Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations

Abstract
1. Introduction
2. Examples of Nuclear Mitochondrial Genes with a Role in Common Diseases
2.1. Parkinson’s Disease
2.2. Alzheimer’s Disease
2.3. Huntington Disease
2.4. Diabetes
2.5. Cancer
2.6. Aging
3. Mitochondrial DNA in Common Diseases
3.1. Mitochondrial DNA Variants and Alzheimer’s Disease (AD)
3.2. Mitochondrial DNA Variants and Parkinson’s Disease (PD)
3.3. Mitochondrial DNA Variants and HD
3.4. Mitochondrial Mutation and Diabetes
3.5. Mitochondrial DNA Variants and Cancer
4. The Relationship between mtDNA Mutations and Aging
4.1. Accumulation of mtDNA Mutations in Normal Aging
4.2. Factors Contributing to the Somatic mtDNA Mutation Rate
4.3. Calculating the mtDNA Mutation Rate with Age
= 16,569 × 1000 × 1.47 × 10−7 × (45 + 280 + 91) = 1013
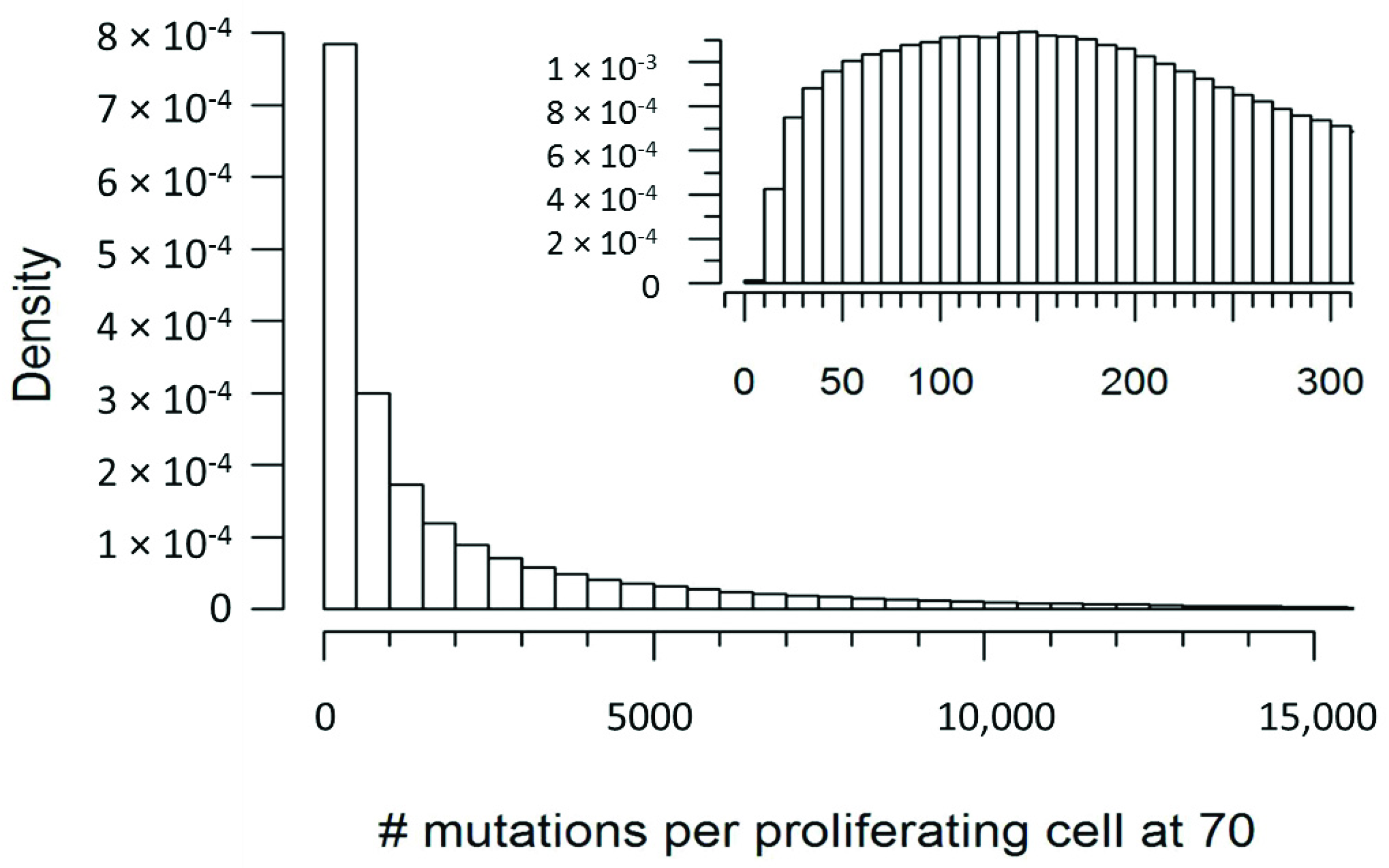
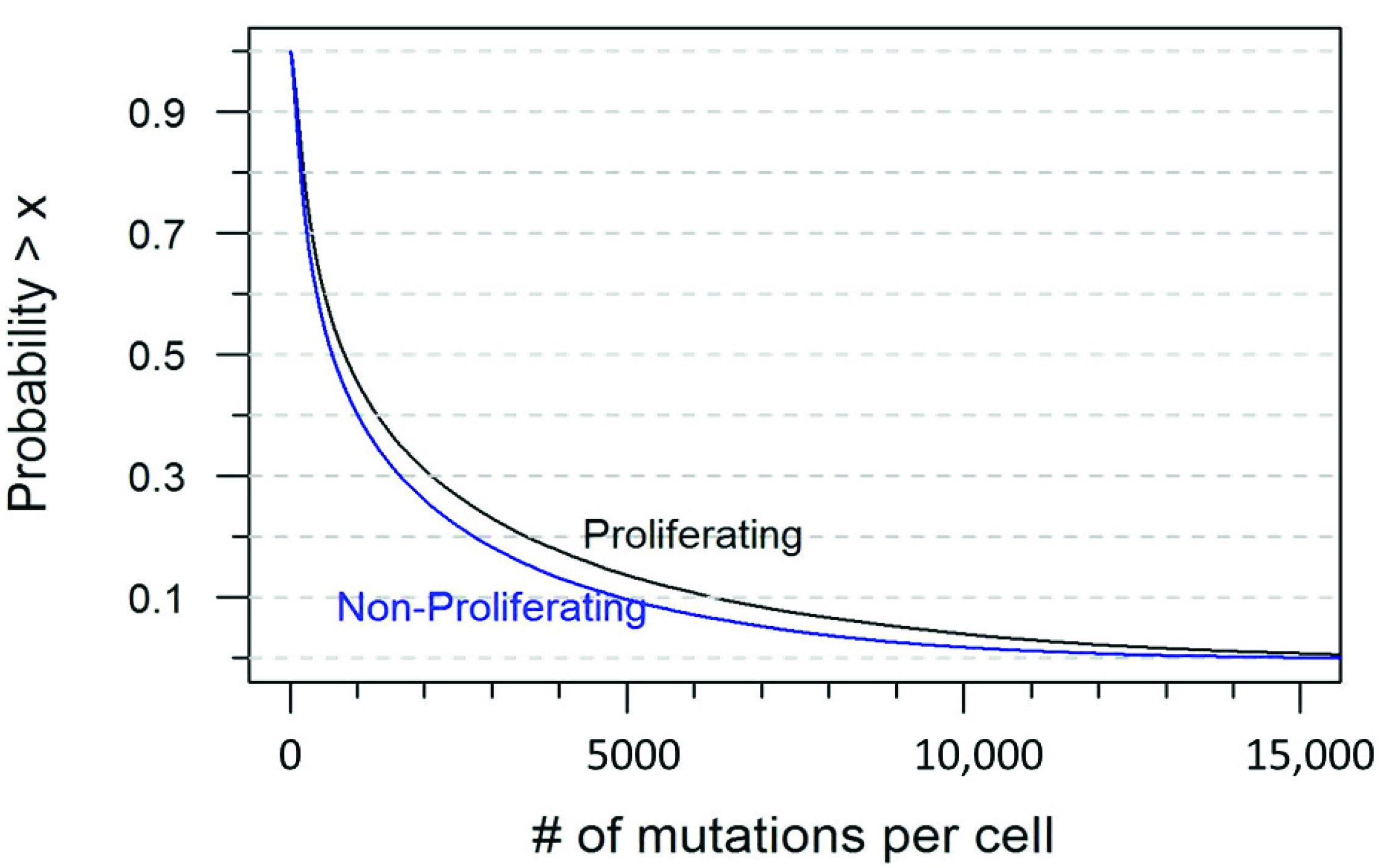
4.4. Simulating mtDNA Mutations
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gray, M.W.; Burger, G.; Lang, B.F. Mitochondrial evolution. Science 1999, 283, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. Int. J. Clin. Exp. Med. 2009, 2, 1–16. [Google Scholar]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.A.; Li, L.; Kakhlon, O.; Myerowitz, R.; Raben, N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015, 11, 385–402. [Google Scholar] [CrossRef]
- Zhang, B.F.; Xing, L.; Cui, P.F.; Wang, F.Z.; Xie, R.L.; Zhang, J.L.; Zhang, M.; He, Y.J.; Lyu, J.Y.; Qiao, J.B.; et al. Mitochondria apoptosis pathway synergistically activated by hierarchical targeted nanoparticles co-delivering siRNA and lonidamine. Biomaterials 2015, 61, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Marin, S.; Chiang, K.; Bassilian, S.; Lee, W.N.; Boros, L.G.; Fernandez-Novell, J.M.; Centelles, J.J.; Medrano, A.; Rodriguez-Gil, J.E.; Cascante, M. Metabolic strategy of boar spermatozoa revealed by a metabolomic characterization. FEBS Lett. 2003, 554, 342–346. [Google Scholar] [CrossRef]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Nunez, M.T. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Shiba-Fukushima, K.; Ishikawa, K.I.; Inoshita, T.; Izawa, N.; Takanashi, M.; Sato, S.; Onodera, O.; Akamatsu, W.; Okano, H.; Imai, Y.; et al. Evidence that phosphorylated ubiquitin signaling is involved in the etiology of Parkinson’s disease. Hum. Mol. Genet. 2017, 26, 3172–3185. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Schapira, A.H.; Taanman, J.W. Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PLoS ONE 2009, 4, e4756. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, A.; Gegg, M.E.; Taanman, J.W.; King, R.H.; Kock, N.; Klein, C.; Schapira, A.H. Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp. Neurol. 2009, 219, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson’s disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605. [Google Scholar] [CrossRef]
