The Ubiquitin Proteasome System Is a Key Regulator of Pluripotent Stem Cell Survival and Motor Neuron Differentiation

 , , , , , , and
, , , , , , and
Abstract
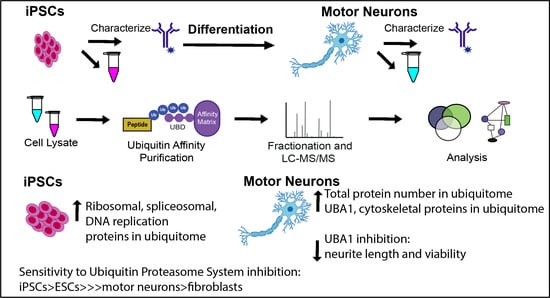
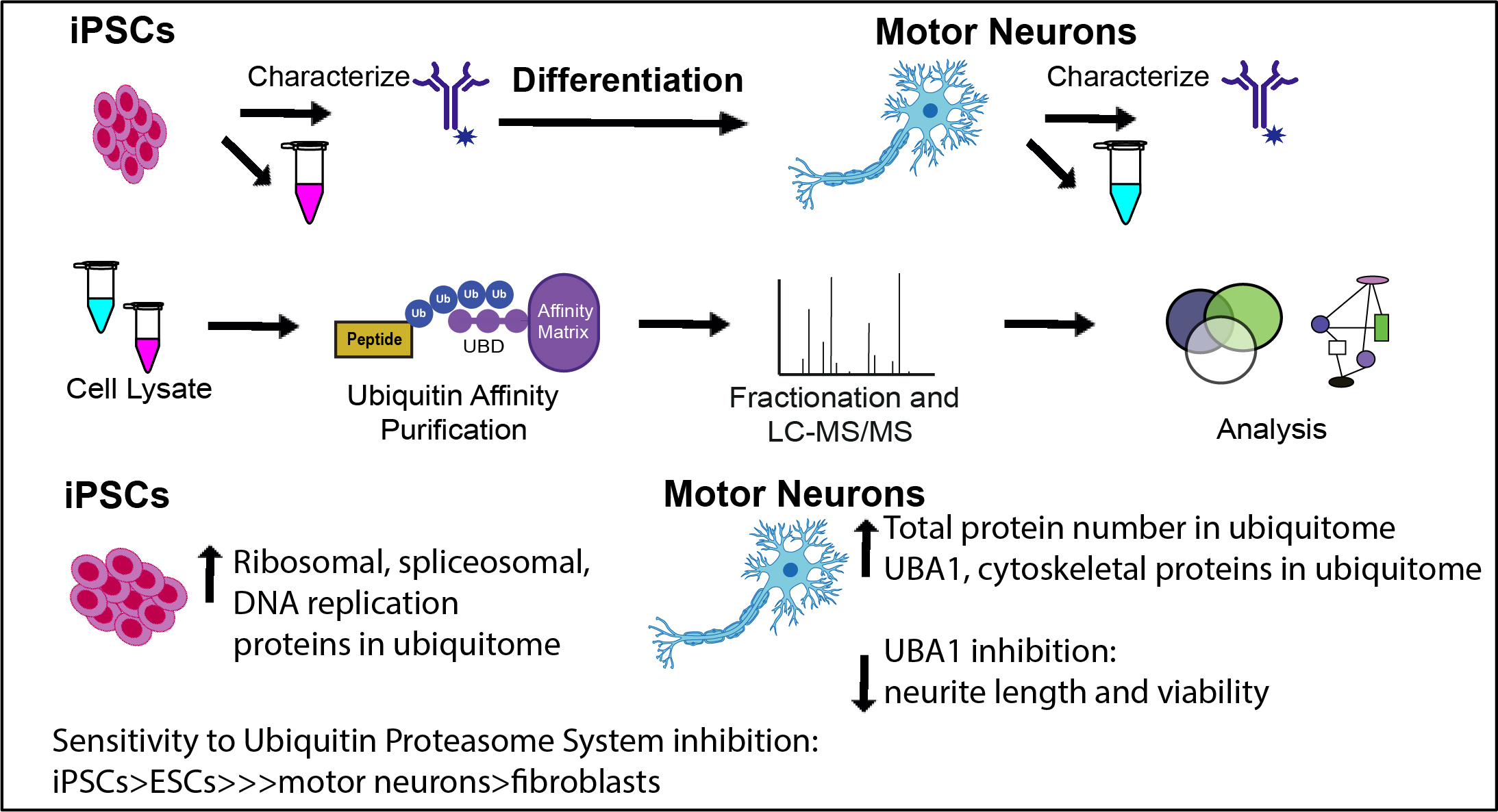{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generation of iPSC-Derived Motor Neurons
2.2. Motor Neuron Differentiation and Characterisation
2.3. Sample Collection
2.4. Ubiquitomics
2.5. Ubiquitin Proteasome System Inhibition
2.6. Statistics
3. Results
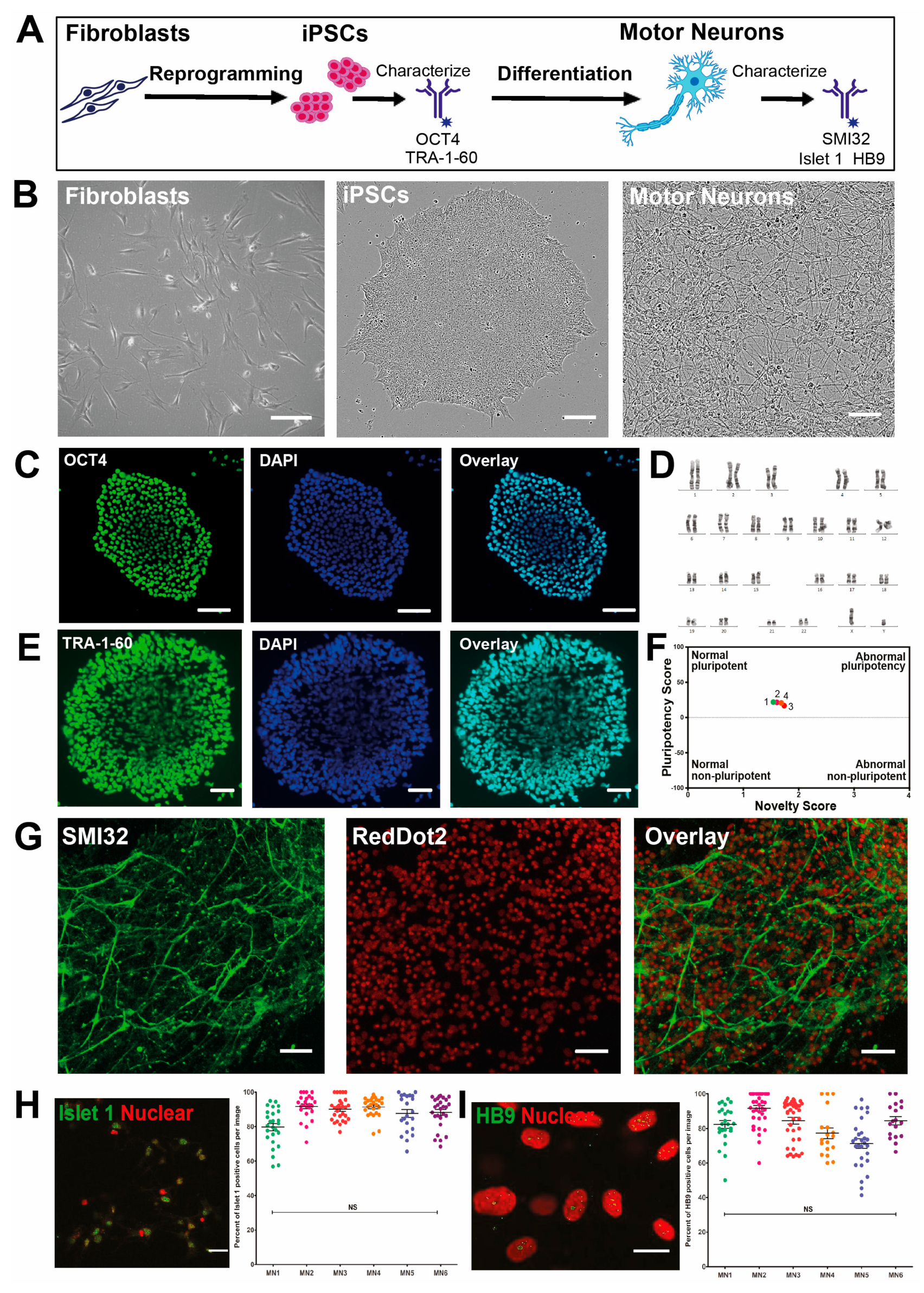
3.1. High Yield Differentiation of Motor Neurons from Fibroblast-Derived iPSCs
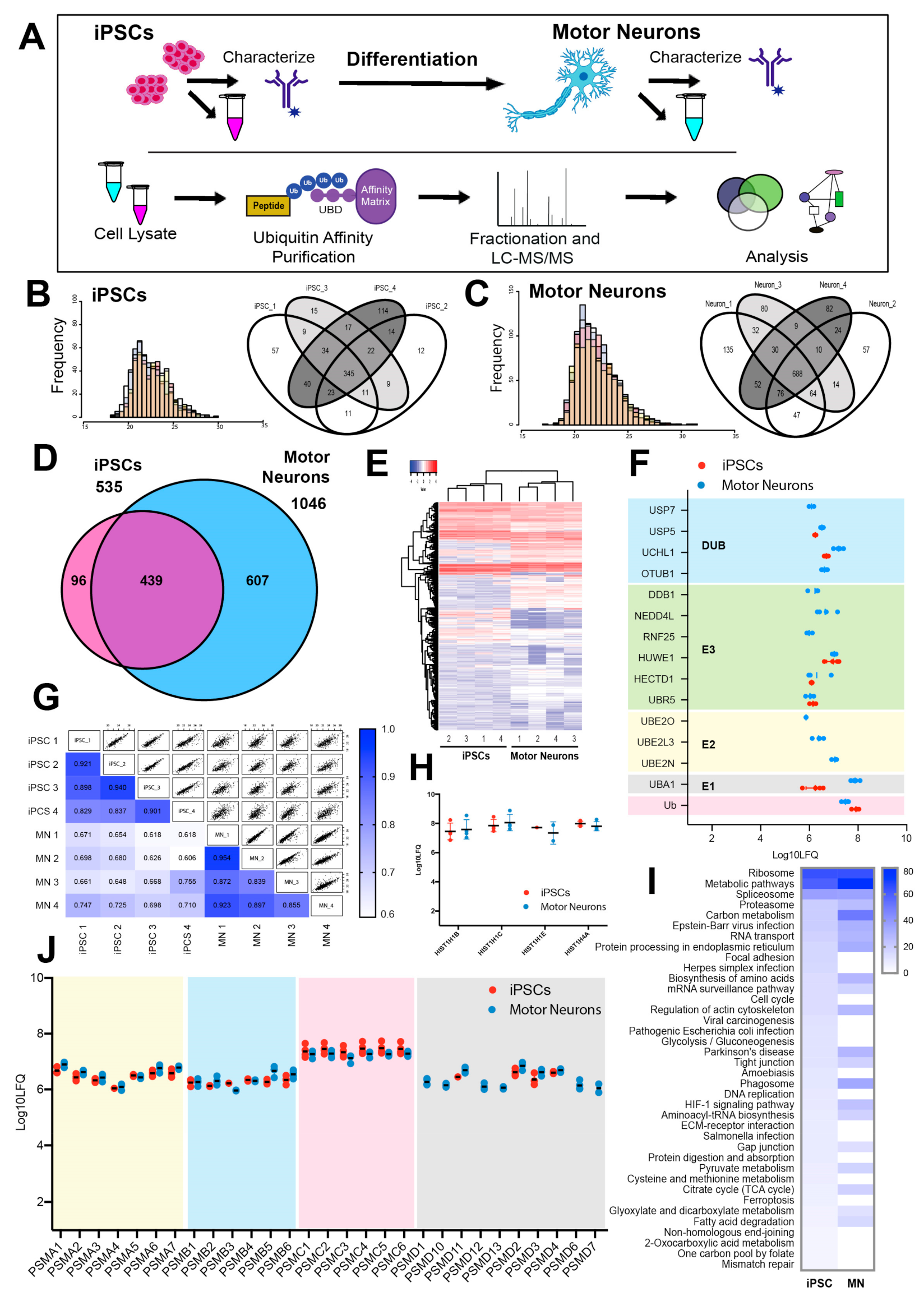
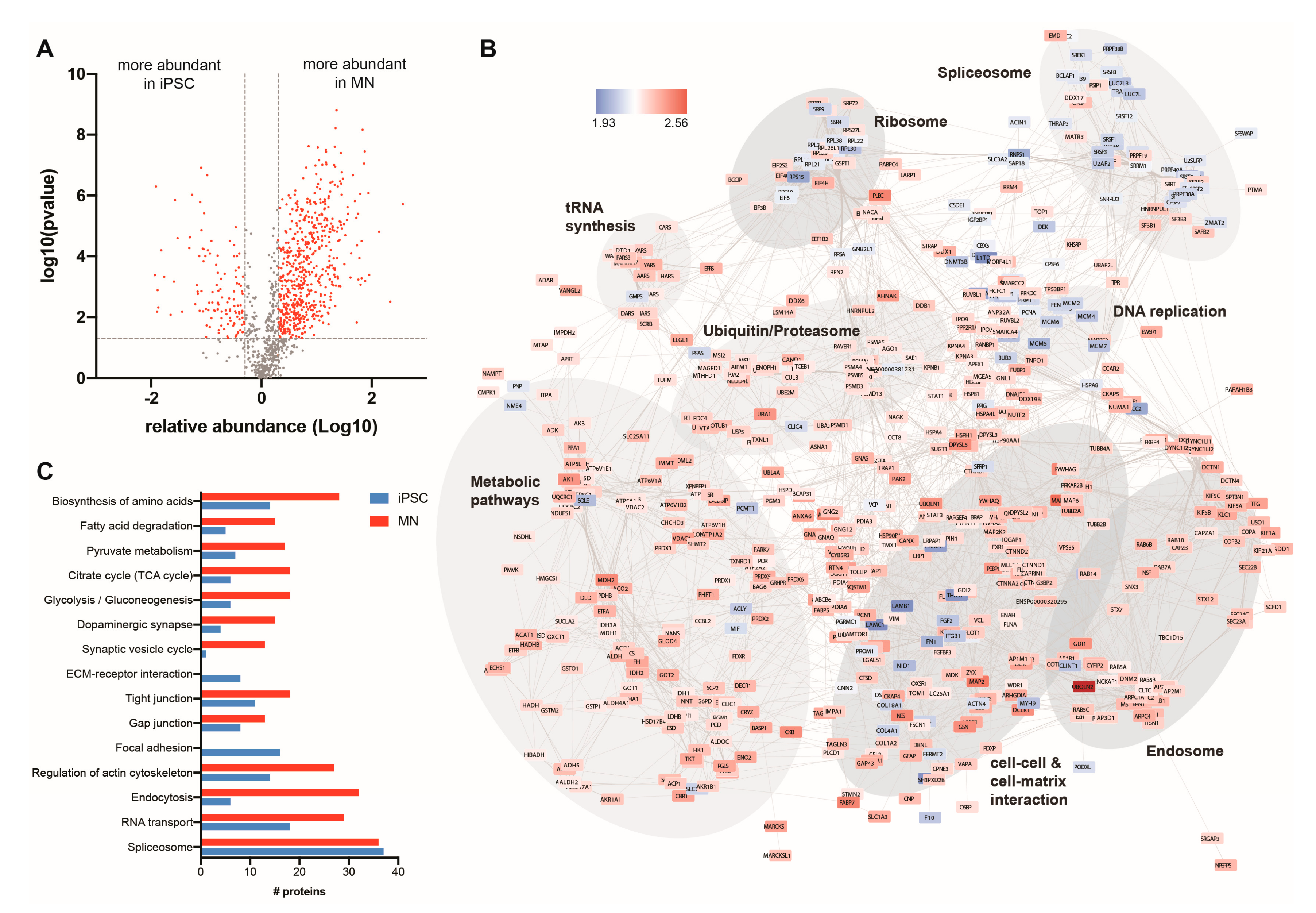
3.2. The Ubiquitin-Modified Proteome (Ubiquitome) of iPSCs and Motor Neurons
3.3. Quantitative Analysis of the Changing Ubiquitome During Motor Neuron Differentiation
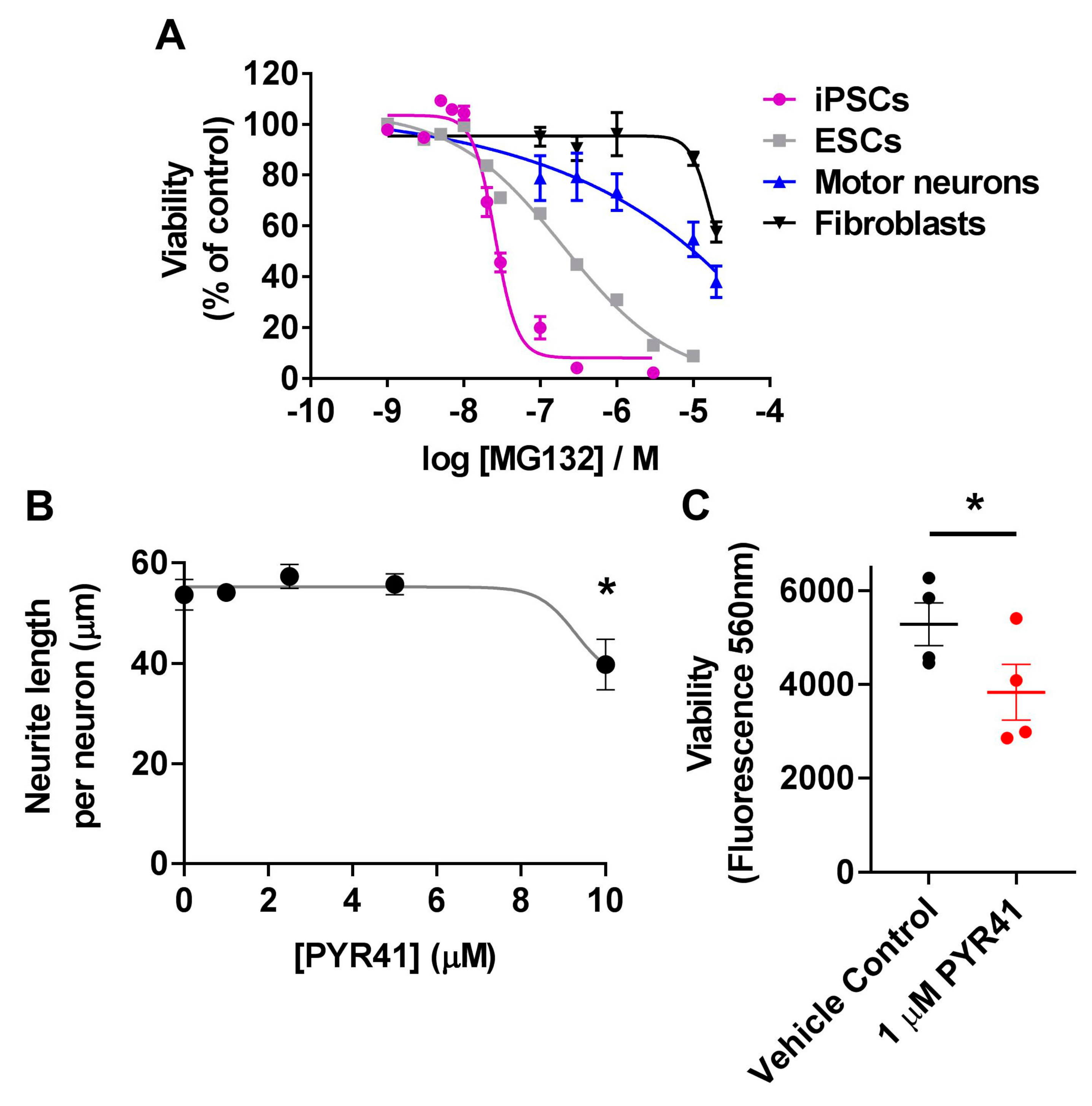
3.4. Differential Sensitivity of iPSCs and Motor Neurons to UPS Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar] [PubMed]
- Paul, S. Dysfunction of the ubiquitin–proteasome system in multiple disease conditions: Therapeutic approaches. BioEssays 2008, 30, 1172–1184. [Google Scholar] [CrossRef]
- Ooi, L.; Sidhu, K.; Poljak, A.; Sutherland, G.; O’Connor, M.D.; Sachdev, P.; Münch, G. Induced pluripotent stem cells as tools for disease modelling and drug discovery in Alzheimer’s disease. J. Neural Transm. 2013, 120, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.; Do-Ha, D.; Muñoz, S.S.; Ooi, L. Common pitfalls of stem cell differentiation: A guide to improving protocols for neurodegenerative disease models and research. Cell. Mol. Life Sci. 2016, 73, 3693–3709. [Google Scholar] [CrossRef]
