FAIM: An Antagonist of Fas-Killing and Beyond


Abstract

1. Introduction
2. The Discovery of FAIM
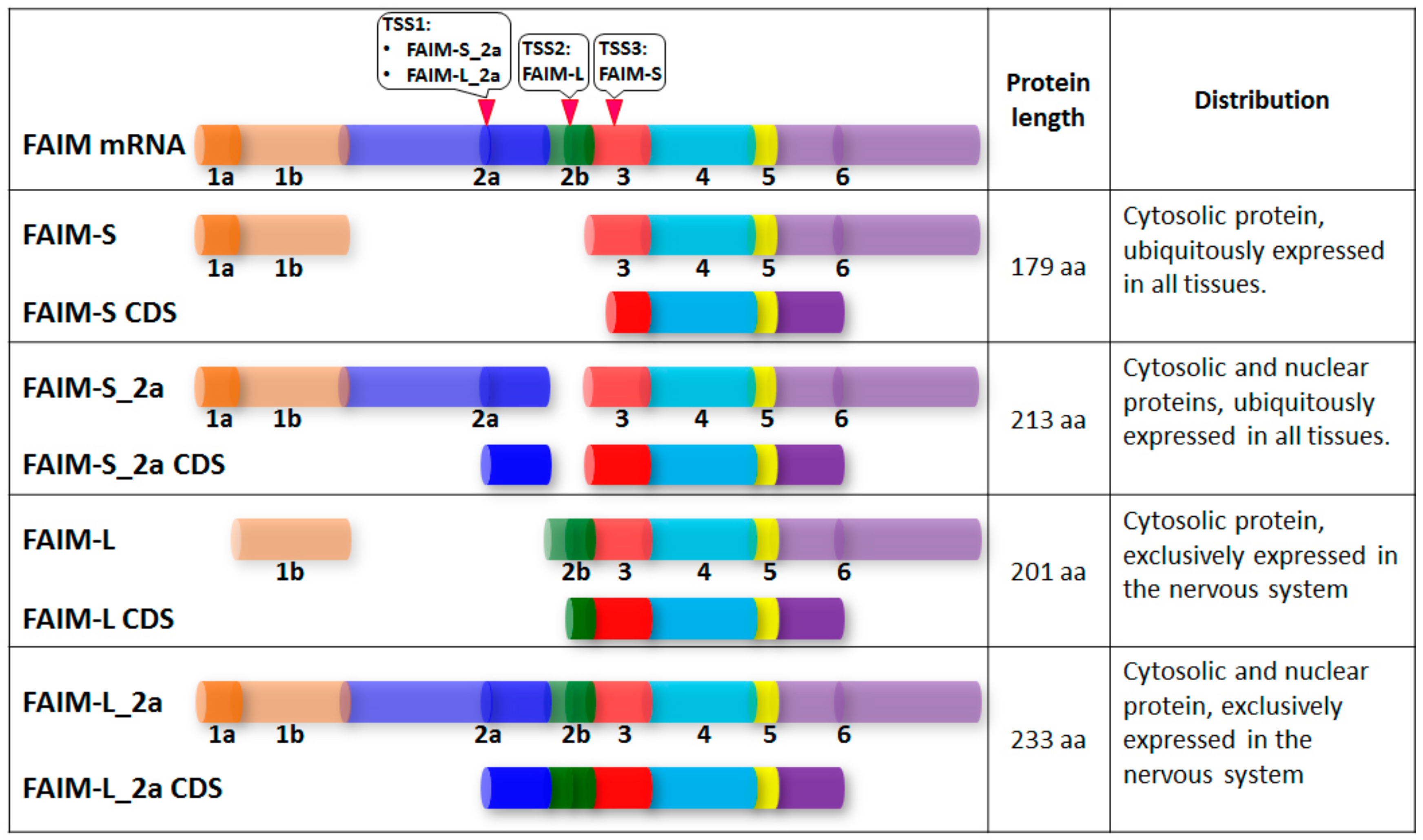
3. Structural Study of FAIM
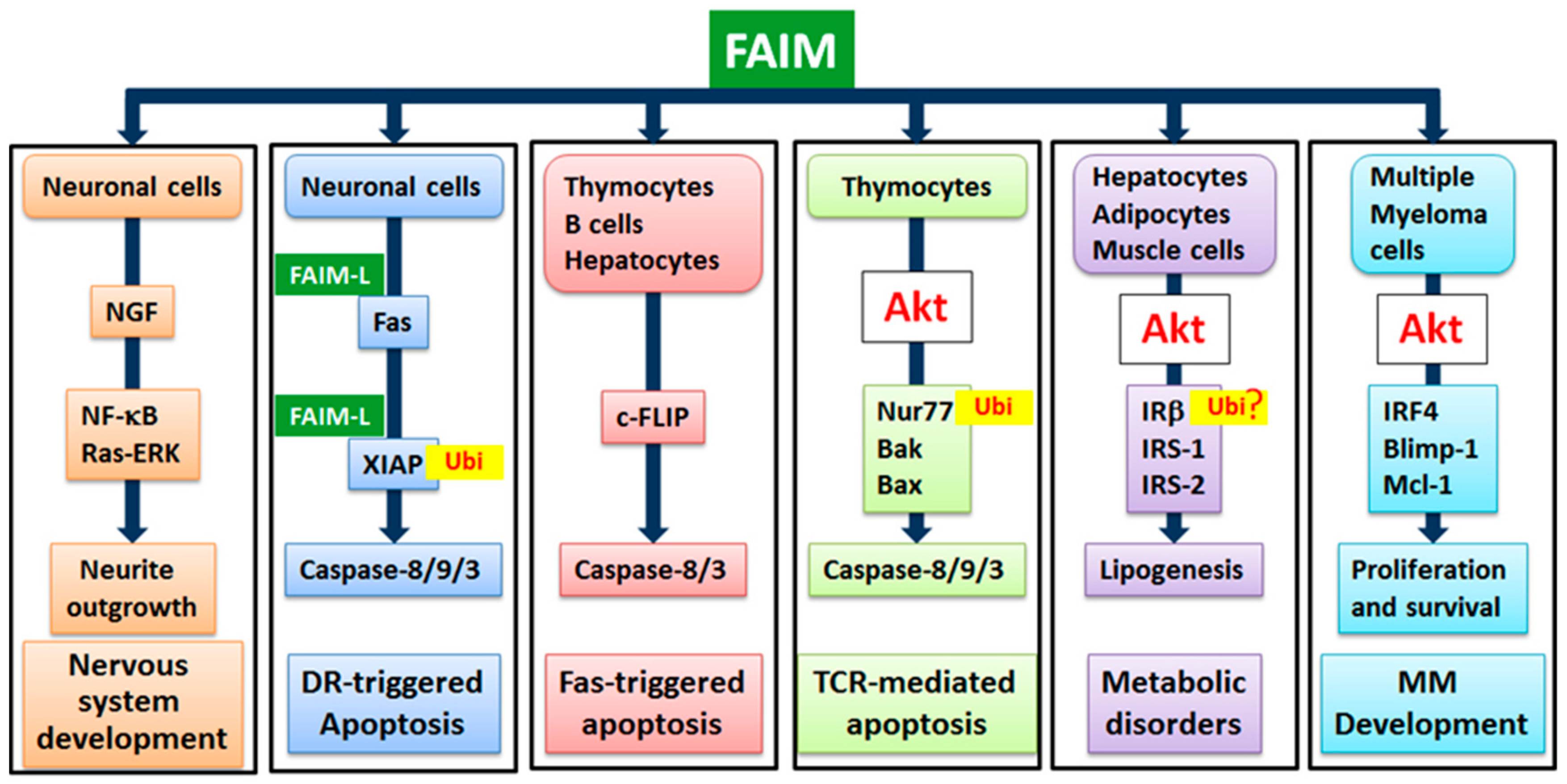
4. Physiological Functions of FAIM
4.1. FAIM’s Role in Fas-Mediated Apoptosis of B Cells, Thymocytes and Hepatocytes
4.2. FAIM’s Role in TCR-Mediated Apoptosis of Thymocytes
4.3. FAIM’s Dual Functions in the Nervous System
4.4. FAIM’s Involvement in Myocardial Infarction
4.5. FAIM’s Role in Insulin Signalling and Energy Homeostasis
5. The Involvement of FAIM in Diseases
5.1. Multiple Myeloma
5.2. Myeloproliferative Diseases
5.3. Other Solid Tumors
5.4. Obesity and Hepatosteatosis
5.5. Alzheimer’s Disease
5.6. Intellectual Disability
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Krammer, P.H. CD95′s deadly mission in the immune system. Nature 2000, 407, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. Embo. J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C.; Schmitz, I.; Krammer, P.H.; Peter, M.E. The role of c-FLIP in modulation of CD95-induced apoptosis. J. Biol. Chem. 1999, 274, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.J.; Fischer, G.M.; Donohoe, T.J.; Colarusso, T.P.; Rothstein, T.L. A novel gene coding for a Fas apoptosis inhibitory molecule (FAIM) isolated from inducibly Fas-resistant B lymphocytes. J. Exp. Med. 1999, 189, 949–956. [Google Scholar] [CrossRef]
- Zhong, X.; Schneider, T.J.; Cabral, D.S.; Donohoe, T.J.; Rothstein, T.L. An alternatively spliced long form of Fas apoptosis inhibitory molecule (FAIM) with tissue-specific expression in the brain. Mol. Immunol. 2001, 38, 65–72. [Google Scholar] [CrossRef]
- Sole, C.; Dolcet, X.; Segura, M.F.; Gutierrez, H.; Diaz-Meco, M.T.; Gozzelino, R.; Sanchis, D.; Bayascas, J.R.; Gallego, C.; Moscat, J.; et al. The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-κB signaling. J. Cell Biol. 2004, 167, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Coccia, E.; Calleja-Yague, I.; Planells-Ferrer, L.; Sanuy, B.; Sanz, B.; Lopez-Soriano, J.; Moubarak, R.S.; Munell, F.; Barneda-Zahonero, B.; Comella, J.X.; et al. Identification and characterization of new isoforms of human fas apoptotic inhibitory molecule (FAIM). PLoS ONE 2017, 12, e0185327. [Google Scholar] [CrossRef]
- Hemond, M.; Rothstein, T.L.; Wagner, G. Fas apoptosis inhibitory molecule contains a novel beta-sandwich in contact with a partially ordered domain. J. Mol. Biol. 2009, 386, 1024–1037. [Google Scholar] [CrossRef]