- Princely Abudu, Y.; Pankiv, S.; Mathai, B.J.; Hakon Lystad, A.; Bindesboll, C.; Brenne, H.B.; Yoke Wui Ng, M.; Thiede, B.; Yamamoto, A.; Mutugi Nthiga, T.; et al. NIPSNAP1 and NIPSNAP2 Act as “Eat Me” Signals for Mitophagy. Dev. Cell 2019. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar] [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 6078–6090. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Morozova, K.N.; Suldina, L.A.; Malankhanova, T.B.; Grigor’eva, E.V.; Zakian, S.M.; Kiseleva, E.; Malakhova, A.A. Introducing an expanded CAG tract into the huntingtin gene causes a wide spectrum of ultrastructural defects in cultured human cells. PLoS ONE 2018, 13, e0204735. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef]
- Khalil, B.; El Fissi, N.; Aouane, A.; Cabirol-Pol, M.J.; Rival, T.; Lievens, J.C. PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 2015, 6, e1617. [Google Scholar] [CrossRef]
- Zorzano, A.; Liesa, M.; Palacin, M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int. J. Biochem. Cell Biol. 2009, 41, 1846–1854. [Google Scholar] [CrossRef]
- Parra, V.; Verdejo, H.E.; Iglewski, M.; Del Campo, A.; Troncoso, R.; Jones, D.; Zhu, Y.; Kuzmicic, J.; Pennanen, C.; Lopez-Crisosto, C.; et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes 2014, 63, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Joung, K.H.; Ryu, M.J.; Kim, S.J.; Kim, H.; Chung, H.K.; Lee, M.H.; Lee, S.E.; Choi, M.J.; Chang, J.Y.; et al. Disruption of CR6-interacting factor-1 (CRIF1) in mouse islet beta cells leads to mitochondrial diabetes with progressive beta cell failure. Diabetologia 2015, 58, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Serrano, M.; Camafeita, E.; Lopez, J.A.; Rubio, M.A.; Breton, I.; Garcia-Consuegra, I.; Garcia-Santos, E.; Lago, J.; Sanchez-Pernaute, A.; Torres, A.; et al. Differential proteomic and oxidative profiles unveil dysfunctional protein import to adipocyte mitochondria in obesity-associated aging and diabetes. Redox Biol. 2017, 11, 415–428. [Google Scholar] [CrossRef]
- Choi, J.; Ravipati, A.; Nimmagadda, V.; Schubert, M.; Castellani, R.J.; Russell, J.W. Potential roles of PINK1 for increased PGC-1alpha-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion 2014, 18, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Baysal, B.E.; Maher, E.R. 15 YEARS OF PARAGANGLIOMA: Genetics and mechanism of pheochromocytoma-paraganglioma syndromes characterized by germline SDHB and SDHD mutations. Endocr. -Relat. Cancer 2015, 22, T71–T82. [Google Scholar] [CrossRef]