- Du, Z.-W.; Chen, H.; Liu, H.; Lu, J.; Qian, K.; Huang, C.-L.; Zhong, X.; Fan, F.; Zhang, S.-C. Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 2015, 6, 6626. [Google Scholar] [CrossRef]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced Pluripotent Stem Cells Generated from Patients with ALS Can Be Differentiated into Motor Neurons. Science 2008, 321, 1218. [Google Scholar] [CrossRef]
- Qu, Q.; Li, D.; Louis, K.R.; Li, X.; Yang, H.; Sun, Q.; Crandall, S.R.; Tsang, S.; Zhou, J.; Cox, C.L.; et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat. Commun. 2014, 5, 3499. [Google Scholar] [CrossRef] [PubMed]
- Arber, S.; Han, B.; Mendelsohn, M.; Smith, M.; Jessell, T.M.; Sockanathan, S. Requirement for the Homeobox Gene Hb9 in the Consolidation of Motor Neuron Identity. Neuron 1999, 23, 659–674. [Google Scholar] [CrossRef]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Chen, W.; Hong, S.-T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Farrawell, N.E.; Lambert-Smith, I.; Mitchell, K.; McKenna, J.; McAlary, L.; Ciryam, P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J. Cell. Sci. 2018, 131, jcs209122. [Google Scholar] [CrossRef]
- Dlamini, N.; Josifova, D.J.; Paine, S.M.L.; Wraige, E.; Pitt, M.; Murphy, A.J.; King, A.; Buk, S.; Smith, F.; Abbs, S.; et al. Clinical and neuropathological features of X-linked spinal muscular atrophy (SMAX2) associated with a novel mutation in the UBA1 gene. Neuromuscul. Disord. 2013, 23, 391–398. [Google Scholar] [CrossRef]
- Ramser, J.; Ahearn, M.E.; Lenski, C.; Yariz, K.O.; Hellebrand, H.; von Rhein, M.; Clark, R.D.; Schmutzler, R.K.; Lichtner, P.; Hoffman, E.P.; et al. Rare Missense and Synonymous Variants in UBE1 Are Associated with X-Linked Infantile Spinal Muscular Atrophy. Am. J. Hum. Genet. 2008, 82, 188–193. [Google Scholar] [CrossRef]
