A Versatile Strategy to Reduce UGA-Selenocysteine Recoding Efficiency of the Ribosome Using CRISPR-Cas9-Viral-Like-Particles Targeting Selenocysteine-tRNA[Ser]Sec Gene

 , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Production of Viral-Like-Particles
2.3. Transduction Procedure
2.4. Genomic DNA Extraction and Analysis
2.5. Total RNA Extraction and Analysis by Northern Blot or RT-qPCR
2.6. Protein Extraction and Analysis by Western Blot
3. Results
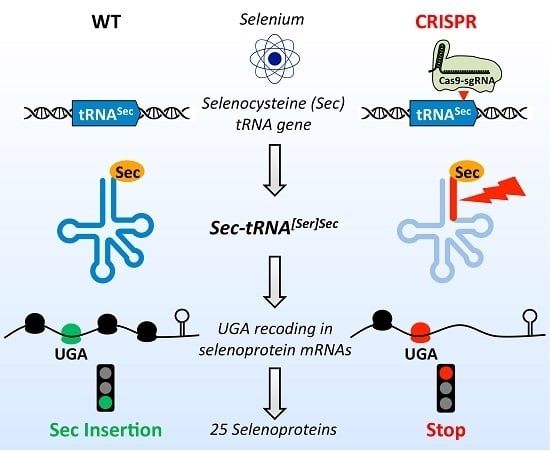
3.1. Design of a CRISPR-Cas9-Viral-Like-Particles Targeting the Sec-tRNA[Ser]Sec (TRNAU1) Gene
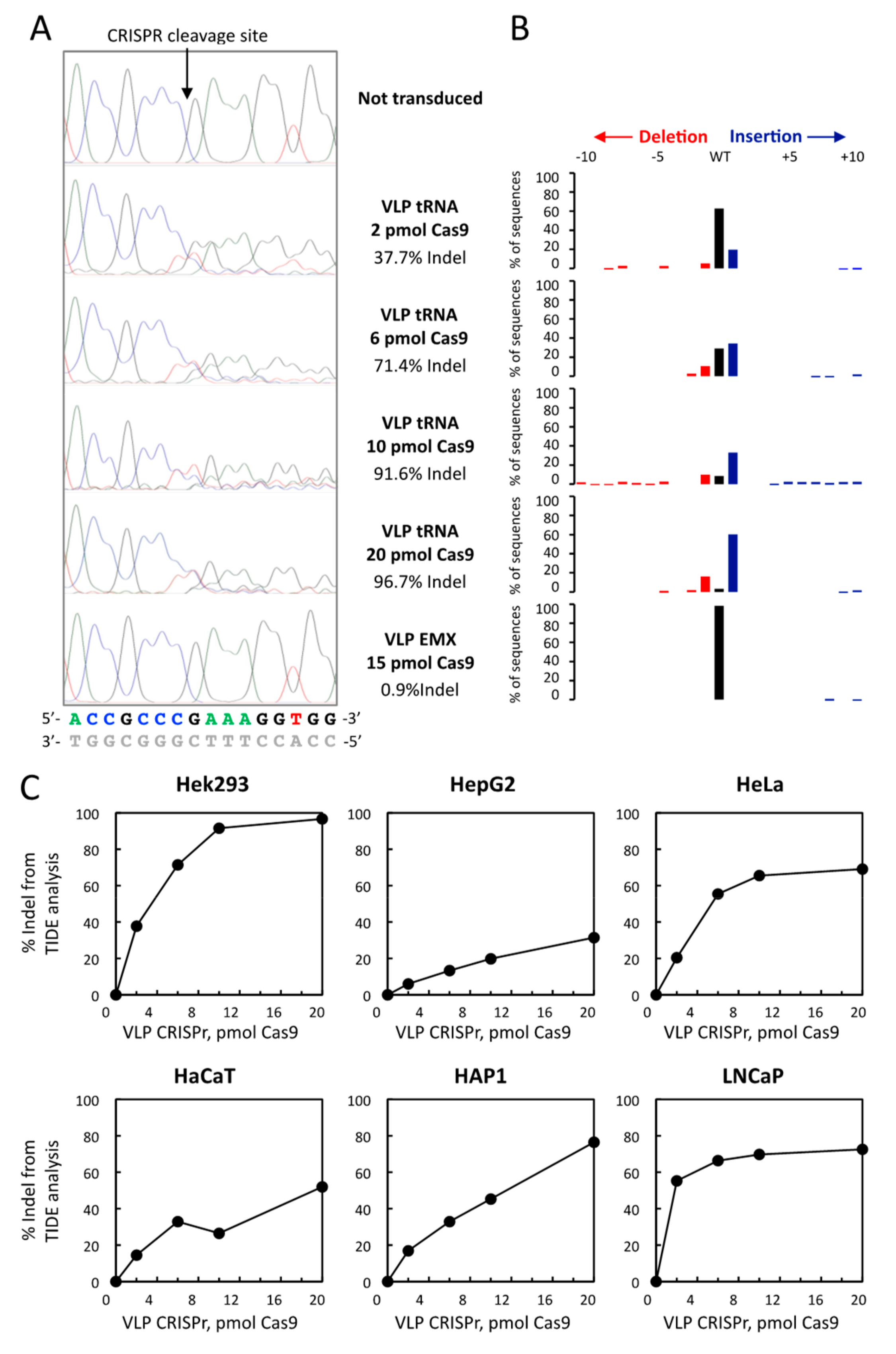
3.2. CRISPR-Cas9-VLPs Induced Mutations in TRNAU1 Gene with High Efficiency in Various Cell Lines
3.3. Comparison of TIDE Analysis with the Sequencing of Indel in the Hek293 and HAP1 Cell Lines
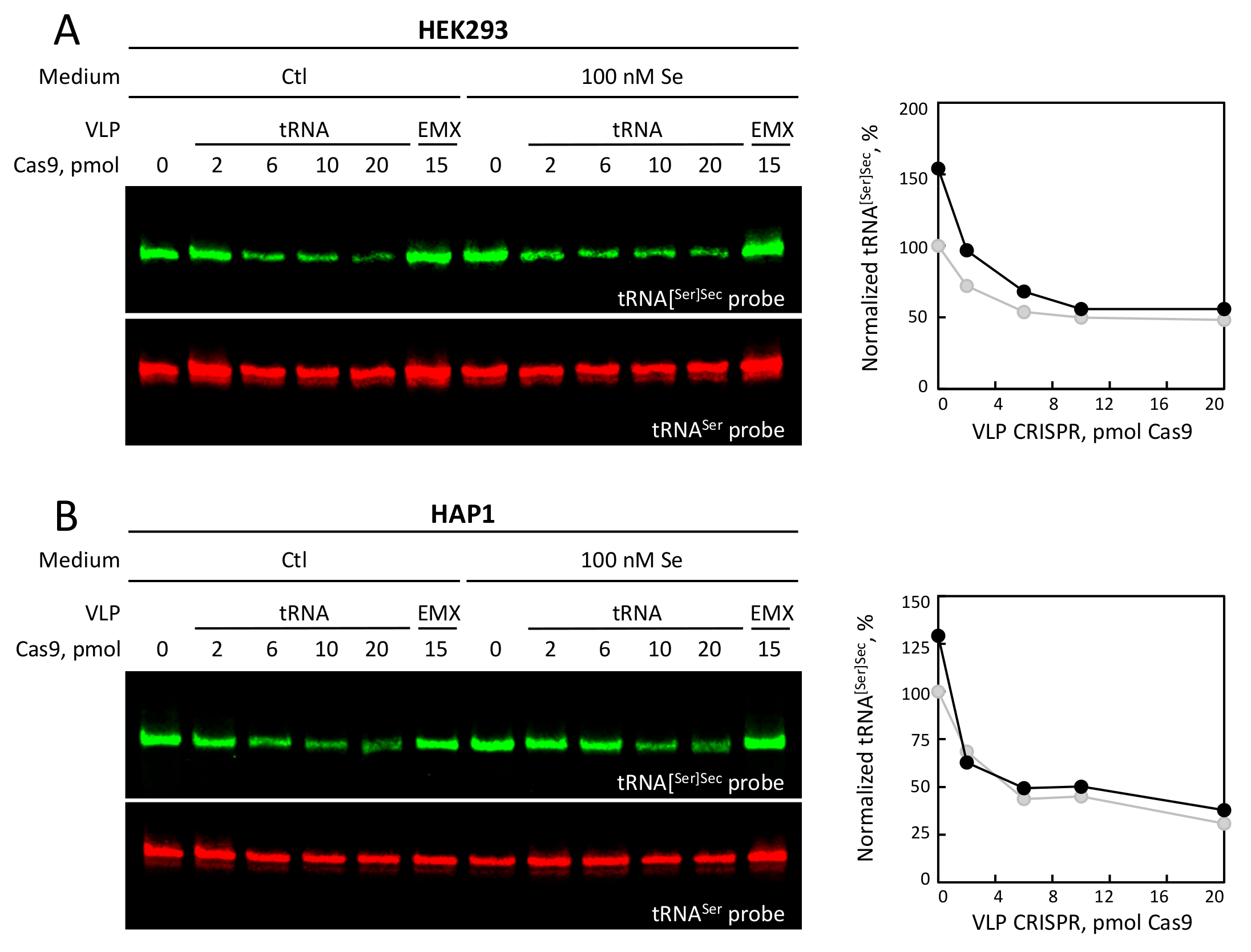
3.4. Selenium Levels and tRNA VLP Treatment Altered the Levels of tRNA[Ser]Sec
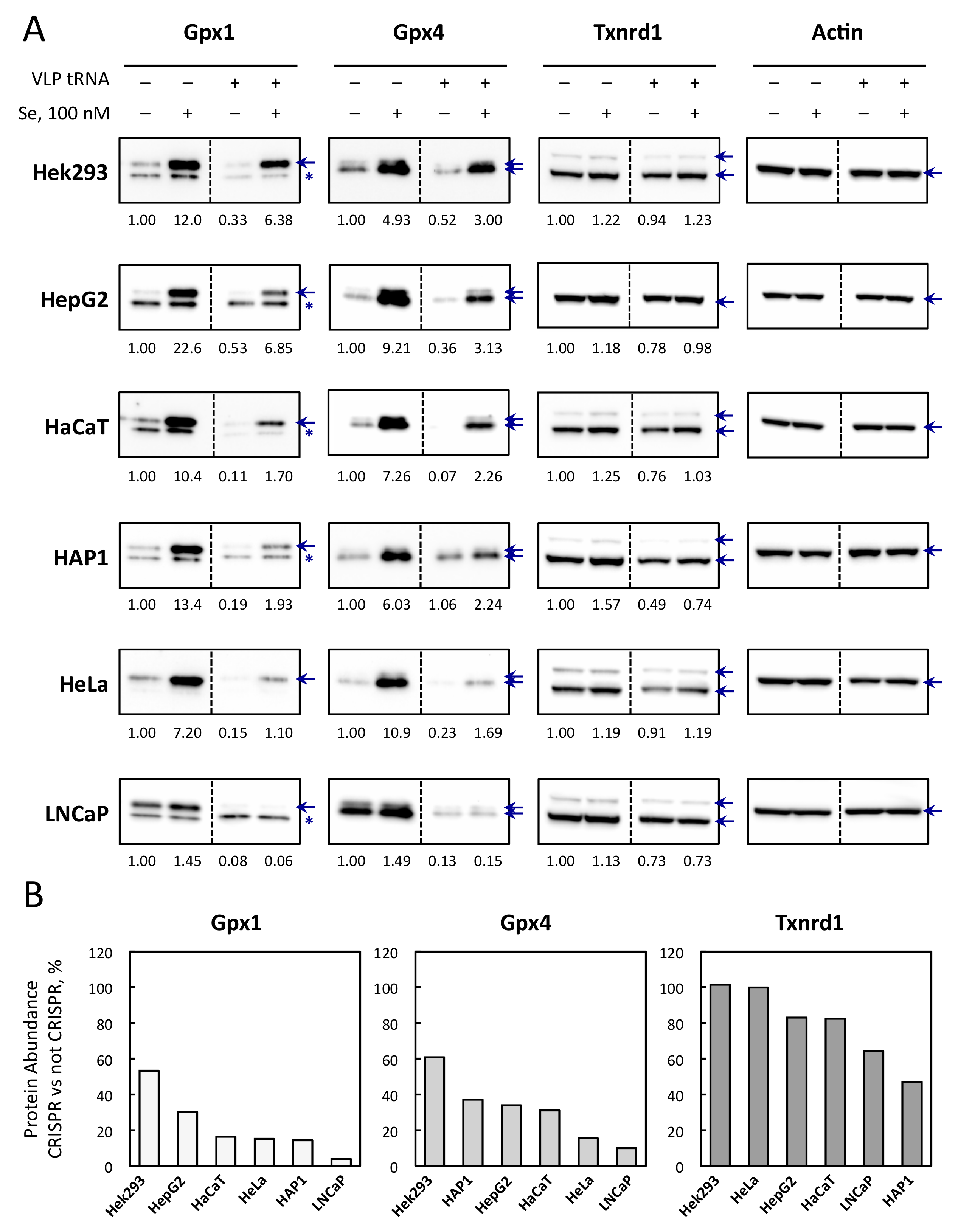
3.5. Selenoprotein Levels Are Differently Affected by CRISPR-Cas9-VLPs Amongst Cell Lines
3.6. Selenoprotein mRNA Levels Are Not Affected by tRNA[Ser]Sec Down-Regulation in Hek293 and HAP1
3.7. The Overexpression of tRNA[Ser]Sec Allowed the Recovery of Selenoprotein Expression in VLP Treated Cells
4. Discussion
4.1. Development of a Novel Method to Produce Cells Lines With Reduced Selenoprotein Levels
4.2. The Cas9-Induced Mutations in TRNAU1 Gene Lead to a Selective Down-Regulation of Selenoproteins to a Level Which Is Similar to Selenium Deficiency
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vindry, C.; Ohlmann, T.; Chavatte, L. Translation regulation of mammalian selenoproteins. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Lee, B.J.; Tsuji, P.A.; Tobe, R.; Park, J.M.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L. Selenocysteine tRNA[Ser]Sec: From nonsense suppressor trna to the quintessential constituent in selenoprotein biosynthesis. In Selenium: Its Molecular Biology and Role in Human Health, 4th ed.; Hatfield, D.L., Schweizer, U., Tsuji, P.A., Gladyshev, V.N., Eds.; Springer Science+Business Media: Berlin, Germany, 2016; pp. 3–12. [Google Scholar]
