entity. JAMA 1984, 251, 73–79. [Google Scholar] [CrossRef]
- Weakley, F.L.; Turnbull, R.B. Recognition of regional ileitis in the operating room. Dis. Colon Rectum 1971, 14, 17–23. [Google Scholar] [CrossRef]
- Sheehan, A.L.; Warren, B.F.; Gear, M.W.; Shepherd, N.A. Fat-wrapping in Crohn’s disease: Pathological basis and relevance to surgical practice. Br. J. Surg. 1992, 79, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, W.; Gong, J.; Zuo, L.; Zhang, W.; Gu, L.; Guo, Z.; Cao, L.; Li, L.; Li, J. Influence of exclusive enteral nutrition therapy on visceral fat in patients with Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 1568–1574. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Chamaillard, M.; Gonzalez, F.; Beclin, E.; Decourcelle, C.; Antunes, L.; Gay, J.; Neut, C.; Colombel, J.F.; Desreumaux, P. Mesenteric fat in Crohn’s disease: A pathogenetic hallmark or an innocent bystander? Gut 2007, 56, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obesity Reviews 2010, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ponemone, V.; Keshavarzian, A.; Brand, M.I.; Saclarides, T.; Abcarian, H.; Cabay, R.J.; Fletcher, E.; Larsen, B.; Durstine, L.J.; Fantuzzi, G.; et al. Apoptosis and Inflammation: Role of Adipokines in Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2010, 1, e1. [Google Scholar] [CrossRef] [PubMed]
- Kredel, L.I.; Batra, A.; Stroh, T.; Kuhl, A.A.; Zeitz, M.; Erben, U.; Siegmund, B. Adipokines from local fat cells shape the macrophage compartment of the creeping fat in Crohn’s disease. Gut 2013, 62, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Heimesaat, M.M.; Bereswill, S.; Fischer, A.; Glauben, R.; Kunkel, D.; Scheffold, A.; Erben, U.; Kuhl, A.; Loddenkemper, C.; et al. Mesenteric fat-control site for bacterial translocation in colitis? Mucosal Immunol. 2012, 5, 580–591. [Google Scholar] [CrossRef]
- Duchmann, R.; Kaiser, I.; Hermann, E.; Mayet, W.; Ewe, K.; Meyer zum Buschenfelde, K.H. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 102, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 8, 1268–1273. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Sedman, P.C.; Macfie, J.; Sagar, P.; Mitchell, C.J.; May, J.; Mancey-Jones, B.; Johnstone, D. The prevalence of gut translocation in humans. Gastroenterology 1994, 107, 643–649. [Google Scholar] [CrossRef]
- Pietsch, J.; Batra, A.; Stroh, T.; Fedke, I.; Glauben, R.; Okur, B.; Zeitz, M.; Siegmund, B. Toll-like receptor expression and response to specific stimulation in adipocytes and preadipocytes: On the role of fat in inflammation. Ann. N. Y. Acad. Sci. 2006, 1072, 407–409. [Google Scholar] [CrossRef]
- Lin, Y.; Lee, H.; Berg, A.H.; Lisanti, M.P.; Shapiro, L.; Scherer, P.E. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J. Biol. Chem. 2000, 11, 24255–24263. [Google Scholar] [CrossRef] [PubMed]
- Schaeffler, A.; Gross, P.; Buettner, R.; Bollheimer, C.; Buechler, C.; Neumeier, M.; Kopp, A.; Schoelmerich, J.; Falk, W. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-κB pathway in adipocytes links nutritional signalling with innate immunity. Immunology 2009, 126, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Kopp, A.; Buechler, C.; Neumeier, M.; Weigert, J.; Aslanidis, C.; Scholmerich, J.; Schaffler, A. Innate immunity and adipocyte function: Ligand-specific activation of multiple Toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity 2009, 17, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Stroh, T.; Batra, A.; Glauben, R.; Fedke, I.; Erben, U.; Kroese, A.; Heimesaat, M.M.; Bereswill, S.; Girardin, S.; Zeitz, M.; et al. Nucleotide Oligomerization Domains 1 and 2: Regulation of Expression and Function in Preadipocytes. J. Immunol. 2008, 181, 3620–3627. [Google Scholar] [CrossRef] [PubMed]