- Huo, J.; Xu, S.; Guo, K.; Zeng, Q.; Lam, K.P. Genetic deletion of faim reveals its role in modulating c-FLIP expression during CD95-mediated apoptosis of lymphocytes and hepatocytes. Cell Death Differ. 2009, 16, 1062–1070. [Google Scholar] [CrossRef]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schroter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Thompson, C.B. Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 2002, 109 Suppl 1, S97–S107. [Google Scholar] [CrossRef]
- Van Meerwijk, J.P.; Marguerat, S.; Lees, R.K.; Germain, R.N.; Fowlkes, B.J.; MacDonald, H.R. Quantitative impact of thymic clonal deletion on the T cell repertoire. J. Exp. Med. 1997, 185, 377–383. [Google Scholar] [CrossRef]
- Werlen, G.; Hausmann, B.; Naeher, D.; Palmer, E. Signaling life and death in the thymus, Timing is everything. Science 2003, 299, 1859–1863. [Google Scholar] [CrossRef]
- Huo, J.; Xu, S.; Lam, K.P. Fas apoptosis inhibitory molecule regulates T cell receptor-mediated apoptosis of thymocytes by modulating Akt activation and Nur77 expression. J. Biol. Chem. 2010, 285, 11827–11835. [Google Scholar] [CrossRef]
- Woronicz, J.D.; Calnan, B.; Ngo, V.; Winoto, A. Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature 1994, 367, 277–281. [Google Scholar] [CrossRef]
- Liu, Z.G.; Smith, S.W.; McLaughlin, K.A.; Schwartz, L.M.; Osborne, B.A. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature 1994, 367, 281–284. [Google Scholar] [CrossRef]
- Planells-Ferrer, L.; Urresti, J.; Coccia, E.; Galenkamp, K.M.; Calleja-Yague, I.; Lopez-Soriano, J.; Carriba, P.; Barneda-Zahonero, B.; Segura, M.F.; Comella, J.X. Fas apoptosis inhibitory molecules: More than death-receptor antagonists in the nervous system. J. Neurochem. 2016, 139, 11–21. [Google Scholar] [CrossRef]
- Segura, M.F.; Sole, C.; Pascual, M.; Moubarak, R.S.; Perez-Garcia, M.J.; Gozzelino, R.; Iglesias, V.; Badiola, N.; Bayascas, J.R.; Llecha, N.; et al. The long form of Fas apoptotic inhibitory molecule is expressed specifically in neurons and protects them against death receptor-triggered apoptosis. J. Neurosci. 2007, 27, 11228–11241. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J. Biol. Chem. 2006, 281, 3254–3260. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Ashwell, J.D. IAPs, what’s in a name? Mol. Cell 2008, 30, 123–135. [Google Scholar] [CrossRef]