- Bulteau, A.-L.; Chavatte, L. Update on selenoprotein biosynthesis. Antioxid. Redox Signal. 2015, 23, 775–794. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Berry, M.J. Eukaryotic selenoprotein synthesis: mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life 2008, 60, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef] [PubMed]
- Allmang, C.; Krol, A. Selenoprotein synthesis: UGA does not end the story. Biochimie 2006, 88, 1561–1571. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.M.; Copeland, P.R. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003, 23, 17–40. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Latreche, L.; Jean-Jean, O.; Driscoll, D.M.; Chavatte, L. Novel structural determinants in human SECIS elements modulate the translational recoding of UGA as selenocysteine. Nucleic Acids Res. 2009, 37, 5868–5880. [Google Scholar] [CrossRef]
- Berry, M.J.; Banu, L.; Harney, J.W.; Larsen, P.R. Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J. 1993, 12, 3315–3322. [Google Scholar] [CrossRef]
- Touat-Hamici, Z.; Legrain, Y.; Bulteau, A.-L.; Chavatte, L. Selective up-regulation of human selenoproteins in response to oxidative stress. J. Biol. Chem. 2014, 289, 14750–14761. [Google Scholar] [CrossRef]
- Legrain, Y.; Touat-Hamici, Z.; Chavatte, L. Interplay between selenium levels, selenoprotein expression, and replicative senescence in WI-38 human fibroblasts. J. Biol. Chem. 2014, 289, 6299–6310. [Google Scholar] [CrossRef] [PubMed]
- Latreche, L.; Duhieu, S.; Touat-Hamici, Z.; Jean-Jean, O.; Chavatte, L. The differential expression of glutathione peroxidase 1 and 4 depends on the nature of the SECIS element. RNA Biol. 2012, 9, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Chavatte, L.; Brown, B.A.; Driscoll, D.M. Ribosomal protein L30 is a component of the UGA-selenocysteine recoding machinery in eukaryotes. Nat. Struct. Mol. Biol. 2005, 12, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Rebsch, C.M.; Kinzy, S.A.; Fletcher, J.E.; Copeland, P.R. Efficiency of mammalian selenocysteine incorporation. J. Biol. Chem. 2004, 279, 37852–37859. [Google Scholar] [CrossRef] [PubMed]