- Kredel, L.; Batra, A.; Siegmund, B. Role of fat and adipokines in intestinal inflammation. Curr. Opin. Gastroenterol. 2014, 30, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Zeitz, M.; Siegmund, B. Adipokine signaling in inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1897–1905. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Zhou, H.; Li, Y.; Song, Y.L. NOD1 activation induces innate immune responses and insulin resistance in human adipocytes. Diabetes Metab. 2012, 38, 538–543. [Google Scholar] [CrossRef] [PubMed]
- McGovern, D.P.; Hysi, P.; Ahmad, T.; van Heel, D.A.; Moffatt, M.F.; Carey, A.; Cookson, W.O.; Jewell, D.P. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 2005, 14, 1245–1250. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassul, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Charriere, G.; Cousin, B.; Arnaud, E.; Andre, M.; Bacou, F.; Penicaud, L.; Casteilla, L. Preadipocyte conversion to macrophage. Evidence of plasticity. J. Biol. Chem. 2003, 278, 9850–9855. [Google Scholar] [CrossRef]
- Cousin, B.; Munoz, O.; Andre, M.; Fontanilles, A.M.; Dani, C.; Cousin, J.L.; Laharrague, P.; Casteilla, L.; Penicaud, L. A role for preadipocytes as macrophage-like cells. FASEB J. 1999, 13, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Karrasch, T.; Schaeffler, A. Adipokines and the role of visceral adipose tissue in inflammatory bowel disease. Ann. Gastroenterol. 2016, 29, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Pietsch, J.; Fedke, I.; Glauben, R.; Okur, B.; Stroh, T.; Zeitz, M.; Siegmund, B. Leptin-dependent toll-like receptor expression and responsiveness in preadipocytes and adipocytes. Am. J. Pathol. 2007, 170, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Barbier, M.; Vidal, H.; Desreumaux, P.; Dubuguoy, L.; Bourreille, A.; Colombel, J.F.; Cherbut, C.; Galmiche, J.P. Overexpression of leptin mRNA in mesenteric adipose tissue in inflammatory bowel diseases. Gastroenterol. Clin. Biol. 2003, 1221, 987–991. [Google Scholar] [CrossRef]
- Nishi, Y.; Isomoto, H.; Ueno, H.; Ohnita, K.; Wen, C.Y.; Takeshima, F.; Mishima, R.; Nakazato, M.; Kohno, S. Plasma leptin and ghrelin concentrations in patients with Crohn’s disease. World J. Gastroenterol. 2005, 11, 7314–7317. [Google Scholar] [CrossRef]
- Zulian, A.; Cancello, R.; Micheletto, G.; Gentilini, D.; Gilardini, L.; Danelli, P.; Invitti, C. Visceral adipocytes: Old actors in obesity and new protagonists in Crohn’s disease? Gut 2012, 61, 86–94. [Google Scholar] [CrossRef]
- Coope, A.; Pascoal, L.; da Silva, F.A.; Botezelli, J.D.; Ayrizono, M.L.; Milanski, M.; Camargo, M.G.; Planell, N.; Portovedo, M.; Dias, C.B.; et al. Transcriptional and molecular pathways activated in mesenteric adipose tissue and intestinal mucosa of Crohn’s disease patients. Int. J. Inflam. 2017, 2017, 7646859. [Google Scholar] [CrossRef]
- Silverberg, M.S.; Satsangi, J.; Ahmad, T.; Arnott, I.D.; Bernstein, C.N.; Brant, S.R.; Caprilli, R.; Colombel, J.F.; Gasche, C.; Geboes, K.; et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can. J. Gastroenterol. 2005, 19 (Suppl. A), 5A–36A. [Google Scholar] [CrossRef]
- Geboes, K.; Colombel, J.F.; Greenstein, A.; Jewell, D.P.; Sandborn, W.J.; Vatn, M.H.; Warren, B.; Riddell, R.H. Indeterminate colitis: A review of the concept—What’s in a name? Inflamm. Bowel Dis. 2008, 14, 850–857. [Google Scholar] [CrossRef]
- Cleynen, I.; Boucher, G.; Jostins, L.; Schumm, L.P.; Zeissig, S.; Ahmad, T.; Andersen, V.; Andrews, J.M.; Annese, V.; Brand, S.; et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: A genetic association study. Lancet 2016, 387, 156–167. [Google Scholar] [CrossRef]