- Powis, R.A.; Karyka, E.; Boyd, P.; Côme, J.; Jones, R.A.; Zheng, Y.; Szunyogova, E.; Groen, E.J.N.; Hunter, G.; Thomson, D.; et al. Systemic restoration of UBA1 ameliorates disease in spinal muscular atrophy. JCI Insight 2016, 1, e87908. [Google Scholar] [CrossRef]
- Groen, E.J.N.; Talbot, K.; Gillingwater, T.H. Advances in therapy for spinal muscular atrophy: Promises and challenges. Nat. Rev. Neurol. 2018, 14, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Brockington, A.; Ning, K.; Heath, P.R.; Wood, E.; Kirby, J.; Fusi, N.; Lawrence, N.; Wharton, S.B.; Ince, P.G.; Shaw, P.J. Unravelling the enigma of selective vulnerability in neurodegeneration: Motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013, 125, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Markowetz, F.; Unwin, R.D.; Leek, J.T.; Airoldi, E.M.; MacArthur, B.D.; Lachmann, A.; Rozov, R.; Ma’ayan, A.; Boyer, L.A.; et al. Systems-level dynamic analyses of fate change in murine embryonic stem cells. Nature 2009, 462, 358. Available online: https://www.nature.com/articles/nature08575#supplementary-information (accessed on 5 June 2019). [CrossRef] [PubMed]
- Pardo, M.; Lang, B.; Yu, L.; Prosser, H.; Bradley, A.; Babu, M.M.; Choudhary, J. An Expanded Oct4 Interaction Network: Implications for Stem Cell Biology, Development, and Disease. Cell Stem Cell 2010, 6, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.M.; Aranda-Orgilles, B.; Strikoudis, A.; Apostolou, E.; Loizou, E.; Moran-Crusio, K.; Farnsworth, C.L.; Koller, A.A.; Dasgupta, R.; Silva, J.C.; et al. Regulation of Pluripotency and Cellular Reprogramming by the Ubiquitin-Proteasome System. Cell Stem Cell 2012, 11, 783–798. [Google Scholar] [CrossRef]
- Balez, R.; Steiner, N.; Engel, M.; Muñoz, S.S.; Lum, J.S.; Wu, Y.; Wang, D.; Vallotton, P.; Sachdev, P.; O’Connor, M.; et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci. Rep. 2016, 6, 31450. Available online: https://www.nature.com/articles/srep31450#supplementary-information (accessed on 5 June 2019). [CrossRef]
