Identification of Keratin 23 as a Hepatitis C Virus-Induced Host Factor in the Human Liver

 ,
,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Compounds and Reagents
2.3. Plasmids
2.4. Production of Viruses and Pseudoparticles
2.5. Western Blotting
2.6. Dot Blot
2.7. Immunofluorescence Analysis
2.8. Real-Time Quantitative PCR
2.9. In Vitro Transcription and Electroporation of Huh-7.5 Cells
2.10. HCV Infection Assays
2.11. Luciferase Assays
2.12. Generation of CRISPR/Cas9 Knockout Cells
2.13. Statistical Methods
3. Results
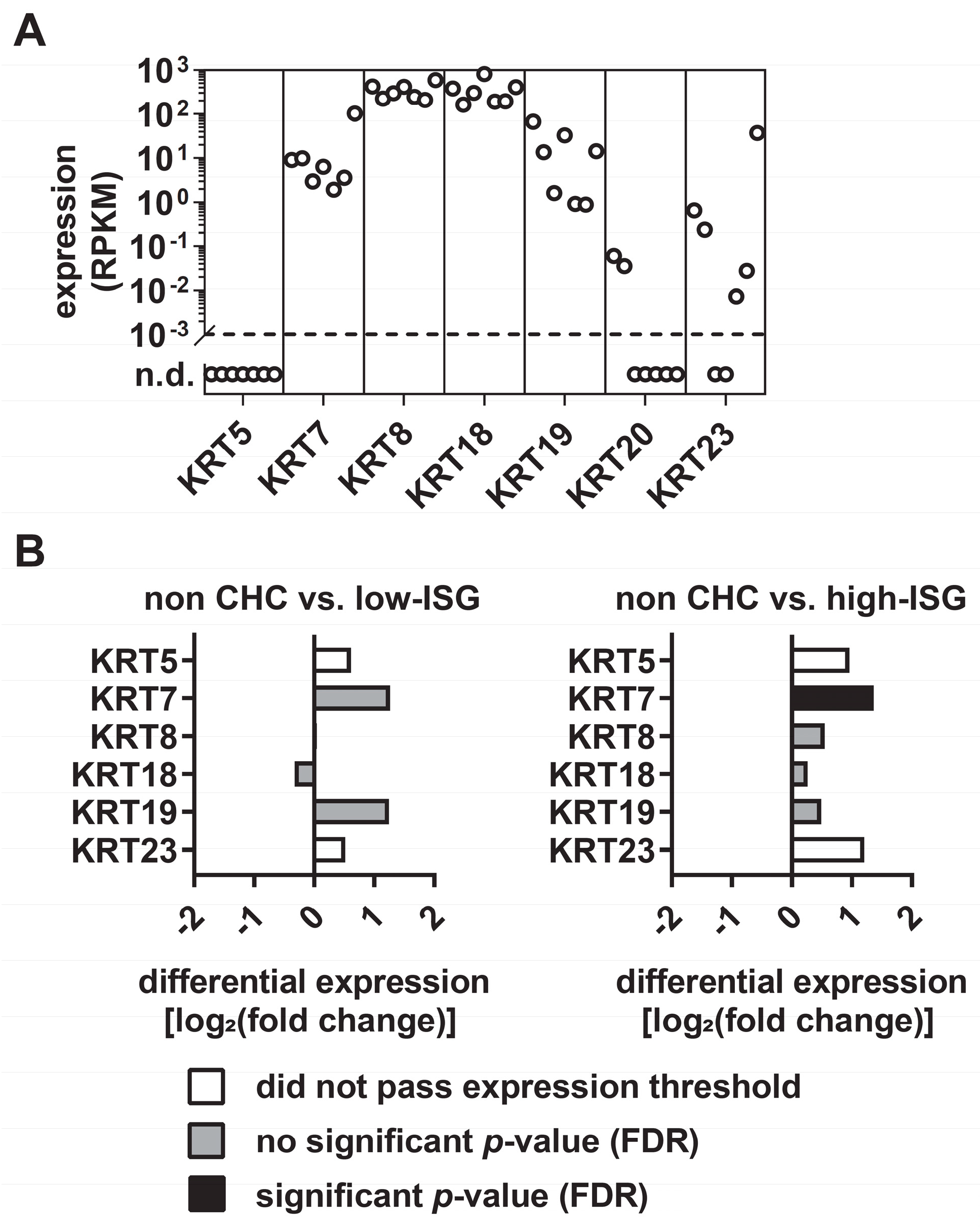
3.1. Differential Expression Levels of KRTs in Primary Human Hepatocytes
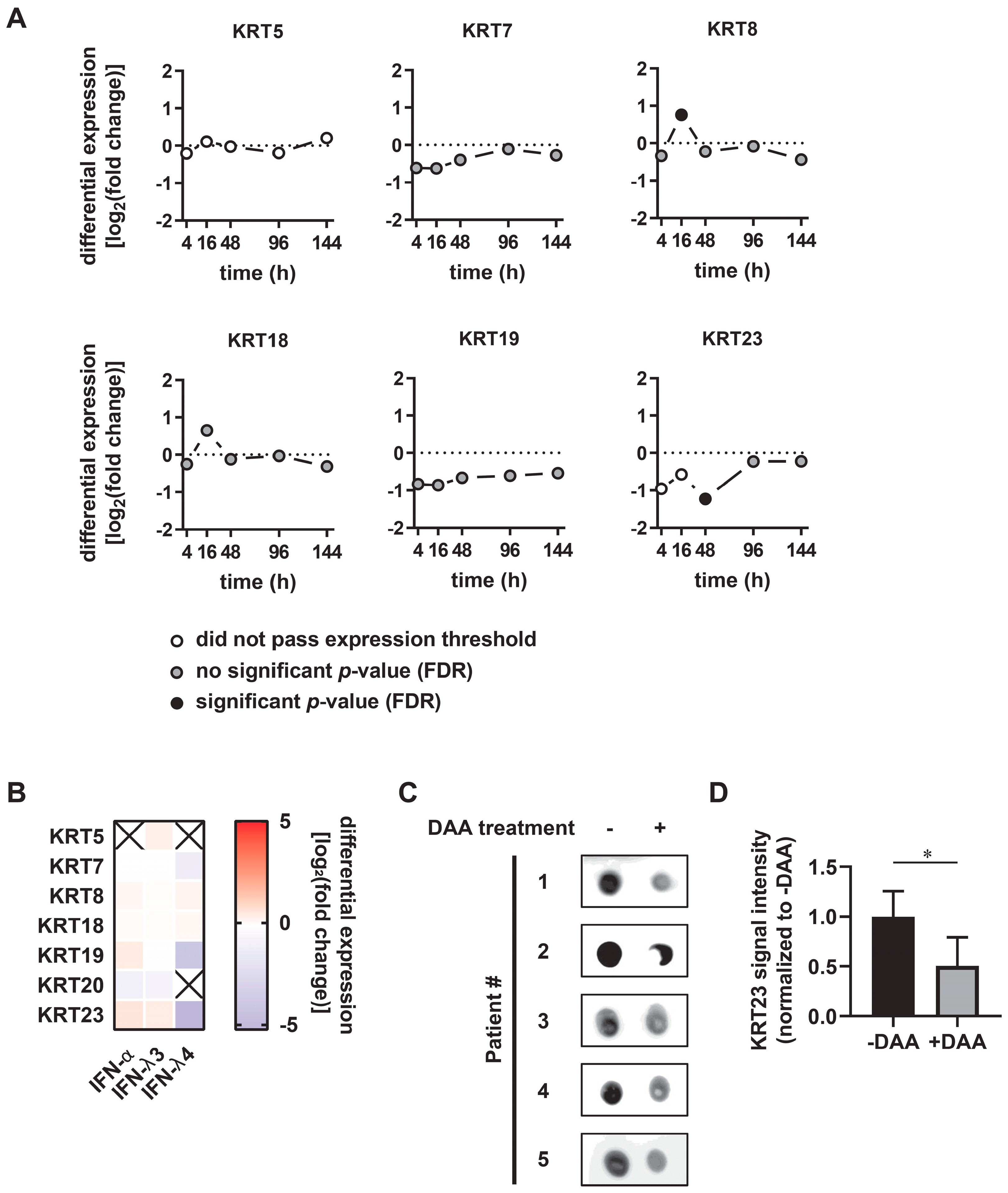
3.2. KRT23 Expression is Induced Ex Vivo in PHHs upon HCV Challenge
3.3. KRT23 Plasma Levels Decrease Concomitant with HCV Clearance upon Anti-HCV Therapy
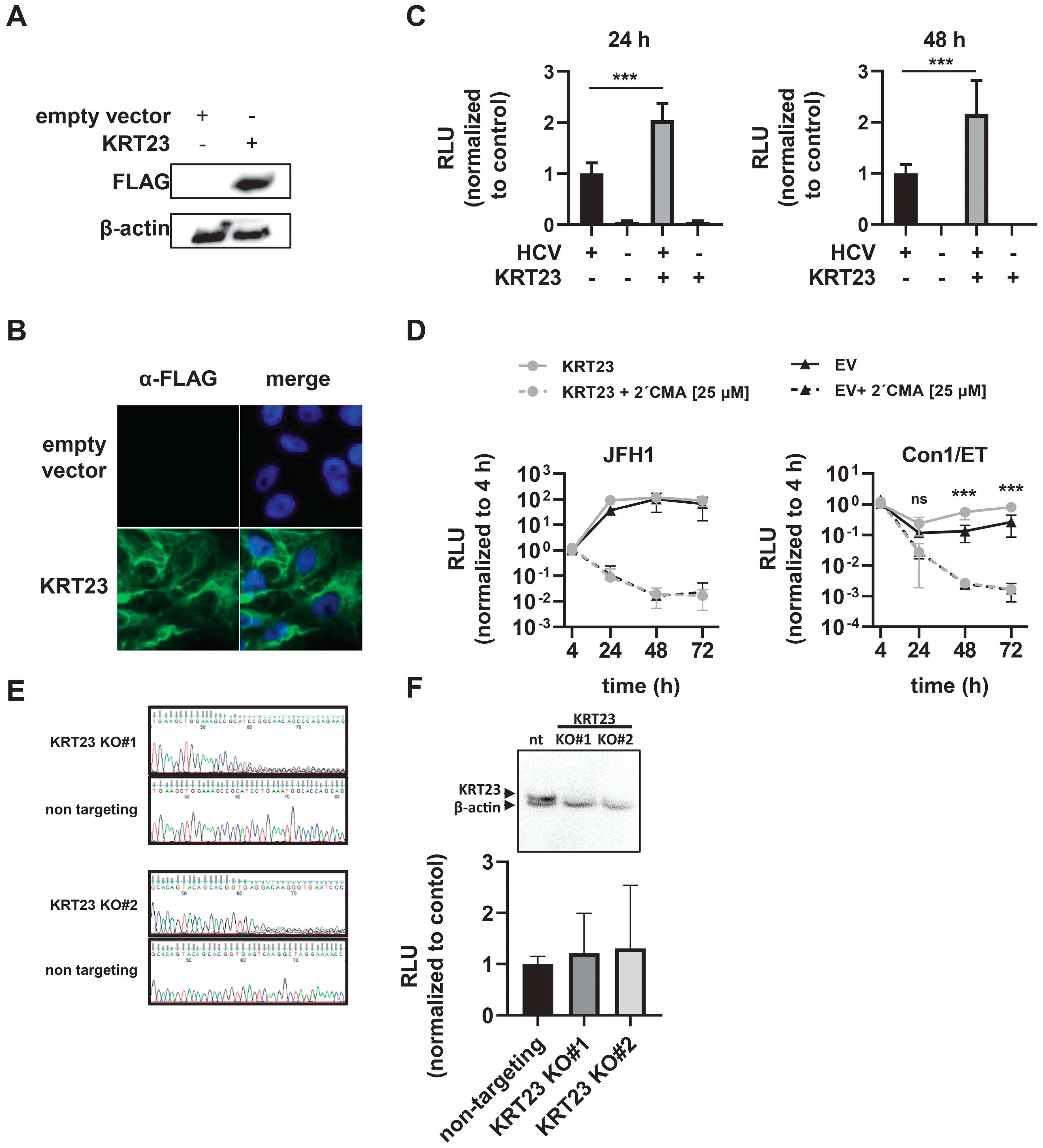
3.4. Ectopically-Expressed KRT23 Facilitates the HCV Life Cycle Progression
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KRT23 KO Oligos | |