- Kredel, L.I.; Jodicke, L.J.; Scheffold, A.; Grone, J.; Glauben, R.; Erben, U.; Kuhl, A.A.; Siegmund, B. T-cell Composition in Ileal and Colonic Creeping Fat—Separating Ileal from Colonic Crohn’s Disease. J. Crohns Colitis 2019, 13, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vazquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Nielsen, J. From next-generation sequencing to systematic modeling of the gut microbiome. Front. Genet. 2015, 6, 219. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.G.; Coffey, J.C.; McDermott, K.; Cotter, P.D.; Cabrera-Rubio, R.; Kiely, P.A.; Dunne, C.P. The Human Mesenteric Lymph Node Microbiome Differentiates Between Crohn’s Disease and Ulcerative Colitis. J. Crohns Colitis 2019, 13, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zulian, A.; Cancello, R.; Ruocco, C.; Gentilini, D.; Di Blasio, A.M.; Danelli, P.; Micheletto, G.; Cesana, E.; Invitti, C. Differences in visceral fat and fat bacterial colonization between ulcerative colitis and Crohn’s disease. An In Vivo and In Vitro study. PLoS ONE 2013, 8, e78495. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, B.; Sennello, J.A.; Jones-Carson, J.; Gamboni-Robertson, F.; Lehr, H.A.; Batra, A.; Fedke, I.; Zeitz, M.; Fantuzzi, G. Leptin receptor expression on T lymphocytes modulates chronic intestinal inflammation in mice. Gut 2004, 53, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Halaas, J.L.; Horvath, C.M.; Darnell, J.E.; Stoffel, M.; Friedman, J.M. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat. Genet. 1996, 14, 95–97. [Google Scholar] [CrossRef]
- Bluher, S.; Shah, S.; Mantzoros, C.S. Leptin Deficiency: Clinical Implications and Opportunities for Therapeutic Interventions. J. Investig. Med. 2009, 57, 784–788. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Matarese, G.; Lord, G.M.; Keogh, J.M.; Lawrence, E.; Agwu, C.; Sanna, V.; Jebb, S.A.; Perna, F.; Fontana, S.; et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Invest. 2002, 110, 1093–1103. [Google Scholar] [CrossRef]
- Raso, G.M.; Pacilio, M.; Esposito, E.; Coppola, A.; Di Carlo, R.; Meli, R. Leptin potentiates IFN-ɣ-induced expression of nitric oxide synthase and cyclo-oxygenase-2 in murine macrophage J774A.1. Br. J. Pharmacol. 2002, 137, 799–804. [Google Scholar] [CrossRef]
- Caldefie-Chezet, F.; Poulin, A.; Vasson, M.P. Leptin regulates functional capacities of polymorphonuclear neutrophils. Free Radic. Res. 2003, 37, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Okur, B.; Glauben, R.; Erben, U.; Ihbe, J.; Stroh, T.; Fedke, I.; Chang, H.D.; Zeitz, M.; Siegmund, B. Leptin: A critical regulator of CD4+ T-cell polarization In Vitro and In Vivo. Endocrinology 2010, 151, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Bloom, S.R.; Lechler, R.I. Leptin inhibits the anti-CD3-driven proliferation of peripheral blood T cells but enhances the production of proinflammatory cytokines. J. Leukoc. Biol. 2002, 72, 330–338. [Google Scholar] [PubMed]
- Sitaraman, S.; Liu, X.; Charrier, L.; Gu, L.H.; Ziegler, T.R.; Gewirtz, A.; Merlin, D. Colonic leptin: Source of a novel proinflammatory cytokine involved in IBD. FASEB J. 2004, 18, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Valentini, L.; Wirth, E.K.; Schweizer, U.; Hengstermann, S.; Schaper, L.; Koernicke, T.; Dietz, E.; Norman, K.; Buning, C.; Winklhofer-Roob, B.M.; et al. Circulating adipokines and the protective effects of hyperinsulinemia in inflammatory bowel disease. Nutrition 2009, 25, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Karmiris, K.; Koutroubakis, I.E.; Xidakis, C.; Polychronaki, M.; Voudouri, T.; Kouroumalis, E.A. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm. Bowel Dis. 2006, 12, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, V.S.; Milanski, M.; Fagundes, J.J.; Torsoni, A.S.; Ayrizono, M.L.; Nunez, C.E.; Dias, C.B.; Meirelles, L.R.; Dalal, S.; Coy, C.S.; et al. Serum levels and mesenteric fat tissue expression of adiponectin and leptin in patients with Crohn’s disease. Clin. Exp. Immunol. 2012, 170, 358–364. [Google Scholar] [CrossRef]