- Zavacki, A.M.; Mansell, J.B.; Chung, M.; Klimovitsky, B.; Harney, J.W.; Berry, M.J. Coupled tRNA(Sec)-dependent assembly of the selenocysteine decoding apparatus. Mol. Cell 2003, 11, 773–781. [Google Scholar] [CrossRef]
- Tujebajeva, R.M.; Copeland, P.R.; Xu, X.M.; Carlson, B.A.; Harney, J.W.; Driscoll, D.M.; Hatfield, D.L.; Berry, M.J. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 2000, 1, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Fagegaltier, D.; Hubert, N.; Yamada, K.; Mizutani, T.; Carbon, P.; Krol, A. Characterization of mSelB, a novel mammalian elongation factor for selenoprotein translation. EMBO J. 2000, 19, 4796–4805. [Google Scholar] [CrossRef]
- Copeland, P.R.; Fletcher, J.E.; Carlson, B.A.; Hatfield, D.L.; Driscoll, D.M. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 2000, 19, 306–314. [Google Scholar] [CrossRef]
- Copeland, P.R.; Driscoll, D.M. Purification, redox sensitivity, and RNA binding properties of SECIS-binding protein 2, a protein involved in selenoprotein biosynthesis. J. Biol. Chem. 1999, 274, 25447–25454. [Google Scholar] [CrossRef]
- Santesmasses, D.; Mariotti, M.; Guigo, R. Computational identification of the selenocysteine tRNA (tRNASec) in genomes. PLoS Comput. Biol. 2017, 13, e1005383. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Gladyshev, V.N. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Bosl, M.R.; Takaku, K.; Oshima, M.; Nishimura, S.; Taketo, M.M. Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc. Natl. Acad. Sci. USA 1997, 94, 5531–5534. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A. Selenocysteine tRNA[Ser]Sec mouse models for elucidating roles of selenoproteins in health and development. In Selenium: Its Molecular Biology and Role in Human Health, 4th ed.; Hatfield, D.L., Schweizer, U., Tsuji, P.A., Gladyshev, V.N., Eds.; Springer Science+Business Media: Berlin, Germany, 2016; pp. 555–566. [Google Scholar]
