New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach

 ,
,  , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients, BRCA1/2 Testing and Human Ethics
2.2. Next-Generation Sequencing Analysis
2.3. In Silico Analysis
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
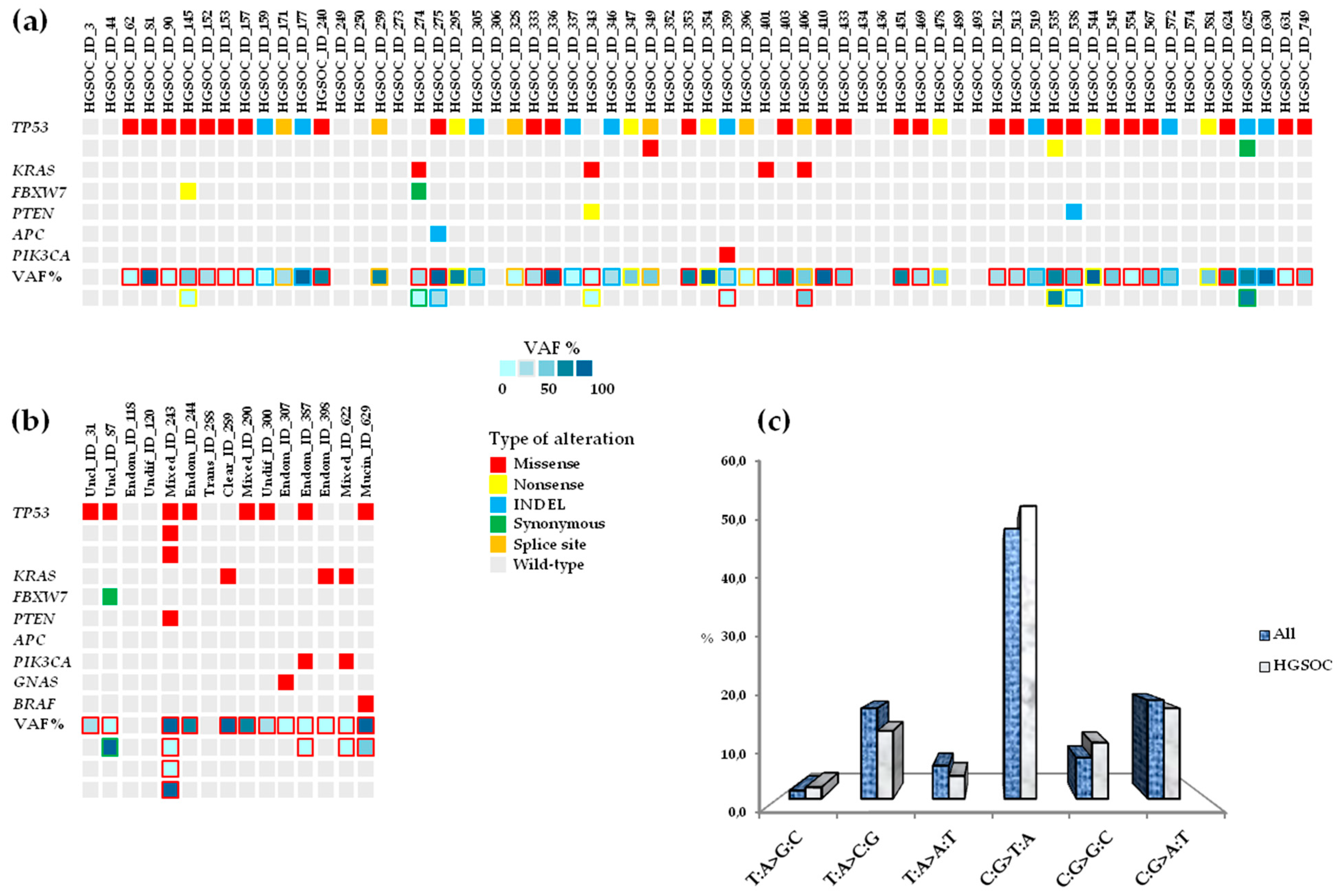
3.2. Landscape of Somatic Mutations in Advanced Ovarian Cancer
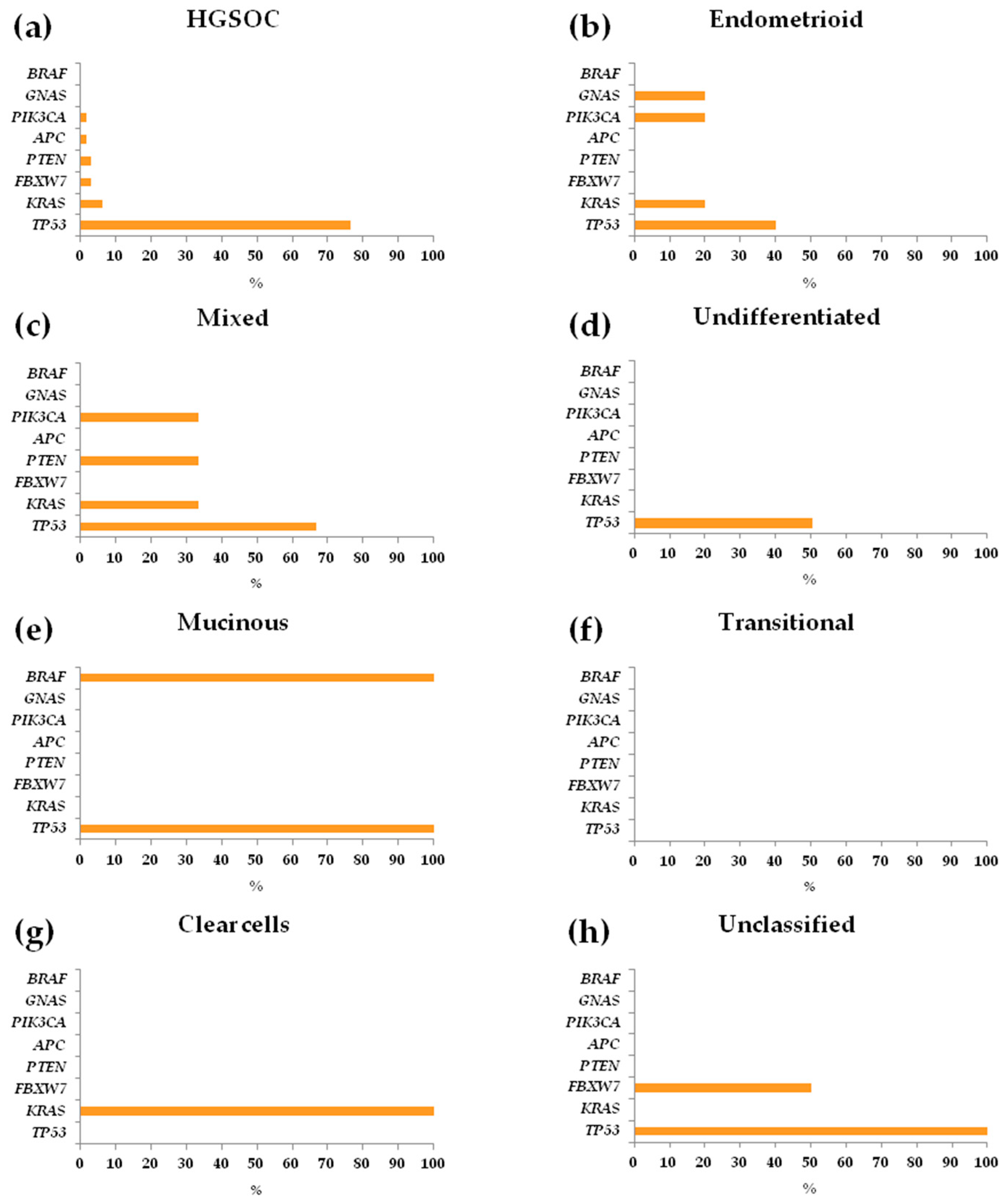
3.3. Repertoire and Distribution of Somatic Mutations in HGSOC and non-HGSOC
3.4. Concomitant Mutated Driver Genes in HGSOC and Impact on Clinical Outcome
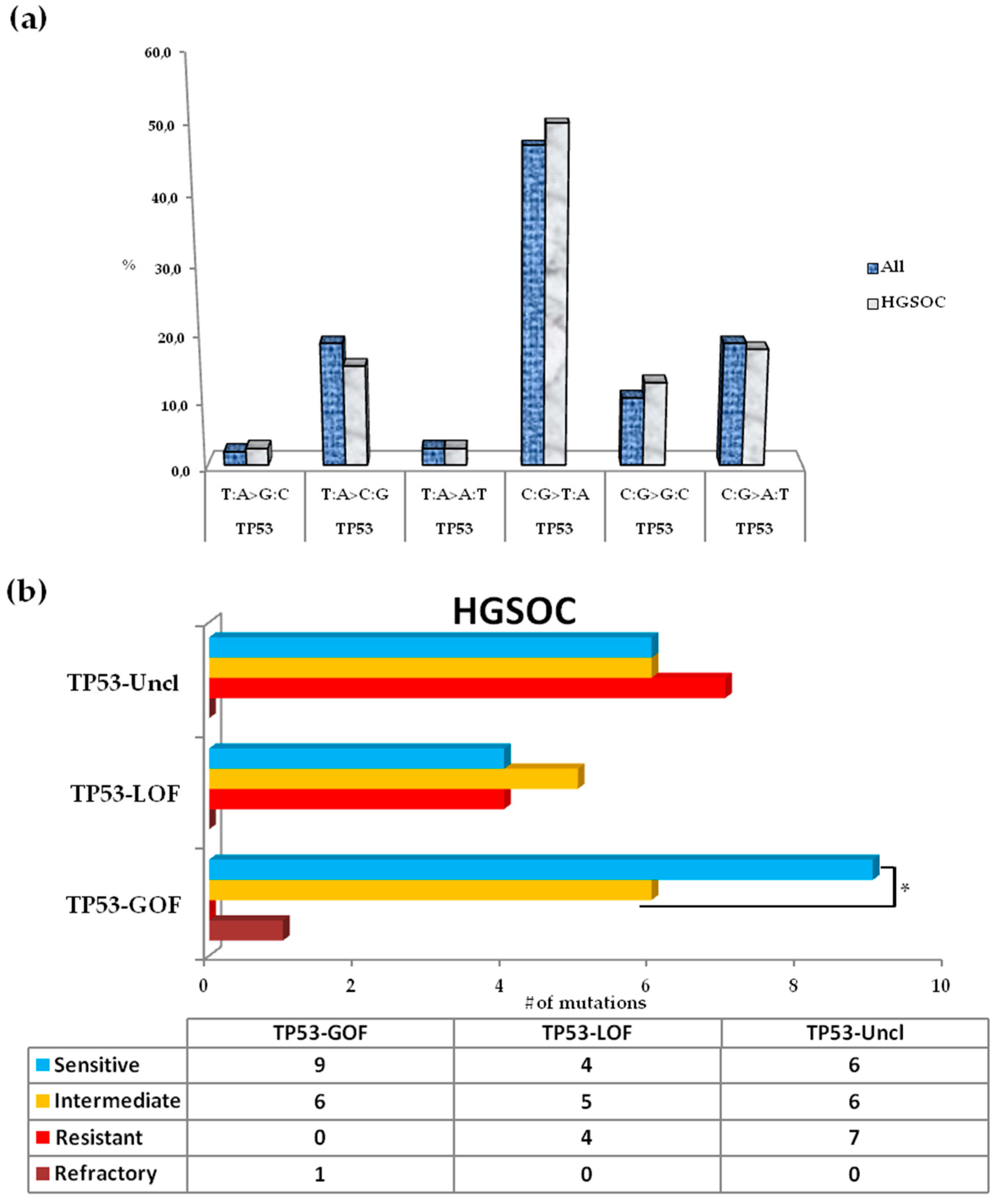
3.5. Somatic Spectrum of TP53 Mutations in Patients with HGSOC and Impact on Clinical Outcome
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Mortality Database Health Statistics and Information Systems; WHO: Geneva, Switzerland, 2017; Available online: http://www.who.int/healthinfo/statistics/mortality_rawdata/en/ (accessed on 15 October 2017).
- Raja, F.A.; Counsell, N.; Colombo, N.; Pfisterer, J.; du Bois, A.; Parmar, M.K.; Vergote, I.B.; Gonzalez-Martin, A.; Alberts, D.S.; Plante, M.; et al. Platinum versus platinum-combination chemotherapy in platinum-sensitive recurrent ovarian cancer: A meta-analysis using individual patient data. Ann. Oncol. 2013, 24, 3028–3034. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Bristow, R.E.; Chi, D.S.; Cliby, W.A. Role of aggressive surgical cytoreduction in advanced ovarian cancer. J. Gynecol. Oncol. 2015, 26, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, N.S.; Larry Maxwell, G.; Miller, A.; Hamilton, C.A.; Rungruang, B.; Rodriguez, N.; Richard, S.D.; Krivak, T.C.; Fowler, J.M.; Mutch, D.G.; et al. Predictive modeling for determination of microscopic residual disease at primary cytoreduction: An NRG Oncology/Gynecologic Oncology Group 182 Study. Gynecol. Oncol. 2018, 148, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Torres, D.; Wang, C.; Kumar, A.; Bakkum-Gamez, J.N.; Weaver, A.L.; McGreen, M.E.; Konecny, G.E.; Goode, E.L.; Cliby, W.A. Factors that influence survival in high-grade serous ovarian cancer: A complex relationship between molecular subtype, disease dissemination, and operability. Gynecol. Oncol. 2018, 150, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.M. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Palmirotta, R.; Silvestris, E.; D’Oronzo, S.; Cardascia, A.; Silvestris, F. Ovarian cancer: Novel molecular aspects for clinical assessment. Crit. Rev. Oncol. Hematol. 2017, 117, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Matsumura, N.; Mandai, M.; Yoshihara, K.; Tanabe, H.; Nakai, H.; Yamanoi, K.; Abiko, K.; Yoshioka, Y.; Hamanishi, J.; et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. Am. J. Pathol. 2016, 186, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Bodurka, D.C.; Deavers, M.T.; Tian, C.; Sun, C.C.; Malpica, A.; Coleman, R.L.; Lu, K.H.; Sood, A.K.; Birrer, M.J.; Ozols, R.; et al. Reclassification of serous ovarian carcinoma by a 2-tier system: A Gynecologic Oncology Group Study. Cancer 2012, 118, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Malpica, A.; Deavers, M.T.; Lu, K.; Bodurka, D.C.; Atkinson, E.N.; Gershenson, D.M.; Silva, E.G. Grading ovarian serous carcinoma using a two-tier system. Am. J. Surg. Pathol. 2004, 28, 496–504. [Google Scholar] [CrossRef]