- Chouliaras, G.; Panayotou, I.; Margoni, D.; Mantzou, E.; Pervanidou, P.; Manios, Y.; Chrousos, G.P.; Roma, E. Circulating leptin and adiponectin and their relation to glucose metabolism in children with Crohn’s disease and ulcerative colitis. Pediatr. Res. 2013, 74, 420–426. [Google Scholar] [CrossRef]
- Biesiada, G.; Czepiel, J.; Ptak-Belowska, A.; Targosz, A.; Krzysiek-Maczka, G.; Strzalka, M.; Konturek, S.J.; Brzozowski, T.; Mach, T. Expression and release of leptin and proinflammatory cytokines in patients with ulcerative colitis and infectious diarrhea. J. Physiol. Pharmacol. 2012, 63, 471–481. [Google Scholar]
- Paul, G.; Schaffler, A.; Neumeier, M.; Furst, A.; Bataille, F.; Buechler, C.; Muller-Ladner, U.; Scholmerich, J.; Rogler, G.; Herfarth, H. Profiling adipocytokine secretion from creeping fat in Crohn’s disease. Inflamm. Bowel Dis. 2006, 12, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Gambero, A.; Marostica, M.; Abdalla Saad, M.J.; Pedrazzoli, J. Mesenteric adipose tissue alterations resulting from experimental reactivated colitis. Inflamm. Bowel Dis. 2007, 13, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Robinson, K.; Prins, J.; Venkatesh, B. Clinical review: Adiponectin biology and its role in inflammation and critical illness. Crit. Care 2011, 15, 221. [Google Scholar] [CrossRef] [PubMed]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef] [PubMed]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun. 1999, 257, 79–83. [Google Scholar] [CrossRef]
- Han, S.H.; Sakuma, I.; Shin, E.K.; Koh, K.K. Antiatherosclerotic and anti-insulin resistance effects of adiponectin: Basic and clinical studies. Prog. Cardiovasc. Dis. 2009, 52, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Song, G.G. Association of circulating resistin, leptin, adiponectin and visfatin levels with Behcet disease: A meta-analysis. Clin. Exp. Dermatol. 2018, 43, 536–545. [Google Scholar] [CrossRef]
- Lee, Y.H.; Song, G.G. Meta-analysis of circulating adiponectin, leptin, and resistin levels in systemic sclerosis. Z. Rheumatol. 2017, 76, 789–797. [Google Scholar] [CrossRef]
- Kyriakou, A.; Patsatsi, A.; Sotiriadis, D.; Goulis, D.G. Serum Leptin, Resistin, and Adiponectin Concentrations in Psoriasis: A Meta-Analysis of Observational Studies. Dermatology 2017, 23, 378–389. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kiyohara, T.; Murayama, Y.; Kihara, S.; Okamoto, Y.; Funahashi, T.; Ito, T.; Nezu, R.; Tsusui, S.; Miyagawa, J.I.; et al. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn’s disease. Gut 2005, 54, 789–796. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Hawkins, M.; Combs, T.P.; Rajala, M.W.; Doebber, T.; Berger, J.P.; Wagner, J.A.; Wu, M.; Knopps, A.; Xiang, A.H.; et al. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J. Biol. Chem. 2004, 279, 12152–12162. [Google Scholar] [CrossRef] [PubMed]
- Engl, J.; Bobbert, T.; Ciardi, C.; Laimer, M.; Tatarczyk, T.; Kaser, S.; Weiss, H.; Molnar, C.; Tilg, H.; Patsch, J.R.; et al. Effects of pronounced weight loss on adiponectin oligomer composition and metabolic parameters. Obesity 2007, 15, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Olszanecka-Glinianowicz, M.; Handzlik-Orlik, G.; Orlik, B.; Chudek, J. Adipokines in the pathogenesis of idiopathic inflammatory bowel disease. Endokrynol. Pol. 2013, 64, 226–231. [Google Scholar] [PubMed]
- Bokarewa, M.; Nagaev, I.; Dahlberg, L.; Smith, U.; Tarkowski, A. Resistin, an adipokine with potent proinflammatory properties. J. Immunol. 2005, 174, 5789–5795. [Google Scholar] [CrossRef] [PubMed]