- Baysal, B.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Drovdlic, C.M.; Savul, S.A.; McLeod, D.R.; Yee, H.A.; Brackmann, D.E.; Slattery, W.H., 3rd; Myers, E.N.; et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J. Med. Genet. 2002, 39, 178–183. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019. [Google Scholar] [CrossRef]
- Zhou, J.; Chong, S.Y.; Lim, A.; Singh, B.K.; Sinha, R.A.; Salmon, A.B.; Yen, P.M. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 2017, 9, 583–599. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Oliveira, M.P.; Khamoui, A.V.; Aparicio, R.; Rera, M.; Rossiter, H.B.; Walker, D.W. Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat. Commun. 2017, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [PubMed]
- Fesahat, F.; Houshmand, M.; Panahi, M.S.; Gharagozli, K.; Mirzajani, F. Do haplogroups H and U act to increase the penetrance of Alzheimer’s disease? Cell Mol. Neurobiol. 2007, 27, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Maruszak, A.; Safranow, K.; Branicki, W.; Gaweda-Walerych, K.; Pospiech, E.; Gabryelewicz, T.; Canter, J.A.; Barcikowska, M.; Zekanowski, C. The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. J. Alzheimers Dis. 2011, 27, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Coto, E.; Gomez, J.; Alonso, B.; Corao, A.I.; Diaz, M.; Menendez, M.; Martinez, C.; Calatayud, M.T.; Moris, G.; Alvarez, V. Late-onset Alzheimer’s disease is associated with mitochondrial DNA 7028C/haplogroup H and D310 poly-C tract heteroplasmy. Neurogenetics 2011, 12, 345–346. [Google Scholar] [CrossRef]
- Santoro, A.; Balbi, V.; Balducci, E.; Pirazzini, C.; Rosini, F.; Tavano, F.; Achilli, A.; Siviero, P.; Minicuci, N.; Bellavista, E.; et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE 2010, 5, e12037. [Google Scholar] [CrossRef]
- Bi, R.; Zhang, W.; Yu, D.; Li, X.; Wang, H.Z.; Hu, Q.X.; Zhang, C.; Lu, W.; Ni, J.; Fang, Y.; et al. Mitochondrial DNA haplogroup B5 confers genetic susceptibility to Alzheimer’s disease in Han Chinese. Neurobiol. Aging 2015, 36, 1604 e7–1604 e16. [Google Scholar] [CrossRef]
- Tranah, G.J.; Nalls, M.A.; Katzman, S.M.; Yokoyama, J.S.; Lam, E.T.; Zhao, Y.; Mooney, S.; Thomas, F.; Newman, A.B.; Liu, Y.; et al. Mitochondrial DNA sequence variation associated with dementia and cognitive function in the elderly. J. Alzheimers Dis. 2012, 32, 357–372. [Google Scholar] [CrossRef]
- Tanaka, N.; Goto, Y.; Akanuma, J.; Kato, M.; Kinoshita, T.; Yamashita, F.; Tanaka, M.; Asada, T. Mitochondrial DNA variants in a Japanese population of patients with Alzheimer’s disease. Mitochondrion 2010, 10, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Ratnaike, T.E.; De Gruyter, H.L.; Jaros, E.; Turnbull, D.M. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol. Aging 2012, 33, 2210–2214. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Mellick, G.D.; Rowe, D.B.; Halliday, G.M.; Jones, M.M.; Manwaring, N.; Vandebona, H.; Silburn, P.A.; Wang, J.J.; Mitchell, P.; et al. Mitochondrial DNA haplogroups J and K are not protective for Parkinson’s disease in the Australian community. Mov. Disord. 2009, 24, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Marelli, C.; Achilli, A.; Goldwurm, S.; Pezzoli, G.; Barone, P.; Pellecchia, M.T.; Stanzione, P.; Brusa, L.; Bentivoglio, A.R.; et al. Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson’s disease in Italians. Eur. J. Hum. Genet. 2005, 13, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Hudson, G.; Nalls, M.; Evans, J.R.; Breen, D.P.; Winder-Rhodes, S.; Morrison, K.E.; Morris, H.R.; Williams-Gray, C.H.; Barker, R.A.; Singleton, A.B.; et al. Two-stage association study and meta-analysis of mitochondrial DNA variants in Parkinson’s disease. Neurology 2013, 80, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.W.; Chuang, J.H.; Chen, J.B.; Tiao, M.M.; Wang, P.W.; Huang, S.T.; Huang, T.L.; Lee, W.C.; Weng, S.W.; Huang, P.H.; et al. Mitochondrial DNA variants as genetic risk factors for Parkinson’s disease. Eur. J. Neurol. 2016, 23, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Pulst, S.M.; Sutton, J.P.; Browne, S.E.; Beal, M.F.; Johns, D.R. Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology 1999, 53, 1787–1793. [Google Scholar] [CrossRef]
- Smigrodzki, R.; Parks, J.; Parker, W.D. High frequency of mitochondrial complex I mutations in Parkinson’s disease and aging. Neurobiol. Aging 2004, 25, 1273–1281. [Google Scholar] [CrossRef]
- Coxhead, J.; Kurzawa-Akanbi, M.; Hussain, R.; Pyle, A.; Chinnery, P.; Hudson, G. Somatic mtDNA variation is an important component of Parkinson’s disease. Neurobiol. Aging 2016, 38, 217 e211–217 e216. [Google Scholar] [CrossRef]
- Purevsuren, J.; Fukao, T.; Hasegawa, Y.; Kobayashi, H.; Li, H.; Mushimoto, Y.; Fukuda, S.; Yamaguchi, S. Clinical and molecular aspects of Japanese patients with mitochondrial trifunctional protein deficiency. Mol. Genet. Metab. 2009, 98, 372–377. [Google Scholar] [CrossRef]
- Hong, Y.B.; Lee, J.H.; Park, J.M.; Choi, Y.R.; Hyun, Y.S.; Yoon, B.R.; Yoo, J.H.; Koo, H.; Jung, S.C.; Chung, K.W.; et al. A compound heterozygous mutation in HADHB gene causes an axonal Charcot-Marie-tooth disease. BMC Med. Genet. 2013, 14, 125. [Google Scholar] [CrossRef] [PubMed]
- Slone, J.; Peng, Y.; Chamberlin, A.; Harris, B.; Kaylor, J.; McDonald, M.T.; Lemmon, M.; El-Dairi, M.A.; Tchapyjnikov, D.; Gonzalez-Krellwitz, L.A.; et al. Biallelic mutations in FDXR cause neurodegeneration associated with inflammation. J. Hum. Genet. 