- Engel, M.; Balez, R.; Muñoz, S.S.; Cabral-da-Silva, M.C.; Stevens, C.H.; Bax, M.; Do-Ha, D.; Sidhu, K.; Sachdev, P.; Ooi, L. Viral-free generation and characterization of a human induced pluripotent stem cell line from dermal fibroblasts. Stem Cell Res. 2018, 32, 135–138. [Google Scholar] [CrossRef]
- Muñoz, S.S.; Balez, R.; Berg, T.; Engel, M.; Bax, M.; Do-Ha, D.; Stevens, C.H.; Greenough, M.; Bush, A.; Ooi, L. Generation and characterization of human induced pluripotent stem cell lines from a familial Alzheimer’s disease PSEN1 A246E patient and a non-demented family member bearing wild-type PSEN1. Stem Cell Res. 2018, 31, 227–230. [Google Scholar] [CrossRef]
- Müller, F.-J.; Brändl, B.; Loring, J.F. Assessment of human pluripotent stem cells with PluriTest. In StemBook; Harvard Stem Cell Institute: Cambridge, MA, USA, 2008. [Google Scholar]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.L.; Sullivan, G.J.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803. [Google Scholar] [CrossRef]
- Zeineddine, R.; Pundavela, J.F.; Corcoran, L.; Stewart, E.M.; Dzung, D.-H.; Bax, M.; Guillemin, G.; Vine, K.L.; Hatters, D.M.; Ecroyd, H.; et al. SOD1 protein aggregates stimulate macropinocytosis in neurons to facilitate their propagation. Mol. Neurodegener. 2015, 10, 1–17. [Google Scholar] [CrossRef]
- Nagarajan, S.R.; Brandon, A.E.; McKenna, J.A.; Shtein, H.C.; Nguyen, T.Q.; Suryana, E.; Poronnik, P.; Cooney, G.J.; Saunders, D.N.; Hoy, A.J. Insulin and diet-induced changes in the ubiquitin-modified proteome of rat liver. PLoS ONE 2017, 12, e0174431. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Martens, W.L.; Poronnik, P.; Saunders, D. Hypothesis-Driven Sonification of Proteomic Data Distributions Indicating Neurodegredation in Amyotrophic Lateral Sclerosis. In Proceedings of the 22nd International Conference on Auditory Display—ICAD 2016, Canberra, Australia, 3–7 July 2016; The International Community for Auditory Display: Canberra, Australia, 2016; Volumes 21–27. [Google Scholar]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinf. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Yang, Y.; Kitagaki, J.; Dai, R.-M.; Tsai, Y.C.; Lorick, K.L.; Ludwig, R.L.; Pierre, S.A.; Jensen, J.P.; Davydov, I.V.; Oberoi, P.; et al. Inhibitors of Ubiquitin-Activating Enzyme (E1), a New Class of Potential Cancer Therapeutics. Cancer Res. 2007, 67, 9472. [Google Scholar] [CrossRef]
- Chen, C.; Meng, Y.; Wang, L.; Wang, H.-X.; Tian, C.; Pang, G.-D.; Li, H.-H.; Du, J. Ubiquitin-activating enzyme E1 inhibitor PYR41 attenuates angiotensin II-induced activation of dendritic cells via the IκBa/NF-κB and MKP1/ERK/STAT1 pathways. Immunology 2014, 142, 307–319. [Google Scholar] [CrossRef]
- Saez, I.; Koyuncu, S.; Gutierrez-Garcia, R.; Dieterich, C.; Vilchez, D. Insights into the ubiquitin-proteasome system of human embryonic stem cells. Sci. Rep. 2018, 8, 4092. [Google Scholar] [CrossRef]
- Munoz, J.; Low, T.Y.; Kok, Y.J.; Chin, A.; Frese, C.K.; Ding, V.; Choo, A.; Heck, A.J.R. The quantitative proteomes of human-induced pluripotent stem cells and embryonic stem cells. Mol. Syst. Biol. 2014, 7, 550. [Google Scholar] [CrossRef]
- Kim, Y.-D.; Lee, J.; Kim, H.-S.; Lee, M.-O.; Son, M.-Y.; Yoo, C.H.; Choi, J.-K.; Lee, S.C.; Cho, Y.S. The unique spliceosome signature of human pluripotent stem cells is mediated by SNRPA1, SNRPD1, and PNN. Stem Cell Res. 2017, 22, 43–53. [Google Scholar] [CrossRef]