- Theocharidou, E.; Balaska, A.; Vogiatzis, K.; Tellis, C.C.; Gossios, T.D.; Athyros, V.G.; Tselepis, A.D.; Karagiannis, A. Hypertrophic Mesenteric Adipose Tissue May Play a Role in Atherogenesis in Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2016, 22, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- Bostrom, E.A.; Ekstedt, M.; Kechagias, S.; Sjowall, C.; Bokarewa, M.I.; Almer, S. Resistin is associated with breach of tolerance and anti-nuclear antibodies in patients with hepatobiliary inflammation. Scand. J. Immunol. 2011, 74, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Karmiris, K.; Koutroubakis, I.E.; Xidakis, C.; Polychronaki, M.; Kouroumalis, E.A. The effect of infliximab on circulating levels of leptin, adiponectin and resistin in patients with inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2007, 19, 789–794. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Garcia-Unzueta, M.T.; Gonzalez-Juanatey, C.; Miranda-Filloy, J.A.; Vazquez-Rodriguez, T.R.; De Matias, J.M.; Martin, J.; Dessein, P.H.; Llorca, J. Anti-TNF-alpha therapy modulates resistin in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2008, 26, 311–316. [Google Scholar]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef]
- Chang, Y.C.; Chang, T.J.; Lee, W.J.; Chuang, L.M. The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism 2010, 59, 93–99. [Google Scholar] [CrossRef]
- Luk, T.; Malam, Z.; Marshall, J.C. Pre-B cell colony-enhancing factor (PBEF)/visfatin: A novel mediator of innate immunity. J. Leukoc. Biol. 2008, 83, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Starr, A.E.; Deeke, S.A.; Ning, Z.; Chiang, C.K.; Zhang, X.; Mottawea, W.; Singleton, R.; Benchimol, E.; Wen, M.; Mack, D.R.; et al. Proteomic analysis of ascending colon biopsies from a paediatric inflammatory bowel disease inception cohort identifies protein biomarkers that differentiate Crohn’s disease from UC. Gut 2017, 66, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [PubMed]
- Gerner, R.R.; Klepsch, V.; Macheiner, S.; Arnhard, K.; Adolph, T.E.; Grander, C.; Wieser, V.; Pfister, A.; Moser, P.; Hermann-Kleiter, N.; et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut 2017, 67, 1813–1823. [Google Scholar] [CrossRef] [PubMed]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef]
- Mariani, F.; Roncucci, L. Chemerin/chemR23 axis in inflammation onset and resolution. Inflamm. Res. 2015, 64, 85–95. [Google Scholar] [CrossRef]
- Wittamer, V.; Bondue, B.; Guillabert, A.; Vassart, G.; Parmentier, M.; Communi, D. Neutrophil-mediated maturation of chemerin: A link between innate and adaptive immunity. J. Immunol. 2005, 175, 487–493. [Google Scholar] [CrossRef]
- Lin, Y.; Yang, X.; Yue, W.; Xu, X.; Li, B.; Zou, L.; He, R. Chemerin aggravates DSS-induced colitis by suppressing M2 macrophage polarization. Cell Mol. Immunol. 2014, 11, 355–366. [Google Scholar] [CrossRef]
- Dranse, H.J.; Rourke, J.L.; Stadnyk, A.W.; Sinal, C.J. Local chemerin levels are positively associated with DSS-induced colitis but constitutive loss of CMKLR1 does not protect against development of colitis. Physiol. Rep. 2015, 3, e12497. [Google Scholar] [CrossRef]
- Weigert, J.; Obermeier, F.; Neumeier, M.; Wanninger, J.; Filarsky, M.; Bauer, S.; Aslanidis, C.; Rogler, G.; Ott, C.; Schaffler, A.; et al. Circulating levels of chemerin and adiponectin are higher in ulcerative colitis and chemerin is elevated in Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Terzoudis, S.; Malliaraki, N.; Damilakis, J.; Dimitriadou, D.A.; Zavos, C.; Koutroubakis, I.E. Chemerin, visfatin, and vaspin serum levels in relation to bone mineral density in patients with inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2016, 28, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Waluga, M.; Hartleb, M.; Boryczka, G.; Kukla, M.; Zwirska-Korczala, K. Serum adipokines in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6912–6917. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, L.; Lin, T.R.; Chai, B.; Fan, Y.; Gantz, I.; Mulholland, M.W. Inhibition of Adipogenesis by Ghrelin. Mol. Biol. Cell 2004, 15, 2484–2491. [Google Scholar] [CrossRef]