- Jost, P.J.; Grabow, S.; Gray, D.; McKenzie, M.D.; Nachbur, U.; Huang, D.C.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J.; Strasser, A.; et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009, 460, 1035–1039. [Google Scholar] [CrossRef]
- Moubarak, R.S.; Planells-Ferrer, L.; Urresti, J.; Reix, S.; Segura, M.F.; Carriba, P.; Marques-Fernandez, F.; Sole, C.; Llecha-Cano, N.; Lopez-Soriano, J.; et al. FAIM-L is an IAP-binding protein that inhibits XIAP ubiquitinylation and protects from Fas-induced apoptosis. J. Neurosci. 2013, 33, 19262–19275. [Google Scholar] [CrossRef]
- Martinez-Marmol, R.; Barneda-Zahonero, B.; Soto, D.; Andres, R.M.; Coccia, E.; Gasull, X.; Planells-Ferrer, L.; Moubarak, R.S.; Soriano, E.; Comella, J.X. FAIM-L regulation of XIAP degradation modulates Synaptic Long-Term Depression and Axon Degeneration. Sci. Rep. 2016, 6, 35775. [Google Scholar] [CrossRef]
- Williams, A.R.; Trachtenberg, B.; Velazquez, D.L.; McNiece, I.; Altman, P.; Rouy, D.; Mendizabal, A.M.; Pattany, P.M.; Lopera, G.A.; Fishman, J.; Zambrano, J.P.; Heldman, A.W.; Hare, J.M. Intramyocardial stem cell injection in patients with ischemic cardiomyopathy, Functional recovery and reverse remodeling. Circ. Res. 2011, 108, 792–796. [Google Scholar] [CrossRef]
- Tse, H.F.; Kwong, Y.L.; Chan, J.K.; Lo, G.; Ho, C.L.; Lau, C.P. Angiogenesis in ischaemic myocardium by intramyocardial autologous bone marrow mononuclear cell implantation. Lancet 2003, 361, 47–49. [Google Scholar] [CrossRef]
- Liu, X.; Hu, D.; Zeng, Z.; Zhu, W.; Zhang, N.; Yu, H.; Chen, H.; Wang, K.; Wang, Y.; Wang, L.; et al. SRT1720 promotes survival of aged human mesenchymal stem cells via FAIM: A pharmacological strategy to improve stem cell-based therapy for rat myocardial infarction. Cell Death Dis. 2017, 8, e2731. [Google Scholar] [CrossRef]
- Huo, J.; Ma, Y.; Liu, J.J.; Ho, Y.S.; Liu, S.; Soh, L.Y.; Chen, S.; Xu, S.; Han, W.; Hong, A.; et al. Loss of Fas apoptosis inhibitory molecule leads to spontaneous obesity and hepatosteatosis. Cell Death Dis. 2016, 7, e2091. [Google Scholar] [CrossRef]
- White, M.F.; Kahn, C.R. The insulin signaling system. J. Biol. Chem. 1994, 269, 1–4. [Google Scholar]
- Ramos, F.J.; Langlais, P.R.; Hu, D.; Dong, L.Q.; Liu, F. Grb10 mediates insulin-stimulated degradation of the insulin receptor: A mechanism of negative regulation. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1262–E1266. [Google Scholar] [CrossRef]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Iida, S.; Rao, P.H.; Butler, M.; Corradini, P.; Boccadoro, M.; Klein, B.; Chaganti, R.S.; Dalla-Favera, R. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 1997, 17, 226–230. [Google Scholar] [CrossRef]