2018, 63, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; McMackin, M.; Nguyen, A.; Cortopassi, G. Parkin deficiency accelerates consequences of mitochondrial DNA deletions and Parkinsonism. Neurobiol. Dis. 2017, 100, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 2014, 70, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Banoei, M.M.; Houshmand, M.; Panahi, M.S.; Shariati, P.; Rostami, M.; Manshadi, M.D.; Majidizadeh, T. Huntington’s disease and mitochondrial DNA deletions: Event or regular mechanism for mutant huntingtin protein and CAG repeats expansion?! Cell Mol. Neurobiol. 2007, 27, 867–875. [Google Scholar] [CrossRef]
- Petersen, M.H.; Budtz-Jorgensen, E.; Sorensen, S.A.; Nielsen, J.E.; Hjermind, L.E.; Vinther-Jensen, T.; Nielsen, S.M.; Norremolle, A. Reduction in mitochondrial DNA copy number in peripheral leukocytes after onset of Huntington’s disease. Mitochondrion 2014, 17, 14–21. [Google Scholar] [CrossRef]
- Mousavizadeh, K.; Rajabi, P.; Alaee, M.; Dadgar, S.; Houshmand, M. Usage of mitochondrial D-loop variation to predict risk for Huntington disease. Mitochondrial DNA 2015, 26, 579–582. [Google Scholar] [CrossRef]
- Kasraie, S.; Houshmand, M.; Banoei, M.M.; Ahari, S.E.; Panahi, M.S.; Shariati, P.; Bahar, M.; Moin, M. Investigation of tRNA(Leu/Lys) and ATPase 6 genes mutations in Huntington’s disease. Cell Mol. Neurobiol. 2008, 28, 933–938. [Google Scholar] [CrossRef]
- Naing, A.; Kenchaiah, M.; Krishnan, B.; Mir, F.; Charnley, A.; Egan, C.; Bano, G. Maternally inherited diabetes and deafness (MIDD): Diagnosis and management. J. Diabetes Its Complicat. 2014, 28, 542–546. [Google Scholar] [CrossRef]
- Goto, Y.; Nonaka, I.; Horai, S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Emrick, L.T.; Hsu, J.W.; Chanprasert, S.; Jahoor, F.; Scaglia, F.; Craigen, W.J. Glucose metabolism derangements in adults with the MELAS m.3243A>G mutation. Mitochondrion 2014, 18, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Bendall, K.E.; Sykes, B.C. Length heteroplasmy in the first hypervariable segment of the human mtDNA control region. Am. J. Hum. Genet. 1995, 57, 248–256. [Google Scholar] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Chen, G.; He, J.; Zhang, F.; Li, M.; Wang, Q.; Zhou, H.; Lyu, J.; Bai, Y. Mitochondrial common deletion is elevated in blood of breast cancer patients mediated by oxidative stress. Mitochondrion 2016, 26, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Li, S.H.; Lin, J.C.; Wu, C.C.; Yeh, D.C.; Wei, Y.H. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat. Res. 2004, 547, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zheng, L.; Liu, W.; Wang, X.; Wang, Z.; French, A.J.; Kang, D.; Chen, L.; Thibodeau, S.N. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res. 2011, 71, 2978–2987. [Google Scholar] [CrossRef]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef]