- Itoh, Y.; Chiba, S.; Sekine, S.; Yokoyama, S. Crystal structure of human selenocysteine tRNA. Nucleic Acids Res. 2009, 37, 6259–6268. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.K.; Matsufuji, T.; Matsufuji, S.; Carlson, B.A.; Kim, S.S.; Hatfield, D.L.; Lee, B.J. Methylation of the ribosyl moiety at position 34 of selenocysteine tRNA[Ser]Sec is governed by both primary and tertiary structure. RNA 2000, 6, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Chittum, H.S.; Baek, H.J.; Diamond, A.M.; Fernandez-Salguero, P.; Gonzalez, F.; Ohama, T.; Hatfield, D.L.; Kuehn, M.; Lee, B.J. Selenocysteine tRNA[Ser]Sec levels and selenium-dependent glutathione peroxidase activity in mouse embryonic stem cells heterozygous for a targeted mutation in the tRNA[Ser]Sec gene. Biochemistry 1997, 36, 8634–8639. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, D.; Lee, B.J.; Hampton, L.; Diamond, A.M. Selenium induces changes in the selenocysteine tRNA[Ser]Sec population in mammalian cells. Nucleic Acids Res. 1991, 19, 939–943. [Google Scholar] [CrossRef]
- Carlson, B.A.; Xu, X.M.; Gladyshev, V.N.; Hatfield, D.L. Selective rescue of selenoprotein expression in mice lacking a highly specialized methyl group in selenocysteine tRNA. J. Biol. Chem. 2005, 280, 5542–5548. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Lander, E.S. The Heroes of CRISPR. Cell 2016, 164, 18–28. [Google Scholar] [CrossRef]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massourides, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Arner, E.S.; Berry, M.J.; Brigelius-Flohe, R.; Bruford, E.A.; Burk, R.F.; Carlson, B.A.; Castellano, S.; Chavatte, L.; Conrad, M.; et al. Selenoprotein gene nomenclature. J. Biol. Chem. 2016, 291, 24036–24040. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.A.; Lee, B.J.; Tsuji, P.A.; Copeland, P.R.