| specification | sequence |
| KRT23_Oligo #1_fwd | GTCGTCTCCCACCGGCTGGTGCCATTTCAGGATG GTTTCGAGACGTG |
| KRT23_Oligo #1_rev | CACGTCTCGAAACCATCCTGAAATGGCACCAGCCGGTGGGAGACGAC |
| KRT23_Oligo #2_fwd | GTCGTCTCCCACCGCAGTACAGCACGGTGAGTCAGTTTCGAGACGTG |
| KRT23_Oligo #2_rev | CACGTCTCGAAACTGACTCACCGTGCTGTACTGCGGTGGGAGACGAC |
| KRT23 KO gDNA primers for sequencing | |
| specification | sequence |
| KRT23_gRNA1_left | ACCATGCAGAATCTCAACGAC |
| KRT23_gRNA1_right | TGGAAATTGTCTTCTGGACTCA |
| KRT23_gRNA2_left | ACTGTGCAGAGCAGACAAGGT |
| KRT23_gRNA2_right | GGAAAGTGAGAGTTGTCCCAAG |
References
- Haines, R.L.; Lane, E.B. Keratins and disease at a glance. J. Cell Sci. 2012, 125, 3923–3928. [Google Scholar] [CrossRef] [PubMed]
- McLean, W.H.I.; Moore, C.B.T. Keratin disorders: From gene to therapy. Hum. Mol. Genet. 2011, 20, R189–R197. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.-O.; Darling, J.M.; Krams, S.M.; Esquivel, C.O.; Keeffe, E.B.; Sibley, R.K.; Lee, Y.M.; Wright, T.L.; Omary, M.B. Keratin 8 and 18 mutations are risk factors for developing liver disease of multiple etiologies. Proc. Natl. Acad. Sci. USA 2003, 100, 6063–6068. [Google Scholar] [CrossRef] [PubMed]