- Kang, E.; Wang, X.; Tippner-Hedges, R.; Ma, H.; Folmes, C.D.; Gutierrez, N.M.; Lee, Y.; Van Dyken, C.; Ahmed, R.; Li, Y.; et al. Age-Related Accumulation of Somatic Mitochondrial DNA Mutations in Adult-Derived Human iPSCs. Cell Stem Cell 2016, 18, 625–636. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef]
- Niemann, J.; Johne, C.; Schroder, S.; Koch, F.; Ibrahim, S.M.; Schultz, J.; Tiedge, M.; Baltrusch, S. An mtDNA mutation accelerates liver aging by interfering with the ROS response and mitochondrial life cycle. Free Radic. Biol. Med. 2017, 102, 174–187. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, R.; Nooteboom, M.; Osiewacz, H.D. Control of mitochondrial integrity in ageing and disease. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130439. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, J.F.; Baris, O.R.; Kittelmann, A.; Becker, K.; Rothschild, M.A.; Wiesner, R.J. Catecholamine Metabolism Induces Mitochondrial DNA Deletions and Leads to Severe Adrenal Degeneration during Aging. Neuroendocrinology 2017, 104, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Ahmadieh, N.; Bielas, J.H.; Ericson, N.G.; Van Remmen, H. Skeletal muscle mitochondrial DNA deletions are not increased in CuZn-superoxide dismutase deficient mice. Exp. Gerontol. 2015, 61, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J.; Rosenfeldt, F.L.; Zhang, C.; Linnane, A.W.; Nagley, P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: Lack of change of copy number with age. Nucleic Acids Res. 2003, 31, e61. [Google Scholar] [CrossRef]
- Foote, K.; Reinhold, J.; Yu, E.P.K.; Figg, N.L.; Finigan, A.; Murphy, M.P.; Bennett, M.R. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell 2018, e12773. [Google Scholar] [CrossRef]
- Mengel-From, J.; Thinggaard, M.; Dalgard, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014, 133, 1149–1159. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016, 164, 337–340. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef]
- Bianconi, E.; Piovesan, A.; Facchin, F.; Beraudi, A.; Casadei, R.; Frabetti, F.; Vitale, L.; Pelleri, M.C.; Tassani, S.; Piva, F.; et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013, 40, 463–471. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Med. 2013, 3, a021220. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.; Clayton, D.A. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell 1977, 11, 719–727. [Google Scholar] [CrossRef]
- Oppenheim, R.W. Cell Death during Development of the Nervous System. Annu. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef] [PubMed]
- Slone, J.; Zhang, J.; Huang, T. Experience from the First Live-Birth Derived From Oocyte Nuclear Transfer as a Treatment Strategy for Mitochondrial Diseases. J. Mol. Genet. Med. Int. J. Biomed. Res. 2017, 11, 258. [Google Scholar] [CrossRef]