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L. Selenocysteine tRNA([Ser]Sec), the central component of selenoprotein biosynthesis: isolation, identification, modification, and sequencing. Methods Mol. Biol. 2018, 1661, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Touat-Hamici, Z.; Bulteau, A.L.; Bianga, J.; Jean-Jacques, H.; Szpunar, J.; Lobinski, R.; Chavatte, L. Selenium-regulated hierarchy of human selenoproteome in cancerous and immortalized cells lines. Biochim. Biophys. Acta Gen. Subj. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hopper, A.K.; Huang, H.Y. Quality control pathways for nucleus-encoded eukaryotic trna biosynthesis and subcellular trafficking. Mol. Cell. Biol. 2015, 35, 2052–2058. [Google Scholar] [CrossRef]
- Betat, H.; Morl, M. The CCA-adding enzyme: A central scrutinizer in tRNA quality control. Bioessays 2015, 37, 975–982. [Google Scholar] [CrossRef]
- Lin, Y.C.; Boone, M.; Meuris, L.; Lemmens, I.; Van Roy, N.; Soete, A.; Reumers, J.; Moisse, M.; Plaisance, S.; Drmanac, R.; et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014, 5, 4767. [Google Scholar] [CrossRef]
- Squires, J.E.; Stoytchev, I.; Forry, E.P.; Berry, M.J. SBP2 binding affinity is a major determinant in differential selenoprotein mRNA translation and sensitivity to nonsense-mediated decay. Mol. Cell. Biol. 2007, 27, 7848–7855. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Agostini, M.; Mitchell, C.; Schoenmakers, N.; Papp, L.; Rajanayagam, O.; Padidela, R.; Ceron-Gutierrez, L.; Doffinger, R.; Prevosto, C.; et al. Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans. J. Clin. Investig. 2010, 120, 4220–4235. [Google Scholar] [CrossRef]
- Low, S.C.; Grundner-Culemann, E.; Harney, J.W.; Berry, M.J. SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. EMBO J. 2000, 19, 6882–6890. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Carlson, B.; Agostini, M.; Moran, C.; Rajanayagam, O.; Bochukova, E.; Tobe, R.; Peat, R.; Gevers, E.; Muntoni, F.; et al. Mutation in human selenocysteine transfer RNA selectively disrupts selenoprotein synthesis. J. Clin. Investig. 2016, 126, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Vindry, C.; Ohlmann, T.; Chavatte, L. Selenium metabolism, regulation, and sex differences in mammals. In Selenium, Molecular and Integrative Toxicology, 1st ed.; Michalke, B., Ed.; Springer: New York, NY, USA, 2018; pp. 89–107. [Google Scholar]