- Tajima, E.; Uranishi, M.; Iida, S.; Komatsu, H.; Nitta, M.; Ueda, R. Global real-time quantification/reverse transcription-polymerase chain reaction for detecting proto-oncogenes associated with 14q32 chromosomal translocation in multiple myeloma. Haematologica 2005, 90, 559–562. [Google Scholar]
- Shaffer, A.L.; Emre, N.C.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Uranishi, M.; Iida, S.; Sanda, T.; Ishida, T.; Tajima, E.; Ito, M.; Komatsu, H.; Inagaki, H.; Ueda, R. Multiple myeloma oncogene 1 (MUM1)/interferon regulatory factor 4 (IRF4) upregulates monokine induced by interferon-gamma (MIG) gene expression in B-cell malignancy. Leukemia 2005, 19, 1471–1478. [Google Scholar] [CrossRef]
- Huo, J.; Xu, S.; Lin, B.; Chng, W.J.; Lam, K.P. Fas apoptosis inhibitory molecule is upregulated by IGF-1 signaling and modulates Akt activation and IRF4 expression in multiple myeloma. Leukemia 2013, 27, 1165–1171. [Google Scholar] [CrossRef]
- Turner, C.A., Jr.; Mack, D.H.; Davis, M.M. Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell 1994, 77, 297–306. [Google Scholar] [CrossRef]
- Zhang, B.; Gojo, I.; Fenton, R.G. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 2002, 99, 1885–1893. [Google Scholar] [CrossRef]
- Freund, G.G.; Kulas, D.T.; Mooney, RA. Insulin and IGF-1 increase mitogenesis and glucose metabolism in the multiple myeloma cell line, RPMI 8226. J. Immunol. 1993, 151, 1811–1820. [Google Scholar]
- Chng, W.J.; Kumar, S.; Vanwier, S.; Ahmann, G.; Price-Troska, T.; Henderson, K.; Chung, T.H.; Kim, S.; Mulligan, G.; Bryant, B.; et al. Molecular dissection of hyperdiploid multiple myeloma by gene expression profiling. Cancer Res. 2007, 67, 2982–2989. [Google Scholar] [CrossRef]
- Chng, W.J.; Gualberto, A.; Fonseca, R. IGF-1R is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia 2006, 20, 174–176. [Google Scholar] [CrossRef]
- Economopoulou, C.; Pappa, V.; Kontsioti, F.; Papageorgiou, S.; Kapsimali, V.; Papasteriadi, C.; Economopoulou, P.; Papageorgiou, E.; Dervenoulas, J.; Economopoulos, T. Analysis of apoptosis regulatory genes expression in the bone marrow (BM) of adult de novo myelodysplastic syndromes (MDS). Leuk Res. 2008, 32, 61–69. [Google Scholar] [CrossRef]