- Mylavarapu, S.; Das, A.; Roy, M. Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Front. Oncol. 2018, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.E.; Lu, K.H.; Klimczak, A.M.; Mok, S.C. Recommendations and Choices for BRCA Mutation Carriers at Risk for Ovarian Cancer: A Complicated Decision. Cancers 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzales-Martin, A.; Colombo, N.; Sessa, C.; ESMO Guidelines Working Group. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2013, 24, vi24–vi32. [Google Scholar] [CrossRef] [PubMed]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995–2009: Analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Cecchin, E.; Russo, A.; Campagnutta, E.; Martella, L.; Toffoli, G. Lack of association of CYP1 B1*3 polymorphism and ovarian cancer in a Caucasian population. Int. J. Biol. Markers 2004, 19, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Cecchin, E.; Russo, A.; Corona, G.; Campagnutta, E.; Martella, L.; Boiocchi, M.; Toffoli, G. UGT1A1*28 polymorphism in ovarian cancer patients. Oncol. Rep. 2004, 12, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Cecchin, E.; Canzonieri, V.; Sorio, R.; Giorda, G.; Scalone, S.; De Mattia, E.; Roncato, R.; Gagno, S.; Poletto, E.; et al. Identification of Novel Somatic TP53 Mutations in Patients with High-Grade Serous Ovarian Cancer (HGSOC) Using Next-Generation Sequencing (NGS). Int. J. Mol. Sci. 2018, 19, 1510. [Google Scholar] [CrossRef]
- Brachova, P.; Mueting, S.R.; Carlson, M.J.; Goodheart, M.J.; Button, A.M.; Mott, S.L.; Dai, D.; Thiel, K.W.; Devor, E.J.; Leslie, K.K. TP53 oncomorphic mutations predict resistance to platinum- and taxane-based standard chemotherapy in patients diagnosed with advanced serous ovarian carcinoma. Int. J. Oncol. 2015, 46, 607–618. [Google Scholar] [CrossRef]
- Goodman, A.M.; Choi, M.; Wieduwilt, M.; Mulroney, C.; Costello, C.; Frampton, G.; Miller, V.; Kurzrock, R. Next Generation Sequencing Reveals Potentially Actionable Alterations in the Majority of Patients with Lymphoid Malignancies. JCO Precis Oncol. 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mody, K.; de Abreu, F.B.; Pipas, J.M.; Peterson, J.D.; Gallagher, T.L.; Suriawinata, A.A.; Ripple, G.H.; Hourdequin, K.C.; Smith, K.D.; et al. Molecular profiling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin. Chem. 2014, 60, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Shajani-Yi, Z.; de Abreu, F.B.; Peterson, J.D.; Tsongalis, G.J. Frequency of Somatic TP53 Mutations in Combination with Known Pathogenic Mutations in Colon Adenocarcinoma, Non–Small Cell Lung Carcinoma, and Gliomas as Identified by Next-Generation Sequencing. Neoplasia 2018, 20, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Hirsch, M.; Palescandolo, E.; Kim, E.; Liu, J.; van Hummelen, P.; MacConaill, L.; Drapkin, R.; Hahn, W.C. High throughput interrogation of somatic mutations in high grade serous cancer of the ovary. PLoS ONE 2011, 6, e24433. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Ali, S.M.; Wang, K.; Palmer, G.; Yelensky, R.; Lipson, D.; Miller, V.A.; Zajchowski, D.; Shawver, L.K.; Stephens, P.J. Comprehensive genomic profiling of epithelial ovarian cancer by next generation sequencing-based diagnostic assay reveals new routes to targeted therapies. Gynecol. Oncol. 2013, 130, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Salazar, C.; Campbell, I.G.; Gorringe, K.L. When Is “Type I” Ovarian Cancer Not “Type I”? Indications of an Out-Dated Dichotomy. Front. Oncol. 2018, 8, 654. [Google Scholar] [CrossRef]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef]
- Stewart, C.J.; Leung, Y.; Walsh, M.D.; Walters, R.J.; Young, J.P.; Buchanan, D.D. KRAS mutations in ovarian low-grade endometrioid adenocarcinoma: Association with concurrent endometriosis. Hum. Pathol. 2012, 43, 1177–1183. [Google Scholar] [CrossRef]
- Geyer, J.T.; López-García, M.A.; Sánchez-Estevez, C.; Sarrió, D.; Moreno-Bueno, G.; Franceschetti, I.; Palacios, J.; Oliva, E. Pathogenetic pathways in ovarian endometrioid adenocarcinoma: A molecular study of 29 cases. Am. J. Surg. Pathol. 2009, 33, 1157–1163. [Google Scholar] [CrossRef]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Caramia, F.; Li, J.; Rowley, S.M.; Christie, M.; Allan, P.E.; Stephens, A.N.; Bowtell, D.D.; et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genom. Med. 2015, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Nadal, E.; Chen, G.; Prensner, J.R.; Shiratsuchi, H.; Sam, C.; Zhao, L.; Kalemkerian, G.P.; Brenner, D.; Lin, J.; Reddy, R.M.; et al. KRAS-G12C mutation is associated with poor outcome in surgically resected lung adenocarcinoma. J. Thorac. Oncol. 2014, 9, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef] [PubMed]
- Rachagani, S.; Senapati, S.; Chakraborty, S.; Ponnusamy, M.P.; Kumar, S.; Smith, L.M.; Jain, M.; Batra, S.K. Activated KrasG¹²D is associated with invasion and metastasis of pancreatic cancer cells through inhibition of E-cadherin. Br. J. Cancer. 2011, 104, 1038–1048. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rechsteiner, M.; Zimmermann, A.K.; Wild, P.J.; Caduff, R.; von Teichman, A.; Fink, D.; Moch, H.; Noske, A. TP53 mutations are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Exp. Mol. Pathol. 2013, 95, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nakayama, K.; Ishikawa, N.; Ishikawa, M.; Sultana, R.; Kiyono, T.; Kyo, S. Reconstitution of high-grade serous ovarian carcinoma from primary fallopian tube secretory epithelial cells. Oncotarget 2017, 9, 12609–12619. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, Y.J.; Lee, M.Y.; Ruan, A.; Chen, C.K.; Liu, H.P.; Wang, C.J.; Chao, W.R.; Han, C.P. Multipoint Kras oncogene mutations potentially indicate mucinous carcinoma on the entire spectrum of mucinous ovarian neoplasms. Oncotarget 2016, 7, 82097–82103. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Lee, J.W.; Lee, M.; Kim, H.S.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S.; Seo, J.S. Genomic landscape of ovarian clear cell carcinoma via whole exome sequencing. Gynecol. Oncol. 2018, 148, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.T.; Mao, T.L.; Jones, S.; Veras, E.; Ayhan, A.; Wang, T.L.; Glas, R.; Slamon, D.; Velculescu, V.E.; Kuman, R.J.; et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am. J. Pathol. 2009, 174, 1597–1601. [Google Scholar] [CrossRef] [PubMed]
- López, I.; Oliveira, L.P.; Tucci, P.; Alvarez-Valín, F.; Coudry, A.R.; Marín, M. Different mutation profiles associated to P53 accumulation in colorectal cancer. Gene 2012, 499, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Pfeifer, G.P. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis 2001, 22, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, V.; Schildgen, O. The lonely driver or the orchestra of mutations? How next generation sequencing datasets contradict the concept of single driver checkpoint mutations in solid tumours-NSCLC as a scholarly example. Semin. Cancer Biol. 2018, S1044-579X, 30087. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Wheler, J.J.; Naing, A.; Falchook, G.S.; Hong, D.S.; Stepanek, V.M.; Fu, S.; Piha-Paul, S.A.; Lee, J.J.; Luthra, R.; et al. PIK3CA mutation H1047R is associated with response to PI3K/AKT/mTOR signaling pathway inhibitors in early-phase clinical trials. Cancer Res. 2013, 73, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Li, A.J.; Li, H.G.; Tang, E.J.; Wu, W.; Chen, Y.; Jiang, H.H.; Lin, M.B.; Yin, L. PIK3CA and TP53 mutations predict overall survival of stage II/III colorectal cancer patients. World J. Gastroenterol. 2018, 24, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Jardim, D.L.; Wheler, J.J.; Hess, K.; Tsimberidou, A.M.; Zinner, R.; Janku, F.; Subbiah, V.; Naing, A.; Piha-Paul, S.A.; Westin, S.N.; et al. FBXW7 mutations in patients with advanced cancers: Clinical and molecular characteristics and outcomes with mTOR inhibitors. PLoS ONE 2014, 9, e89388. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, S.; Han, L.; Liu, P.; Li, H.; Ren, X.; Yu, J.; Hao, X. Concurrent somatic mutations in driver genes were significantly correlated with lymph node metastasis and pathological types in solid tumors. Oncotarget 2017, 8, 68746–68757. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef]
- Kotoula, V.; Karavasilis, V.; Zagouri, F.; Kouvatseas, G.; Giannoulatou, E.; Gogas, H.; Lakis, S.; Pentheroudakis, G.; Bobos, M.; Papadopoulou, K.; et al. Effects of TP53 and PIK3CA mutations in early breast cancer: A matter of co-mutation and tumor-infiltrating lymphocytes. Breast Cancer Res. Treat. 2016, 158, 307–321. [Google Scholar] [CrossRef]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef]