- Touat-Hamici, Z.; Legrain, Y.; Sonet, J.; Bulteau, A.-L.; Chavatte, L. Alteration of selenoprotein expression during stress and in aging. In Selenium: Its Molecular Biology and Role in Human Health, 4th ed.; Hatfield, D.L., Schweizer, U., Tsuji, P.A., Gladyshev, V.N., Eds.; Springer Science+Business Media: Berlin, Germany, 2016; pp. 539–551. [Google Scholar]
- Kirchner, S.; Cai, Z.; Rauscher, R.; Kastelic, N.; Anding, M.; Czech, A.; Kleizen, B.; Ostedgaard, L.S.; Braakman, I.; Sheppard, D.N.; et al. Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 2017, 15, e2000779. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vindry, C.; Guillin, O.; Mangeot, P.E.; Ohlmann, T.; Chavatte, L. A Versatile Strategy to Reduce UGA-Selenocysteine Recoding Efficiency of the Ribosome Using CRISPR-Cas9-Viral-Like-Particles Targeting Selenocysteine-tRNA[Ser]Sec Gene. Cells 2019, 8, 574. https://doi.org/10.3390/cells8060574
Vindry C, Guillin O, Mangeot PE, Ohlmann T, Chavatte L. A Versatile Strategy to Reduce UGA-Selenocysteine Recoding Efficiency of the Ribosome Using CRISPR-Cas9-Viral-Like-Particles Targeting Selenocysteine-tRNA[Ser]Sec Gene. Cells. 2019; 8(6):574. https://doi.org/10.3390/cells8060574
Chicago/Turabian StyleVindry, Caroline, Olivia Guillin, Philippe E. Mangeot, Théophile Ohlmann, and Laurent Chavatte. 2019. "A Versatile Strategy to Reduce UGA-Selenocysteine Recoding Efficiency of the Ribosome Using CRISPR-Cas9-Viral-Like-Particles Targeting Selenocysteine-tRNA[Ser]Sec Gene" Cells 8, no. 6: 574. https://doi.org/10.3390/cells8060574
APA StyleVindry, C., Guillin, O., Mangeot, P. E., Ohlmann, T., & Chavatte, L. (2019). A Versatile Strategy to Reduce UGA-Selenocysteine Recoding Efficiency of the Ribosome Using CRISPR-Cas9-Viral-Like-Particles Targeting Selenocysteine-tRNA[Ser]Sec Gene. Cells, 8(6), 574. https://doi.org/10.3390/cells8060574