- Tsuiji, H.; Iguchi, Y.; Furuya, A.; Kataoka, A.; Hatsuta, H.; Atsuta, N.; Tanaka, F.; Hashizume, Y.; Akatsu, H.; Murayama, S.; et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 2013, 5, 221–234. [Google Scholar] [CrossRef]
- Wan, L.; Battle, D.J.; Yong, J.; Gubitz, A.K.; Kolb, S.J.; Wang, J.; Dreyfuss, G. The Survival of Motor Neurons Protein Determines the Capacity for snRNP Assembly: Biochemical Deficiency in Spinal Muscular Atrophy. Mol. Cell. Biol. 2005, 25, 5543–5551. [Google Scholar] [CrossRef]
- Hamilton, A.M.; Zito, K. Breaking It Down: The Ubiquitin Proteasome System in Neuronal Morphogenesis. Neural Plast. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Sugiura, Y.; Han, W.; Gallardo, G.; Khvotchev, M.; Zhang, Y.; Kavalali, E.T.; Südhof, T.C.; Lin, W. Ubiquitin–Synaptobrevin Fusion Protein Causes Degeneration of Presynaptic Motor Terminals in Mice. J. Neurosci. 2015, 35, 11514–11531. [Google Scholar] [CrossRef]
- Yamada, T.; Yang, Y.; Bonni, A. Spatial organization of ubiquitin ligase pathways orchestrates neuronal connectivity. Trends Neurosci. 2013, 36, 218–226. [Google Scholar] [CrossRef]
- Lehman, N.L. The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 2009, 118, 329–347. [Google Scholar] [CrossRef]
- Ma, Q.; Ruan, H.; Peng, L.; Zhang, M.; Gack, M.U.; Yao, W.-D. Proteasome-independent polyubiquitin linkage regulates synapse scaffolding, efficacy, and plasticity. Proc. Natl. Acad. Sci. USA 2017, 114, E8760–E8769. [Google Scholar] [CrossRef]
- Geis, F.K.; Galla, M.; Hoffmann, D.; Kuehle, J.; Zychlinski, D.; Maetzig, T.; Schott, J.W.; Schwarzer, A.; Goffinet, C.; Goff, S.P.; et al. Potent and reversible lentiviral vector restriction in murine induced pluripotent stem cells. Retrovirology 2017, 14, 34. [Google Scholar] [CrossRef]