- Zhou, Z.; He, C.; Wang, J. Regulation mechanism of Fbxw7-related signaling pathways. Oncol. Rep. 2015, 34, 2215–2224. [Google Scholar] [CrossRef]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer. 2018, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Minella, A.C.; Welcker, M.; Clurman, B.E. Ras activity regulates cyclin E degradation by the Fbw7 pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 9649–9654. [Google Scholar] [CrossRef] [PubMed]
- Kitade, S.; Onoyama, I.; Kobayashi, H.; Yagi, H.; Yoshida, S.; Kato, M.; Tsunematsu, R.; Asanoma, K.; Sonoda, K.; Wake, N. FBXW7 is involved in the acquisition of the malignant phenotype in epithelial ovarian tumors. Cancer Sci. 2016, 107, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Hennessy, B.T.; Ng, C.S.; Ju, Z.; Coombes, K.R.; Wolf, J.K.; Sood, A.K.; Levenback, C.F.; Coleman, R.L.; Kavanagh, J.J.; et al. Perifosine plus docetaxel in patients with platinum and taxane resistant or refractory high-grade epithelial ovarian cancer. Gynecol. Oncol. 2012, 126, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009, 16, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Rudd, M.L.; Price, J.C.; Fogoros, S.; Godwin, A.K.; Sgroi, D.C.; Merino, M.J.; Bell, D.W. A unique spectrum of somatic PIK3CA (p110alpha) mutations within primary endometrial carcinomas. Clin. Cancer Res. 2011, 17, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef] [PubMed]
- Cherniack, A.D.; Shen, D.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Ling, S.; Soslow, R.A.; Bruadduys, R.R.; Zuna, R.E.; et al. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef]
- Tang, F.H.; Hsieh, T.H.; Hsu, C.Y.; Lin, H.Y.; Long, C.Y.; Cheng, K.H.; Tsai, E.M. KRAS mutation coupled with p53 loss is sufficient to induce ovarian carcinosarcoma in mice. Int. J. Cancer 2017, 140, 1860–1869. [Google Scholar] [CrossRef]
- Jones, S.; Stransky, N.; McCord, C.L.; Cerami, E.; Lagowski, J.; Kelly, D.; Angiouli, S.D.; Sansen, M.; Kann, L.; Shukla, M.; et al. Genomic analyses of gynecologic carcinosarcomas reveal frequent mutations in chromatin remodeling genes. Nat. Commun. 2014, 5, 5006. [Google Scholar] [CrossRef]
- Kaldawy, A.; Segev, Y.; Lavie, O.; Auslender, R.; Sopik, V.; Narod, S.A. Low-grade serous ovarian cancer: A review. Gynecol. Oncol. 2016, 143, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Gershenson, D.M.; Sun, C.C.; Wong, K.K. Impact of mutational status on survival in low-grade serous carcinoma of the ovary or peritoneum. Br. J. Cancer 2015, 113, 1254–1258. [Google Scholar] [CrossRef] [PubMed]
- Mukohara, T. PI3K mutations in breast cancer: Prognostic and therapeutic implications. Breast Cancer (Dove Med Press) 2015, 7, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Kang, S.; Vogt, P.K. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, R.; Kommoss, S.; Winterhoff, B.J.; Kipp, B.R.; Garcia, J.J.; Voss, J.; Halling, K.; Karnezis, A.; Senz, J.; Yang, W.; et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer 2015, 15, 415. [Google Scholar] [CrossRef] [PubMed]