- Toivola, D.M.; Strnad, P.; Habtezion, A.; Omary, M.B. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010, 20, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ku, N.-O.; Strnad, P.; Bantel, H.; Omary, M.B. Keratins: Biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology 2016, 64, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-S.; Wang, L.; Huang, H.; Nelson, M.; Smith, D.I. Keratin 23 (K23), a novel acidic keratin, is highly induced by histone deacetylase inhibitors during differentiation of pancreatic cancer cells. Genes, Chromosomes Cancer 2001, 30, 123–135. [Google Scholar] [CrossRef]
- Starmann, J.; Fälth, M.; Spindelböck, W.; Lanz, K.-L.; Lackner, C.; Zatloukal, K.; Trauner, M.; Sültmann, H. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS ONE 2012, 7, e46584. [Google Scholar] [CrossRef] [PubMed]
- Guldiken, N.; Kobazi Ensari, G.; Lahiri, P.; Couchy, G.; Preisinger, C.; Liedtke, C.; Zimmermann, H.W.; Ziol, M.; Boor, P.; Zucman-Rossi, J.; et al. Keratin 23 is a stress-inducible marker of mouse and human ductular reaction in liver disease. J. Hepatol. 2016, 65, 552–559. [Google Scholar] [CrossRef]
- Zhong, Q.; An, X.; Yang, Y.-X.; Hu, H.-D.; Ren, H.; Hu, P. Keratin 8 is involved in hepatitis B virus replication. J. Med. Virol. 2014, 86, 687–694. [Google Scholar] [CrossRef]
- Bantel, H.; Lügering, A.; Heidemann, J.; Volkmann, X.; Poremba, C.; Strassburg, C.P.; Manns, M.P.; Schulze-Osthoff, K. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology 2004, 40, 1078–1087. [Google Scholar] [CrossRef]
- Scheel, T.K.H.; Rice, C.M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 2013, 19, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Penin, F.; Dubuisson, J.; Rey, F.A.; Moradpour, D.; Pawlotsky, J.-M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Hepatitis Report 2017. Available online: https://www.who.int/hepatitis/publications/global-hepatitis-report2017/en/ (accessed on 18 June 2019).