- Di Lello, P.; Hymowitz, S.G. Unveiling the Structural and Dynamic Nature of the Ubiquitin Code. Structure 2016, 24, 498–499. [Google Scholar] [CrossRef]
- Vilchez, D.; Boyer, L.; Morantte, I.; Lutz, M.; Merkwirth, C.; Joyce, D.; Spencer, B.; Page, L.; Masliah, E.; Berggren, W.T.; et al. Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 2012, 489, 304–308. [Google Scholar] [CrossRef]
- Alfano, C.; Faggiano, S.; Pastore, A. The Ball and Chain of Polyubiquitin Structures. Trends Biochem. Sci. 2016, 41, 371–385. [Google Scholar] [CrossRef]
- Pontén, F.; Gry, M.; Fagerberg, L.; Lundberg, E.; Asplund, A.; Berglund, L.; Oksvold, P.; Björling, E.; Hober, S.; Kampf, C.; et al. A global view of protein expression in human cells, tissues, and organs. Mol. Syst. Biol. 2009, 5, 337. [Google Scholar] [CrossRef]
- Fort, P.; Kajava, A.V.; Delsuc, F.; Coux, O. Evolution of Proteasome Regulators in Eukaryotes. Genome Biol. Evol. 2015, 7, 1363–1379. [Google Scholar] [CrossRef]
- Myeku, N.; Duff, K.E. Targeting the 26S Proteasome To Protect Against Proteotoxic Diseases. Trends Mol. Med. 2018, 24, 18–29. [Google Scholar] [CrossRef]
- Tamaki, Y.; Shodai, A.; Morimura, T.; Hikiami, R.; Minamiyama, S.; Ayaki, T.; Tooyama, I.; Furukawa, Y.; Takahashi, R.; Urushitani, M. Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci. Rep. 2018, 8, 6030. [Google Scholar] [CrossRef]
- Do-Ha, D.; Buskila, Y.; Ooi, L. Impairments in Motor Neurons, Interneurons and Astrocytes Contribute to Hyperexcitability in ALS: Underlying Mechanisms and Paths to Therapy. Mol. Neurobiol. 2018, 55, 1410–1418. [Google Scholar] [CrossRef]
- Buskila, Y.; Kékesi, O.; Bellot-Saez, A.; Seah, W.; Berg, T.; Trpceski, M.; Yerbury, J.J.; Ooi, L. Dynamic interplay between H-current and M-current controls motoneuron hyperexcitability in amyotrophic lateral sclerosis. Cell Death Dis. 2019, 10, 310. [Google Scholar] [CrossRef]