- Ursem, C.; Atreya, C.E.; Van Loon, K. Emerging treatment options for BRAF-mutant colorectal cancer. Gastrointest Cancer 2018, 8, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Rideout, W.M., III; Shen, J.C.; Spruck, C.H.; Tsai, Y.C. Methylation, mutation and cancer. Bioessays 1992, 14, 33–36. [Google Scholar] [CrossRef]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 Mutations in Human Cancer: Database Reassessment and Prospects for the Next Decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Soussi, T.; Wiman, K.G. TP53: An oncogene in disguise. Cell Death Differ. 2015, 22, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Chun, S.M.; Kim, K.R.; Sohn, I.; Sung, C.O. Clinical Relevance of Gain-Of-Function Mutations of p53 in High-Grade Serous Ovarian Carcinoma. PLoS ONE 2013, 8, e72609. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Xie, L. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget 2017, 8, 97657–97670. [Google Scholar] [CrossRef] [PubMed]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Iwakuma, T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers 2019, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Maru, Y.; Tanaka, N.; Ohira, M.; Itami, M.; Hippo, Y.; Nagase, H. Identification of novelmutations in Japanese ovarian clear cell carcinoma patients using optimized targeted NGS for clinical diagnosis. Gynecol. Oncol. 2017, 144, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.J.; Schlappe, B.A.; Kumar, R.; Olvera, N.; Dao, F.; Abu-Rustum, N.; Aghajanian, C.; DeLair, D.; Hussein, Y.R.; Soslow, R.A.; et al. Massively parallel sequencing analysi of mucinous ovarian carcinomas: Genomic profiling and differential diagnoses. Gynecol. Oncol. 2018, 150, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, F.; Walerych, D.; Del Sal, G. Targeting mutant p53 in cancer: A long road to precision Therapy. FEBS J. 2017, 284, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-TumorEffect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed]
- Babikir, H.A.; Afjei, R.; Paulmurugan, R.; Massoud, T.F. Restoring guardianship of the genome: Anticancer drug strategie sto reverse oncogenic mutant p53 misfolding. Cancer Treat. Rev. 2018, 71, 19–31. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | All, n = 79 | HGSOC, n = 64 |
|---|---|---|
| Age | ||
| Median (range) | 56.3 (31–81.4) | 57.0 (31.0–81.4) |
| CA-125 at diagnosis U/mL | ||
| Median (range) | 855.3 (8.8–13100.0) | 832.8 (37.9–13100.0) |
| Tumor Histology | ||
| Serous | 64 (81.0) | 64 (100.0) |
| Endometrioid | 5 (6.3) | 0 (0.0) |
| Mixed | 3 (3.8) | 0 (0.0) |
| Undifferentiated | 2 (2.5) | 0 (0.0) |
| Mucinous | 1 (1.3) | 0 (0.0) |
| Transitional | 1 (1.3) | 0 (0.0) |
| Clear cells | 1 (1.3) | 0 (0.0) |
| Unclassified | 2 (2.5) | 0 (0.0) |
| FIGO Stage | ||
| IIIB | 3 (3.8) | 1 (1.6) |
| IIIC | 58 (73.4) | 50 (78.1) |
| IV | 18 (22.8) | 13 (20.3) |
| Tumor Grade a | ||
| G2 | 19 (24.0) | 18 (28.1) |
| G3 | 60 (76.0) | 46 (71.9) |
| RD at PDS | ||
| 0 | 40 (50.6) | 33 (51.6) |
| <1 cm | 39 (49.4) | 31 (48.4) |
| Lymph Node involvement | ||
| Negative | 15 (19.0) | 10 (15.6) |