- Kleine, M.; Riemer, M.; Krech, T.; DeTemple, D.; Jäger, M.D.; Lehner, F.; Manns, M.P.; Klempnauer, J.; Borlak, J.; Bektas, H.; et al. Explanted diseased livers-a possible source of metabolic competent primary human hepatocytes. PLoS ONE 2014, 9, e101386. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.; Vieyres, G.; Terczyńska-Dyla, E.; Dijkman, R.; Gad, H.H.; Akhtar, H.; Geffers, R.; Vondran, F.W.R.; Thiel, V.; Kaderali, L. Transcriptome analysis reveals a classical interferon signature induced by IFNλ4 in human primary cells. Gene. Immun. 2015, 16, 414. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Date, T.; Miyamoto, M.; Furusaka, A.; Tokushige, K.; Mizokami, M.; Wakita, T. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 2003, 125, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Hoffmann, S.; Herian, U.; Penin, F.; Bartenschlager, R. Viral and Cellular Determinants of Hepatitis C Virus RNA Replication in Cell Culture. J. Virol. 2003, 77, 3007–3019. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.-G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.-L. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.-S.; et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, R.; Zou, K.; Yu, W.; Guo, W.; Gao, Y.; Li, J.; Li, M.; Tai, Y.; Huang, W.; et al. Keratin 23 promotes telomerase reverse transcriptase expression and human colorectal cancer growth. Cell Death Dis. 2017, 8, e2961. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Regalado-Magdos, A.D.; Stiggelbout, B.; Lee-Kirsch, M.A.; Lieberman, J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 2010, 11, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Irving, W.L.; Rupp, D.; McClure, C.P.; Than, L.M.; Titman, A.; Ball, J.K.; Steinmann, E.; Bartenschlager, R.; Pietschmann, T.; Brown, R.J.P. Development of a high-throughput pyrosequencing assay for monitoring temporal evolution and resistance associated variant emergence in the Hepatitis C virus protease coding-region. Antiviral Res. 2014, 110, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Anggakusuma; Romero-Brey, I.; Berger, C.; Colpitts, C.C.; Boldanova, T.; Engelmann, M.; Todt, D.; Perin, P.M.; Behrendt, P.; Vondran, F.W.R.; et al. Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 2015, 62, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Bruening, J.; Lasswitz, L.; Banse, P.; Kahl, S.; Marinach, C.; Vondran, F.W.; Kaderali, L.; Silvie, O.; Pietschmann, T.; Meissner, F.; et al. Hepatitis C virus enters liver cells using the CD81 receptor complex proteins calpain-5 and CBLB. PLoS Pathog 2018, 14, e1007111. [Google Scholar] [CrossRef] [PubMed]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Chen, S.; Cai, J.; Guo, Y.; Song, Z.; Che, J.; Liu, C.; Wu, C.; Ding, M.; Deng, H. Derivation and Characterization of Hepatic Progenitor Cells from Human Embryonic Stem Cells. PLoS ONE 2009, 4, e6468. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Lienau, T.C.; Tao, G.-Z.; Lazzeroni, L.C.; Stickel, F.; Schuppan, D.; Omary, M.B. Keratin variants associate with progression of fibrosis during chronic hepatitis C infection. Hepatology 2006, 43, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Boldanova, T.; Suslov, A.; Heim, M.H.; Necsulea, A. Transcriptional response to hepatitis C virus infection and interferon-alpha treatment in the human liver. EMBO Mol. Med. 2017, 9, 816–834. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Gu, L.-H.; Coulombe, P.A. Keratin function in skin epithelia: A broadening palette with surprising shades. Curr. Opin. Cell Biol. 2007, 19, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Omary, M.B.; Ku, N.-O.; Strnad, P.; Hanada, S. Toward unraveling the complexity of simple epithelial keratins in human disease. J. Clin. Invest. 2009, 119, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Odena, G.; Chen, J.; Lozano, J.J.; Altamirano, J.; Rodrigo-Torres, D.; Affo, S.; Morales-Ibanez, O.; Matsushita, H.; Zou, J.; Dumitru, R.; et al. LPS-TLR4 Pathway Mediates Ductular Cell Expansion in Alcoholic Hepatitis. Sci. Rep. 2016, 6, 35610. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Brocker, C.N.; Takahashi, S.; Yagai, T.; Kim, T.; Xie, G.; Wang, H.; Qu, A.; Gonzalez, F.J. Keratin 23 is a PPARA-dependent, MYC-amplified oncogene that promotes hepatocyte proliferation. Hepatology 2019. [Google Scholar] [CrossRef]