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E3935–E3943. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Ooi, L.; Blair, I.P.; Ciryam, P.; Dobson, C.M.; Vendruscolo, M. The metastability of the proteome of spinal motor neurons underlies their selective vulnerability in ALS. Neurosci. Lett. 2019, 704, 89–94. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bax, M.; McKenna, J.; Do-Ha, D.; Stevens, C.H.; Higginbottom, S.; Balez, R.; Cabral-da-Silva, M.e.C.; Farrawell, N.E.; Engel, M.; Poronnik, P.; et al. The Ubiquitin Proteasome System Is a Key Regulator of Pluripotent Stem Cell Survival and Motor Neuron Differentiation. Cells 2019, 8, 581. https://doi.org/10.3390/cells8060581
Bax M, McKenna J, Do-Ha D, Stevens CH, Higginbottom S, Balez R, Cabral-da-Silva MeC, Farrawell NE, Engel M, Poronnik P, et al. The Ubiquitin Proteasome System Is a Key Regulator of Pluripotent Stem Cell Survival and Motor Neuron Differentiation. Cells. 2019; 8(6):581. https://doi.org/10.3390/cells8060581
Chicago/Turabian StyleBax, Monique, Jessie McKenna, Dzung Do-Ha, Claire H. Stevens, Sarah Higginbottom, Rachelle Balez, Mauricio e Castro Cabral-da-Silva, Natalie E. Farrawell, Martin Engel, Philip Poronnik, and et al. 2019. "The Ubiquitin Proteasome System Is a Key Regulator of Pluripotent Stem Cell Survival and Motor Neuron Differentiation" Cells 8, no. 6: 581. https://doi.org/10.3390/cells8060581
APA StyleBax, M., McKenna, J., Do-Ha, D., Stevens, C. H., Higginbottom, S., Balez, R., Cabral-da-Silva, M. e. C., Farrawell, N. E., Engel, M., Poronnik, P., Yerbury, J. J., Saunders, D. N., & Ooi, L. (2019). The Ubiquitin Proteasome System Is a Key Regulator of Pluripotent Stem Cell Survival and Motor Neuron Differentiation. Cells, 8(6), 581. https://doi.org/10.3390/cells8060581