| Positive | 42 (53.2) | 34 (53.1) |
| Unknown | 22 (27.8) | 20 (31.2) |
| Treatment | ||
| Carboplatin-Paclitaxel | 56 (70.9) | 45 (70.3) |
| Carboplatin-PDL | 11 (13.9) | 9 (14.1) |
| Carboplatin | 5 (6.3) | 5 (7.8) |
| Other b | 7 (8.9) | 5 (7.8) |
| PFI c, months | ||
| Median (range) | 8.8 (0.0–87.8) | 8.4 (0.0–87.8) |
| TTR c, months | ||
| Median (range) | 14.5 (4.6–93.6) | 14.5 (5.4–93.6) |
| Recurrence | 70 (88.6) | 56 (87.5) |
| OS, months | ||
| Median (range) | 47.7 (8.3–190.4) | 48.2 (13.5–190.4) |
| Deaths | 55 (69.6) | 42 (65.6) |
| Platinum sensitivity c | ||
| Refractory | 7 (8.9) | 3 (4.7) |
| Resistant | 14 (17.7) | 13 (20.3) |
| Intermediate | 20 (25.3) | 19 (29.7) |
| Sensitive | 36 (45.6) | 28 (43.7) |
| Characteristics | n = 64 | TP53 n (%) | p | KRAS n (%) | p | FBXW7 n (%) | p | PTEN n (%) | p | APC n (%) | p | PIK3CA n (%) | p |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tumor Histology | |||||||||||||
| Serous | 64 | 49 (76.6) | - | 4 (6.2) | - | 2 (3.1) | - | 2 (3.1) | - | 1 (1.6) | - | 1 (1.6) | - |
| FIGO Stage a | |||||||||||||
| III | 51 | 39 (76.5) | 1.000 | 1 (2.0) | 0.102 | 2 (3.9) | 1.000 | 2 (3.9) | 1.000 | 1 (2.0) | 1.000 | 1 (2.0) | 1.000 |
| IV | 13 | 10 (76.9) | 2 (15.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||||||
| Tumor Grade | |||||||||||||
| G2 | 18 | 12 (66.7) | 0.326 | 3 (16.7) | 0.064 | 1 (5.5) | 0.487 | 1 (5.5) | 0.487 | 0 (0.0) | 1.000 | 0 (0.0) | 1.000 |
| G3 | 46 | 37 (80.4) | 1 (2.2) | 1 (2.2) | 1 (2.2) | 1 (2.2) | 1 (2.2) | ||||||
| RD at PDS | |||||||||||||
| 0 | 33 | 25 (75.7) | 1.000 | 1 (3.0) | 0.347 | 1 (3.0) | 1.000 | 2 (6.1) | 0.493 | 0 (0.0) | 0.484 | 0 (0.0) | 0.484 |
| <1 cm | 31 | 24 (77.4) | 3 (9.7) | 1 (3.2) | 0 (0.0) | 1 (3.2) | 1 (3.2) | ||||||
| Platinum sensitivity b | |||||||||||||
| Refractory | 3 | 1 (33.3) | 0.017 | 1 (33.3) | 0.202 | 1 (33.3) | 0.019 | 0 (0.0) | 0.660 | 1 (33.3) | <0.001 | 0 (0.0) | 0.502 |
| Resistant | 13 | 12 (92.3) | 0 (0.0) | 0 (0.0) | 1 (7.7) | 0 (0.0) | 0 (0.0) | ||||||
| Intermediate | 19 | 18 (94.7) | 1 (5.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.3) | ||||||
| Sensitive | 28 | 19 (67.8) | 2 (7.1) | 1 (3.6) | 1 (3.6) | 0 (0.0) | 0 (0.0) |
| Median Survival | Univariate | Multivariate # | ||||||
|---|---|---|---|---|---|---|---|---|
| No. of Patients | Time (Months) | HR * | 95% CI * | p* | HR * | 95% CI * | p* | |
| PFIa | ||||||||
| N. of mutated genes | ||||||||
| 0 | 12 | 18.5 | Ref. | - | - | Ref. | - | - |
| 1 | 44 | 8.6 | 1.23 | 0.62–2.43 | 0.559 | 1.35 | 0.63–2.87 | 0.439 |
| >1 | 7 | 8.1 | 2.55 | 1.10–5.92 | 0.029 | 3.10 | 1.13–8.48 | 0.028 |
| TTRa | ||||||||
| N. of mutated genes | ||||||||
| 0 | 12 | 24.2 | Ref. | - | - | Ref. | - | - |
| 1 | 44 | 14.4 | 1.18 | 0.57–2.43 | 0.656 | 1.29 | 0.59–2.85 | 0.523 |
| >1 | 7 | 13.5 | 2.57 | 1.07–6.19 | 0.035 | 3.14 | 1.13–8.74 | 0.028 |
| OS | ||||||||
| N. of mutated genes | ||||||||
| 0 | 12 | 66.0 | Ref. | - | - | Ref. | - | - |
| 1 | 45 | 47.7 | 1.31 | 0.47–3.64 | 0.597 | 1.47 | 0.51–4.23 | 0.469 |