- Rakic, B.; Sagan, S.M.; Noestheden, M.; Bélanger, S.; Nan, X.; Evans, C.L.; Xie, X.S.; Pezacki, J.P. Peroxisome proliferator-activated receptor α antagonism inhibits hepatitis C virus replication. Chem. Biol. 2006, 13, 23–30. [Google Scholar] [CrossRef]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARα activation is essential for HCV core protein–induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 2008, 118, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Yang, Y.; Zhang, X.; Nakajima, T.; Tanaka, N.; Sugiyama, E.; Kamijo, Y.; Lu, Y.; Moriya, K.; Koike, K. Age-dependent PPARα activation induces hepatic sulfatide accumulation in transgenic mice carrying the hepatitis C virus core gene. Glycoconjugate J. 2016, 33, 927–936. [Google Scholar] [CrossRef]
- Higgs; Lerat, H.; Pawlotsky, J.M. Hepatitis C virus-induced activation of β-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683. [Google Scholar] [CrossRef]
- Neumann, A.U.; Lam, N.P.; Dahari, H.; Gretch, D.R.; Wiley, T.E.; Layden, T.J.; Perelson, A.S. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-α therapy. Science 1998, 282, 103–107. [Google Scholar] [CrossRef]
- Liffers, S.-T.; Maghnouj, A.; Munding, J.B.; Jackstadt, R.; Herbrand, U.; Schulenborg, T.; Marcus, K.; Klein-Scory, S.; Schmiegel, W.; Schwarte-Waldhoff, I. Keratin 23, a novel DPC4/Smad4 target gene which binds 14-3-3ε. BMC Cancer 2011, 11, 137. [Google Scholar] [CrossRef]
- Takashima, M.; Kuramitsu, Y.; Yokoyama, Y.; Iizuka, N.; Toda, T.; Sakaida, I.; Okita, K.; Oka, M.; Nakamura, K. Proteomic profiling of heat shock protein 70 family members as biomarkers for hepatitis C virus-related hepatocellular carcinoma. Proteomics 2003, 3, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-M.; Shin, M.-J.; Kim, J.-H.; Oh, J.-W. Proteomic profiling of cellular proteins interacting with the hepatitis C virus core protein. Proteomics 2005, 5, 2227–2237. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-M.; Kim, S.-J.; Kim, J.-H.; Lee, W.; Kim, G.-W.; Lee, K.-H.; Choi, K.-Y.; Oh, J.-W. Interaction of hepatitis C virus core protein with Hsp60 triggers the production of reactive oxygen species and enhances TNF-alpha-mediated apoptosis. Cancer Lett. 2009, 279, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, O.; Fontanes, V.; Raychaudhuri, S.; Loo, R.; Loo, J.; Arumugaswami, V.; Sun, R.; Dasgupta, A.; French, S.W. The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology 2009, 50, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Hayashi, J.; Moriyama, M.; Arakawa, Y.; Hino, O. Hepatitis C virus core protein interacts with 14-3-3 protein and activates the kinase Raf-1. J. Virol. 2000, 74, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.L.; Oelke, A.; Johnson, M.E.; Eilert, K.D.; Simpson, P.C.; Todd, S.C. CD81 associates with 14-3-3 in a redox-regulated palmitoylation-dependent manner. J. Biol. Chem. 2004, 279, 19401–19406. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinast, V.; Leber, S.L.; Brown, R.J.P.; Vieyres, G.; Behrendt, P.; Eßbach, C.; Strnad, P.; Vondran, F.W.R.; Cornberg, M.; Wex, C.; et al. Identification of Keratin 23 as a Hepatitis C Virus-Induced Host Factor in the Human Liver. Cells 2019, 8, 610. https://doi.org/10.3390/cells8060610
Kinast V, Leber SL, Brown RJP, Vieyres G, Behrendt P, Eßbach C, Strnad P, Vondran FWR, Cornberg M, Wex C, et al. Identification of Keratin 23 as a Hepatitis C Virus-Induced Host Factor in the Human Liver. Cells. 2019; 8(6):610. https://doi.org/10.3390/cells8060610
Chicago/Turabian StyleKinast, Volker, Stefan L. Leber, Richard J. P. Brown, Gabrielle Vieyres, Patrick Behrendt, Constanze Eßbach, Pavel Strnad, Florian W. R. Vondran, Markus Cornberg, Cora Wex, and et al. 2019. "Identification of Keratin 23 as a Hepatitis C Virus-Induced Host Factor in the Human Liver" Cells 8, no. 6: 610. https://doi.org/10.3390/cells8060610
APA StyleKinast, V., Leber, S. L., Brown, R. J. P., Vieyres, G., Behrendt, P., Eßbach, C., Strnad, P., Vondran, F. W. R., Cornberg, M., Wex, C., Pietschmann, T., Haybaeck, J., Todt, D., & Steinmann, E. (2019). Identification of Keratin 23 as a Hepatitis C Virus-Induced Host Factor in the Human Liver. Cells, 8(6), 610. https://doi.org/10.3390/cells8060610