- Zeuner, A.; Pedini, F.; Signore, M.; Ruscio, G.; Messina, C.; Tafuri, A.; Girelli, G.; Peschle, C.; De Maria, R. Increased death receptor resistance and FLIPshort expression in polycythemia vera erythroid precursor cells. Blood 2006, 107, 3495–3502. [Google Scholar] [CrossRef]
- Tognon, R.; Gasparotto, E.P.; Leroy, J.M.; Oliveira, G.L.; Neves, R.P.; Carrara, R.C.; Kashima, S.; Covas, D.T.; Santana, M.; Souto, E.X.; et al. Differential expression of apoptosis-related genes from death receptor pathway in chronic myeloproliferative diseases. J. Clin. Pathol. 2011, 64, 75–82. [Google Scholar] [CrossRef]
- Moore, P.S.; Barbi, S.; Donadelli, M.; Costanzo, C.; Bassi, C.; Palmieri, M.; Scarpa, A. Gene expression profiling after treatment with the histone deacetylase inhibitor trichostatin A reveals altered expression of both pro- and anti-apoptotic genes in pancreatic adenocarcinoma cells. Biochim. Biophys. Acta 2004, 1693, 167–176. [Google Scholar] [CrossRef]
- Patron, J.P.; Fendler, A.; Bild, M.; Jung, U.; Muller, H.; Arntzen, M.O.; Piso, C.; Stephan, C.; Thiede, B.; Mollenkopf, H.J.; et al. MiR-133b targets antiapoptotic genes and enhances death receptor-induced apoptosis. PLoS ONE 2012, 7, e35345. [Google Scholar] [CrossRef]
- Ahrens, T.D.; Timme, S.; Hoeppner, J.; Ostendorp, J.; Hembach, S.; Follo, M.; Hopt, UT.; Werner, M.; Busch, H.; Boerries, M.; Lassmann, S. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics 2015, 10, 431–445. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Carriba, P.; Comella, J.X. Amyloid Beta, TNFalpha and FAIM-L: Approaching New Therapeutic Strategies for AD. Front. Neurol. 2014, 5, 276. [Google Scholar] [CrossRef][Green Version]
- Carriba, P.; Jimenez, S.; Navarro, V.; Moreno-Gonzalez, I.; Barneda-Zahonero, B.; Moubarak, R.S.; Lopez-Soriano, J.; Gutierrez, A.; Vitorica, J.; Comella, J.X. Amyloid-beta reduces the expression of neuronal FAIM-L, thereby shifting the inflammatory response mediated by TNFalpha from neuronal protection to death. Cell Death Dis. 2015, 6, e1639. [Google Scholar] [CrossRef]
- Vissers, L.E.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]
- Devanna, P.; van de Vorst, M.; Pfundt, R.; Gilissen, C.; Vernes, S.C. Genome-wide investigation of an ID cohort reveals de novo 3′UTR variants affecting gene expression. Hum. Genet. 2018, 137, 717–721. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Conditions | Tissue/Cell | FAIM Expression and/or Effects | FAIM’s Roles and Mechanisms | Ref. |