| >1 | 7 | 34.0 | 2.58 | 0.91–7.34 | 0.076 | 3.40 | 1.14–10.12 | 0.028 |
| PFIa | ||||||||
| N. of mutated genes | ||||||||
| 0 or 1 b | 56 | 9.0 | Ref. | - | - | Ref. | - | - |
| >1 | 7 | 8.1 | 2.17 | 1.14–4.11 | 0.018 | 2.44 | 1.19–4.99 | 0.015 |
| TTRa | ||||||||
| N. of mutated genes | ||||||||
| 0 or 1 b | 56 | 14.9 | Ref. | - | - | Ref. | - | - |
| >1 | 7 | 13.5 | 2.25 | 1.19–4.26 | 0.012 | 2.54 | 1.27–5.09 | 0.008 |
| OS | ||||||||
| N. of mutated genes | ||||||||
| 0 or 1 b | 57 | 48.8 | Ref. | - | - | Ref. | - | - |
| >1 | 7 | 34.0 | 2.06 | 1.19–3.56 | 0.009 | 2.47 | 1.50–4.07 | <0.001 |
| Characteristics | >1 | 0 or 1 | p | 1 | p | 0 | p |
|---|---|---|---|---|---|---|---|
| n = 7 | n = 57 | (>1 vs. 0 or 1) | n = 45 | (>1 vs. 1) | n = 12 | (>1 vs. 0) | |
| Age | |||||||
| Median (range) | 56.3 (48.5–72.1) | 56.8 (31.0–81.4) | 0.389 * | 56.1(31.0–80.3) | 0.318 * | 58.9 (42.0–81.4) | ns * |
| FIGO Stage a | |||||||
| III | 6 (85.7) | 45 (78.9) | 35 (77.8) | 10 (83.3) | |||
| IV | 1 (14.3) | 12 (21.1) | 10 (22.2) | 2 (16.7) | |||
| Tumor Grade | |||||||
| G2 | 3 (42.9) | 15 (26.3) | 0.391 | 11 (24.4) | 0.369 | 4 (33.3) | 1.000 |
| G3 | 4 (57.1) | 42 (73.7) | 34 (75.6) | 8 (66.7) | |||
| RD at PDS | |||||||
| 0 | 4 (57.1) | 29 (50.9) | 1.000 | 22 (48.9) | 1.000 | 7 (58.3) | 1.000 |
| <1 cm | 3 (42.9) | 28 (49.1) | 23 (51.1) | 5 (41.7) | |||
| Lymph Node involvement | |||||||
| Negative | 2 (28.6) | 8 (14.0) | 0.092 | 6 (13.3) | 0.100 | 2 (16.7) | 0.167 |
| Positive | 1 (14.3) | 33 (57.9) | 26 (57.8) | 7 (58.3) | |||
| Unknown | 4 (57.1) | 16 (28.1) | 13 (28.9) | 3 (25.0) | |||
| Platinum sensitivity b | |||||||
| Refractory | 2 (28.6) | 1 (1.8) | 0.019 | 0 (0.0) | 0.004 | 1 (8.3) | 0.515 |
| Resistant | 1 (14.3) | 12 (21.0) | 10 (22.2) | 2 (16.7) | |||
| Intermediate | 2 (28.6) | 17 (29.8) | 15 (33.3) | 2 (16.7) | |||
| Sensitive | 2 (28.6) | 26 (45.6) | 19 (42.2) | 7 (58.3) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garziera, M.; Roncato, R.; Montico, M.; De Mattia, E.; Gagno, S.; Poletto, E.; Scalone, S.; Canzonieri, V.; Giorda, G.; Sorio, R.; et al. New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells 2019, 8, 584. https://doi.org/10.3390/cells8060584
Garziera M, Roncato R, Montico M, De Mattia E, Gagno S, Poletto E, Scalone S, Canzonieri V, Giorda G, Sorio R, et al. New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells. 2019; 8(6):584. https://doi.org/10.3390/cells8060584
Chicago/Turabian StyleGarziera, Marica, Rossana Roncato, Marcella Montico, Elena De Mattia, Sara Gagno, Elena Poletto, Simona Scalone, Vincenzo Canzonieri, Giorgio Giorda, Roberto Sorio, and et al. 2019. "New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach" Cells 8, no. 6: 584. https://doi.org/10.3390/cells8060584
APA StyleGarziera, M., Roncato, R., Montico, M., De Mattia, E., Gagno, S., Poletto, E., Scalone, S., Canzonieri, V., Giorda, G., Sorio, R., Cecchin, E., & Toffoli, G. (2019). New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells, 8(6), 584. https://doi.org/10.3390/cells8060584