- Tiaka, E.K.; Manolakis, A.C.; Kapsoritakis, A.N.; Potamianos, S.P. Unraveling the link between leptin, ghrelin and different types of colitis. Ann. Gastroenterol. 2011, 24, 20–28. [Google Scholar] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Engel, M.; Burnat, G.; Gaca, P.; Kwiecien, S.; Pajdo, R.; Konturek, S.J. Ghrelin ameliorates colonic inflammation. Role of nitric oxide and sensory nerves. J. Physiol. Pharmacol. 2009, 60, 41–47. [Google Scholar]
- Hosomi, S.; Oshitani, N.; Kamata, N.; Sogawa, M.; Yamagami, H.; Watanabe, K.; Tominaga, K.; Watanabe, T.; Fujiwara, Y.; Maeda, K.; et al. Phenotypical and functional study of ghrelin and its receptor in the pathogenesis of Crohn’s disease. Inflamm. Bowel Dis. 2008, 14, 1205–1213. [Google Scholar] [CrossRef]
- Peracchi, M.; Bardella, M.T.; Caprioli, F.; Massironi, S.; Conte, D.; Valenti, L.; Ronchi, C.; Beck-Peccoz, P.; Arosio, M.; Piodi, L. Circulating ghrelin levels in patients with inflammatory bowel disease. Gut 2006, 55, 432–433. [Google Scholar] [CrossRef]
- Ates, Y.; Degertekin, B.; Erdil, A.; Yaman, H.; Dagalp, K. Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig. Dis. Sci. 2008, 53, 2215–2221. [Google Scholar] [CrossRef]
- Van der Ploeg, L.; Laken, H.; Sharma, S.; Datta, R.; Halem, H.; Dong, J.; Touvay, C.; Teillot, M.; Noonan, P.; Tartaglia, L.; et al. Preclinical gastrointestinal prokinetic efficacy and endocrine effects of the ghrelin mimetic RM-131. Life Sci. 2014, 109, 20–29. [Google Scholar] [CrossRef]
- Shin, A.; Camilleri, M.; Busciglio, I.; Burton, D.; Smith, S.A.; Vella, A.; Ryks, M.; Rhoten, D.; Zinsmeister, A.R. The ghrelin agonist RM-131 accelerates gastric emptying of solids and reduces symptoms in patients with type 1 diabetes mellitus. Clin. Gastroenterol. Hepatol. 2013, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Tabesh, M.; Noroozi, A.; Amini, M.; Feizi, A.; Saraf-Bank, S.; Zare, M. Association of retinol-binding protein 4 with metabolic syndrome in first-degree relatives of type 2 diabetic patients. J. Res. Med. Sci. 2017, 22, 28. [Google Scholar] [PubMed]
- Roma, E.; Krini, M.; Hantzi, E.; Sakka, S.; Panayiotou, I.; Margeli, A.; Papassotiriou, I.; Kanaka-Gantenbein, C. Retinol Binding Protein 4 in children with Inflammatory Bowel Disease: A negative correlation with the disease activity. Hippokratia 2012, 16, 360–365. [Google Scholar] [PubMed]
- Yamawaki, H.; Kuramoto, J.; Kameshima, S.; Usui, T.; Okada, M.; Hara, Y. Omentin, a novel adipocytokine inhibits TNF-induced vascular inflammation in human endothelial cells. Biochem. Biophys. Res. Commun. 2011, 408, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, L.; Liu, L.; Feng, Y.; Lu, L.; Ren, X.; Dong, X.; Sang, W. Serum omentin-1 as a disease activity marker for Crohn’s disease. Dis. Mark. 2014, 2014, 162517. [Google Scholar] [CrossRef] [PubMed]
- Maconi, G.; Greco, S.; Duca, P.; Ardizzone, S.; Massari, A.; Cassinotti, A.; Radice, E.; Porro, G.B. Prevalence and clinical significance of sonographic evidence of mesenteric fat alterations in Crohn’s disease. Inflamm. Bowel Dis. 2008, 14, 1555–1561. [Google Scholar] [CrossRef]
- Uko, V.; Vortia, E.; Achkar, J.P.; Karakas, P.; Fiocchi, C.; Worley, S.; Kay, M.H. Impact of abdominal visceral adipose tissue on disease outcome in pediatric Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 2286–2291. [Google Scholar] [CrossRef]
- Erhayiem, B.; Dhingsa, R.; Hawkey, C.J.; Subramanian, V. Ratio of visceral to subcutaneous fat area is a biomarker of complicated Crohn’s disease. Clin. Gastroenterol. Hepatol. 2011, 9, 684–687. [Google Scholar] [CrossRef]
- Buning, C.; von Kraft, C.; Hermsdorf, M.; Gentz, E.; Wirth, E.K.; Valentini, L.; Haas, V. Visceral Adipose Tissue in Patients with Crohn’s Disease Correlates with Disease Activity, Inflammatory Markers, and Outcome. Inflamm. Bowel Dis. 2015, 21, 2590–2597. [Google Scholar] [CrossRef]