|---|---|---|---|---|
| Multiple myeloma (MM) | IRF4-expressing multiple myeloma cell lines | FAIM was upregulated in IRF4-expressing MM cells. | IRF4-FAIM plays roles in MM progression. | [34] |
| MM patients and MM cell lines | FAIM expression correlates with poorer survival outcomes of newly diagnosed MM patients treated with stem cell transplantation or relapsed MM patients treated in clinical trials with Bortezomib. | FAIM’s diagnostic and prognostic value in MM patients. | [35] | |
| FAIM-IRF4-Akt forward feedback loop for MM development. | |||
| Myeloproliferative diseases (MPD) | CD34 cells and leukocytes from MPD patients | FAIM is elevated in CD34 cells obtained from MPD patients. | FAIM may contribute to MPD pathogenesis. | [43] |
| Prostate cancer | Prostate cancer patients and PC3 cell line | FAIM is one of miR-133b immediate targets. | FAIM may contribute to prostate tumorigenesis and tissue homeostasis. | [45] |
| Esophageal cancers | Esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) cells | Inhibition of histone deacetylases downregulates FAIM expression. | FAIM is one of various genes regulated by inhibition of histone deacetylases in esophageal cancer cells. | [46] |
| Obesity and hepatosteatosis | Human and mouse | FAIM defects lead to non-hyperphagic obesity accompanied by hepatosteatosis, adipocyte hypertrophy, dyslipidaemia, hyperglycaemia and hyperinsulinaemia
| FAIM mediates insulin signaling and plays an essential role in energy homoeostasis.
| [27] |
| Alzheimer’s disease (AD) | Human and mouse |
| FAIM is associated with the progression of AD. | [48] |
| Intellectual disability | Intellectual disability patients | FAIM is down-regulated in intellectually disabled patients. | Unknown | [51] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, J.; Xu, S.; Lam, K.-P. FAIM: An Antagonist of Fas-Killing and Beyond. Cells 2019, 8, 541. https://doi.org/10.3390/cells8060541
Huo J, Xu S, Lam K-P. FAIM: An Antagonist of Fas-Killing and Beyond. Cells. 2019; 8(6):541. https://doi.org/10.3390/cells8060541
Chicago/Turabian StyleHuo, Jianxin, Shengli Xu, and Kong-Peng Lam. 2019. "FAIM: An Antagonist of Fas-Killing and Beyond" Cells 8, no. 6: 541. https://doi.org/10.3390/cells8060541
APA StyleHuo, J., Xu, S., & Lam, K.-P. (2019). FAIM: An Antagonist of Fas-Killing and Beyond. Cells, 8(6), 541. https://doi.org/10.3390/cells8060541