- Poovathingal, S.K.; Gruber, J.; Lakshmanan, L.; Halliwell, B.; Gunawan, R. Is mitochondrial DNA turnover slower than commonly assumed? Biogerontology 2012, 13, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, L.N.; Yee, Z.; Ng, L.F.; Gunawan, R.; Halliwell, B.; Gruber, J. Clonal expansion of mitochondrial DNA deletions is a private mechanism of aging in long-lived animals. Aging Cell 2018, 17, e12814. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C.; et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Investig. 2003, 112, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Fliedner, T.M.; Graessle, D.; Paulsen, C.; Reimers, K. Structure and function of bone marrow hemopoiesis: Mechanisms of response to ionizing radiation exposure. Cancer Biother. Radiopharm. 2002, 17, 405–426. [Google Scholar] [CrossRef] [PubMed]
- Colijn, C.; Mackey, M.C. A mathematical model of hematopoiesis—I. Periodic chronic myelogenous leukemia. J. Theor. Biol. 2005, 237, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Catlin, S.N.; Busque, L.; Gale, R.E.; Guttorp, P.; Abkowitz, J.L. The replication rate of human hematopoietic stem cells in vivo. Blood 2011, 117, 4460–4466. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Johnson, K.A. Fidelity of the human mitochondrial DNA polymerase. J. Biol. Chem. 2006, 281, 36236–36240. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Kubacka, I.; Chinnery, P.F.; Lightowlers, R.N.; Turnbull, D.M.; Howell, N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999, 23, 147. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, A.; Trounce, I.A.; McKenzie, M.; Kelly, R.D.W.; St John, J.C. Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res. 2012, 40, 10124–10138. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Number of Mutations | Probability in a Proliferating Cell | Probability in a Non-Proliferating Cell |
|---|---|---|
| ≤10 | <0.001 | 0.001 |
| ≤50 | 0.031 | 0.050 |
| ≤100 | 0.084 | 0.118 |
| ≤200 | 0.195 | 0.250 |
| ≥500 | 0.606 | 0.550 |
| ≥1000 | 0.455 | 0.402 |
| ≥1500 | 0.370 | 0.318 |
| ≥2000 | 0.311 | 0.261 |
| ≥3000 | 0.231 | 0.183 |
| ≥5000 | 0.137 | 0.097 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Slone, J.; Fei, L.; Huang, T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 2019, 8, 608. https://doi.org/10.3390/cells8060608
Li H, Slone J, Fei L, Huang T. Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells. 2019; 8(6):608. https://doi.org/10.3390/cells8060608
Chicago/Turabian StyleLi, Huanzheng, Jesse Slone, Lin Fei, and Taosheng Huang. 2019. "Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations" Cells 8, no. 6: 608. https://doi.org/10.3390/cells8060608
APA StyleLi, H., Slone, J., Fei, L., & Huang, T. (2019). Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells, 8(6), 608. https://doi.org/10.3390/cells8060608