- Van Der Sloot, K.W.; Joshi, A.D.; Bellavance, D.R.; Gilpin, K.K.; Stewart, K.O.; Lochhead, P.; Garber, J.J.; Giallourakis, C.; Yajnik, V.; Ananthakrishnan, A.N.; et al. Visceral Adiposity, Genetic Susceptibility, and Risk of Complications Among Individuals with Crohn’s Disease. Inflamm. Bowel Dis. 2017, 23, 82–88. [Google Scholar] [CrossRef]
- Frivolt, K.; Hetterich, H.; Schwerd, T.; Hajji, M.S.; Bufler, P.; Coppenrath, E.; Koletzko, S. Increase of Intra-abdominal Adipose Tissue in Pediatric Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Connelly, T.M.; Juza, R.M.; Sangster, W.; Sehgal, R.; Tappouni, R.F.; Messaris, E. Volumetric fat ratio and not body mass index is predictive of ileocolectomy outcomes in Crohn’s disease patients. Dig. Surg. 2014, 31, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wu, X.R.; Remer, E.M.; Lian, L.; Stocchi, L.; Li, Y.; McCullough, A.; Remzi, F.H.; Shen, B. Association between high visceral fat area and postoperative complications in patients with Crohn’s disease following primary surgery. Colorectal Dis. 2016, 18, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Holt, D.Q.; Moore, G.T.; Strauss, B.J.; Hamilton, A.L.; De Cruz, P.; Kamm, M. Visceral adiposity predicts post-operative Crohn’s disease recurrence. Aliment. Pharmacol. Ther. 2017, 45, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, W.; Zuo, L.; Shen, B. The Role of the Mesentery in Crohn’s Disease: The Contributions of Nerves, Vessels, Lymphatics, and Fat to the Pathogenesis and Disease Course. Inflamm. Bowel Dis. 2016, 22, 1483–1495. [Google Scholar] [CrossRef] [PubMed]
- Dignass, A.; Van Assche, G.; Lindsay, J.O.; Lemann, M.; Soderholm, J.; Colombel, J.F.; Danese, S.; D’Hoore, A.; Gassul, M.; Gomollon, F.; et al. The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: Current management. J. Crohns Colitis 2010, 4, 28–62. [Google Scholar] [CrossRef] [PubMed]
- Osterman, M.T.; Kundu, R.; Lichtenstein, G.R.; Lewis, J.D. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: A meta-analysis. Gastroenterology 2006, 130, 1047–1053. [Google Scholar] [CrossRef]
- Holt, D.Q.; Strauss, B.J.; Moore, G.T. Weight and Body Composition Compartments do Not Predict Therapeutic Thiopurine Metabolite Levels in Inflammatory Bowel Disease. Clin. Transl. Gastroenterol. 2016, 7, e199. [Google Scholar] [CrossRef]
- Puig, L. Obesity and psoriasis: Body weight and body mass index influence the response to biological treatment. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1007–1011. [Google Scholar] [CrossRef]
- Kurnool, S.; Nguyen, N.H.; Proudfoot, J.; Dulai, P.S.; Boland, B.S.; Vande Casteele, N.; Evans, E.; Grunvald, E.L.; Zarrinpar, A.; Sandborn, W.; et al. High body mass index is associated with increased risk of treatment failure and surgery in biologic-treated patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2018, 47, 1472–1479. [Google Scholar] [CrossRef]
- Singh, S.; Proudfoot, J.; Xu, R.; Sandborn, W. Obesity and Response to Infliximab in Patients with Inflammatory Bowel Diseases: Pooled Analysis of Individual Participant Data from Clinical Trials. Am. J. Gastroenterol. 2018, 113, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Swainson, M.G.; Batterham, A.M.; Tsakirides, C.; Rutherford, Z.H.; Hind, K. Prediction of whole-body fat percentage and visceral adipose tissue mass from five anthropometric variables. PLoS ONE 2017, 12, e0177175. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Cao, L.; Li, Y.; Cai, X.; Ge, Y.; Zhu, W. Visceral fat is Associated with Mucosal Healing of Infliximab Treatment in Crohn’s Disease. Dis. Colon Rectum 2018, 61, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Eder, P.; Katulska, K.; Krela-Kazmierczak, I.; Stawczyk-Eder, K.; Klimczak, K.; Szymczak, A.; Linke, K.; Lykowska-Szuber, L. The influence of anti-TNF therapy on the magnetic resonance enterographic parameters of Crohn’s disease activity. Abdom. Imaging 2015, 40, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Frivolt, K.; Schwerd, T.; Schatz, S.B.; Freudenberg, F.; Prell, C.; Werkstetter, K.J.; Bufler, P.; Koletzko, S. Hyperadiponectinemia During Infliximab Induction Therapy in Pediatric Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Franchimont, D.; Roland, S.; Gustot, T.; Quertinmont, E.; Toubouti, Y.; Gervy, M.C.; Deviere, J.; Van Gossum, A. Impact of infliximab on serum leptin levels in patients with Crohn’s disease. J. Clin. Endocrinol. Metab. 2005, 90, 3510–3516. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the Global Map of Adipogenesis and Beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Beisner, J.; Wang, G.; Nuding, S.; Oommen, S.T.; Kelly, D.; Parmentier-Decrucq, E.; Dessein, R.; Merour, E.; Chavatte, P.; et al. Peroxisome proliferator-activated receptor gamma activation is required for maintenance of innate antimicrobial immunity in the colon. Proc. Natl. Acad. Sci. USA 2010, 107, 8772–8777. [Google Scholar] [CrossRef]
- Annese, V.; Rogai, F.; Settesoldi, A.; Bagnoli, S. PPARγ in Inflammatory Bowel Disease. PPAR Res. 2012, 2012, 620839. [Google Scholar] [CrossRef]
- Torres, J.; Danese, S.; Colombel, J.F. New therapeutic avenues in ulcerative colitis: Thinking out of the box. Gut 2013, 62, 1642–1652. [Google Scholar] [CrossRef]
- Celinski, K.; Dworzanski, T.; Fornal, R.; Korolczuk, A.; Madro, A.; Brzozowski, T.; Slomka, M. Comparison of anti-inflammatory properties of peroxisome proliferator-activated receptor gamma agonists rosiglitazone and troglitazone in prophylactic treatment of experimental colitis. J. Physiol. Pharmacol. 2013, 64, 587–595. [Google Scholar] [PubMed]
- Lewis, J.D.; Lichtenstein, G.R.; Deren, J.J.; Sands, B.E.; Hanauer, S.B.; Katz, J.A.; Lashner, B.; Present, D.H.; Chuai, S.; Ellenberg, J.H.; et al. Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.J. Revisiting the rosiglitazone story-lessons learned. N. Engl. J. Med. 2010, 363, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Pirat, C.; Farce, A.; Lebegue, N.; Renault, N.; Furman, C.; Millet, R.; Yous, S.; Speca, S.; Berthelot, P.; Desreumaux, P.; et al. Targeting peroxisome proliferator-activated receptors (PPARs): Development of modulators. J. Med. Chem. 2012, 55, 4027–4061. [Google Scholar] [CrossRef] [PubMed]
- Speca, S.; Rousseaux, C.; Dubuquoy, C.; Rieder, F.; Vetuschi, A.; Sferra, R.; Giusti, I.; Bertin, B.; Dubuquoy, L.; Gaudio, E.; et al. Novel PPARgamma Modulator GED-0507-34 Levo Ameliorates Inflammation-driven Intestinal Fibrosis. Inflamm. Bowel Dis. 2016, 22, 279–292. [Google Scholar] [CrossRef]
- U.S. National Library of Medicine. Efficacy and Safety Study of GED-0507-34-Levo for Treatment of UC (SEGMENT); ClinicalTrials gov Identifier NCT02808390; U.S. National Library of Medicine: Bethesda, MA, USA, 2018.



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eder, P.; Adler, M.; Dobrowolska, A.; Kamhieh-Milz, J.; Witowski, J. The Role of Adipose Tissue in the Pathogenesis and Therapeutic Outcomes of Inflammatory Bowel Disease. Cells 2019, 8, 628. https://doi.org/10.3390/cells8060628
Eder P, Adler M, Dobrowolska A, Kamhieh-Milz J, Witowski J. The Role of Adipose Tissue in the Pathogenesis and Therapeutic Outcomes of Inflammatory Bowel Disease. Cells. 2019; 8(6):628. https://doi.org/10.3390/cells8060628
Chicago/Turabian StyleEder, Piotr, Maciej Adler, Agnieszka Dobrowolska, Julian Kamhieh-Milz, and Janusz Witowski. 2019. "The Role of Adipose Tissue in the Pathogenesis and Therapeutic Outcomes of Inflammatory Bowel Disease" Cells 8, no. 6: 628. https://doi.org/10.3390/cells8060628
APA StyleEder, P., Adler, M., Dobrowolska, A., Kamhieh-Milz, J., & Witowski, J. (2019). The Role of Adipose Tissue in the Pathogenesis and Therapeutic Outcomes of Inflammatory Bowel Disease. Cells, 8(6), 628. https://doi.org/10.3390/cells8060628