Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Antibodies and Reagents
2.3. Bacterial Culture Preparation
2.4. Preparation of Macrophages for In Vitro Experiments
2.5. Cells Infection
2.6. Animal Model of Infection
2.7. ELISA for Cytokines
2.8. Western Blot Analysis
2.9. Enumeration of Viable Bacteria
2.10. Immunofluorescence and Confocal Microscopy
2.11. Transmission Electron Microscopy
2.12. Cell Viability Assay
2.13. Histopathological Analysis
2.14. Immunohistochemical Analysis
2.15. Flow Cytometry Analysis
2.16. Statistical Analysis
3. Results
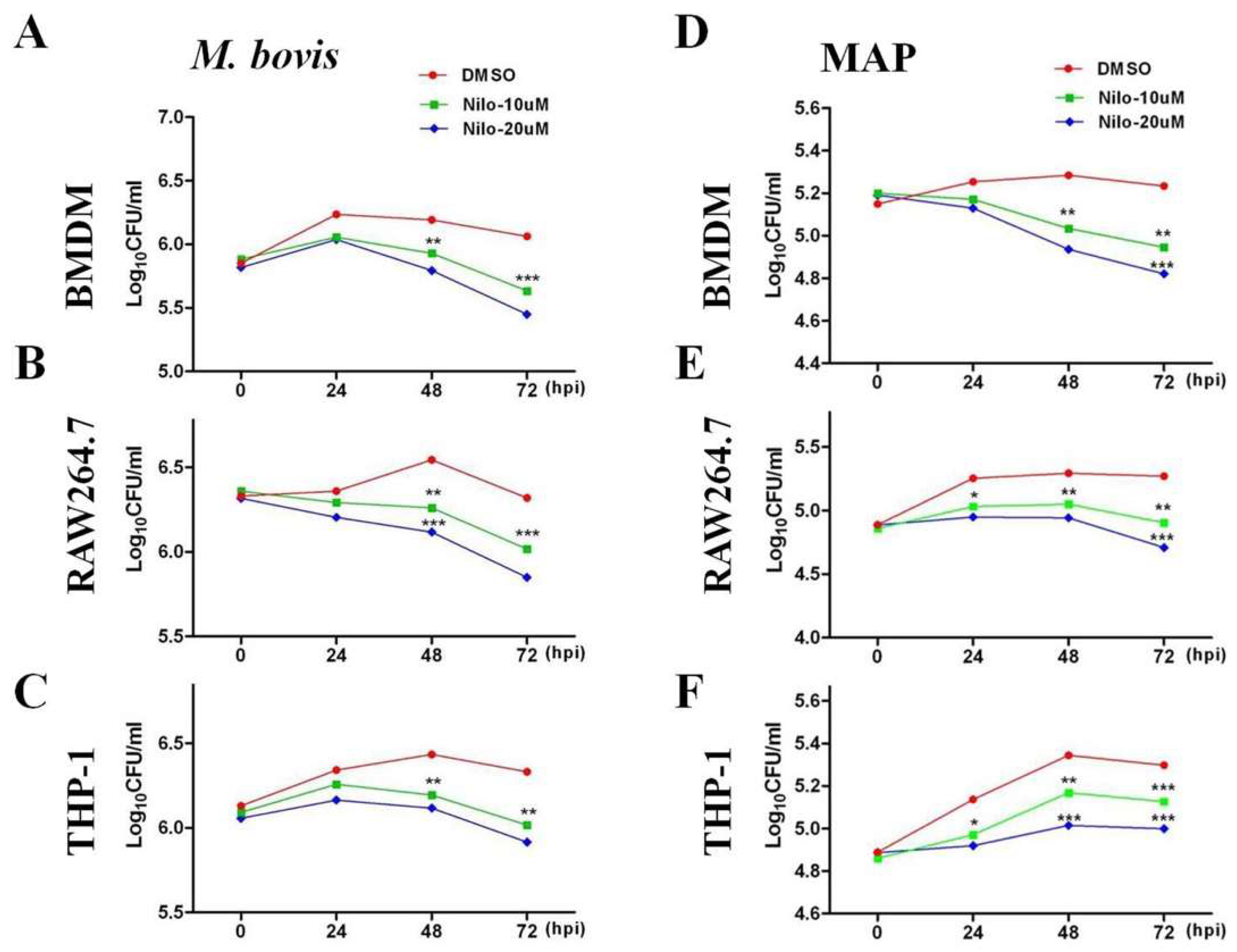
3.1. Nilotinib Inhibits the Intracellular Survival of M. bovis and MAP In Vitro
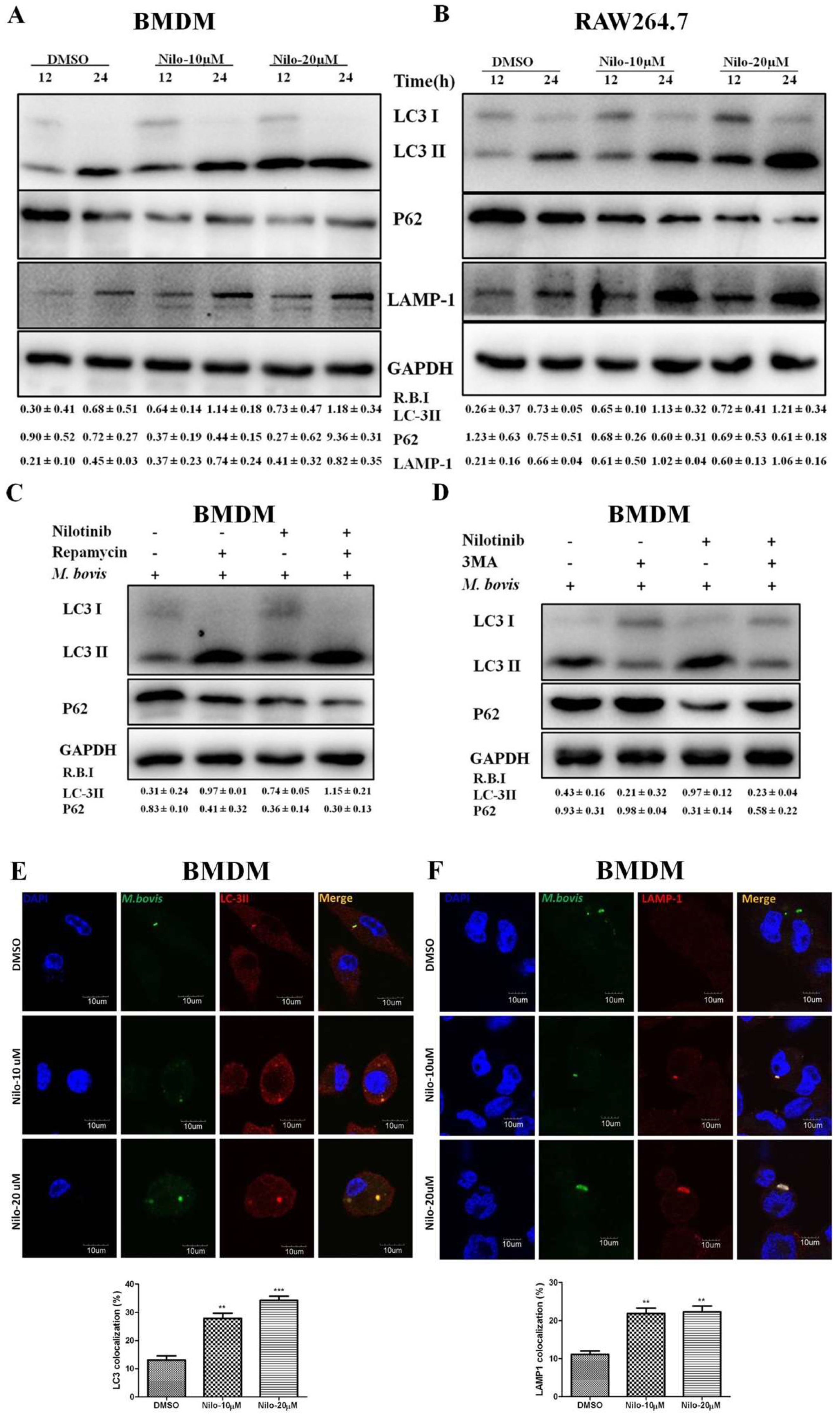
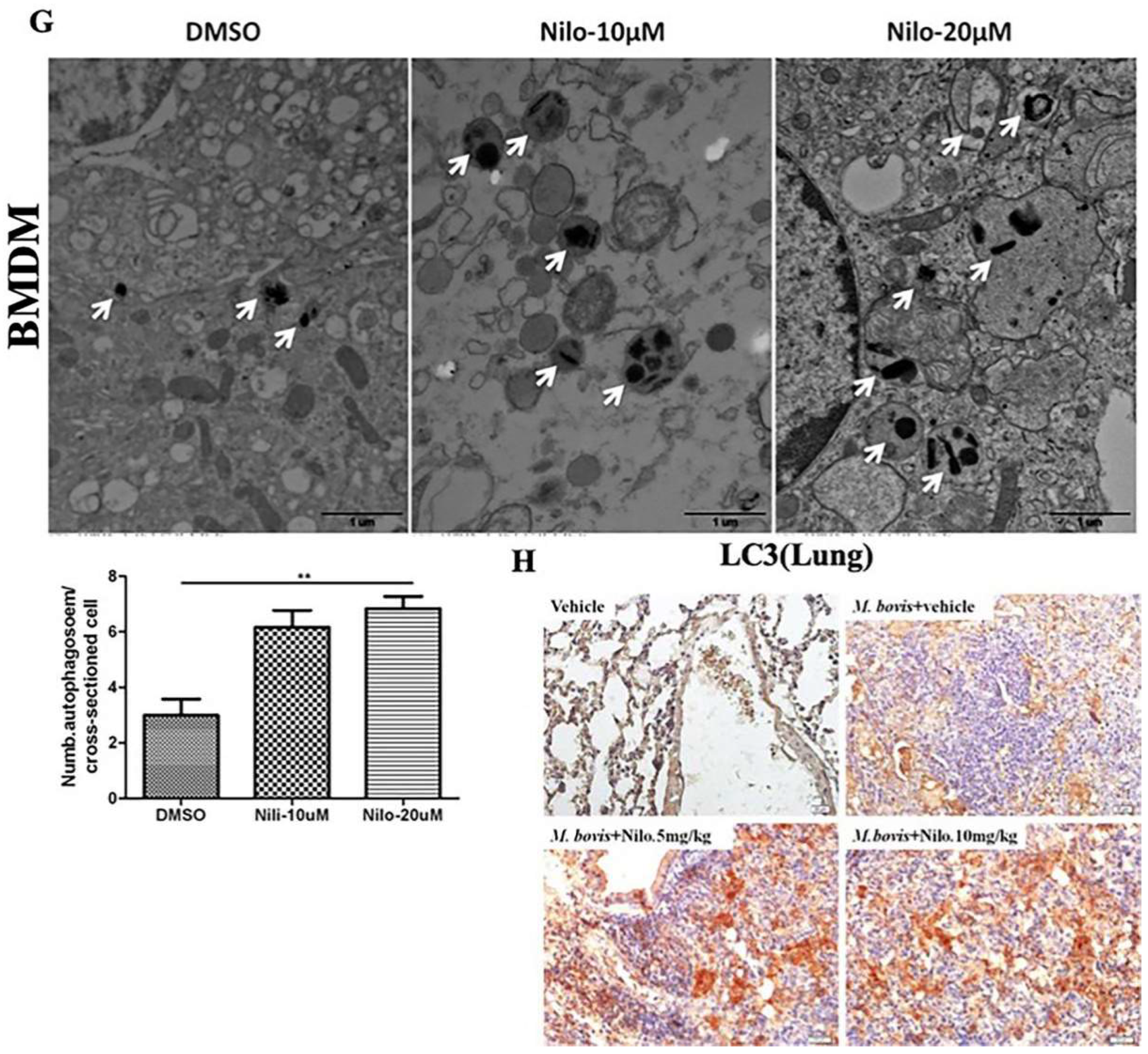
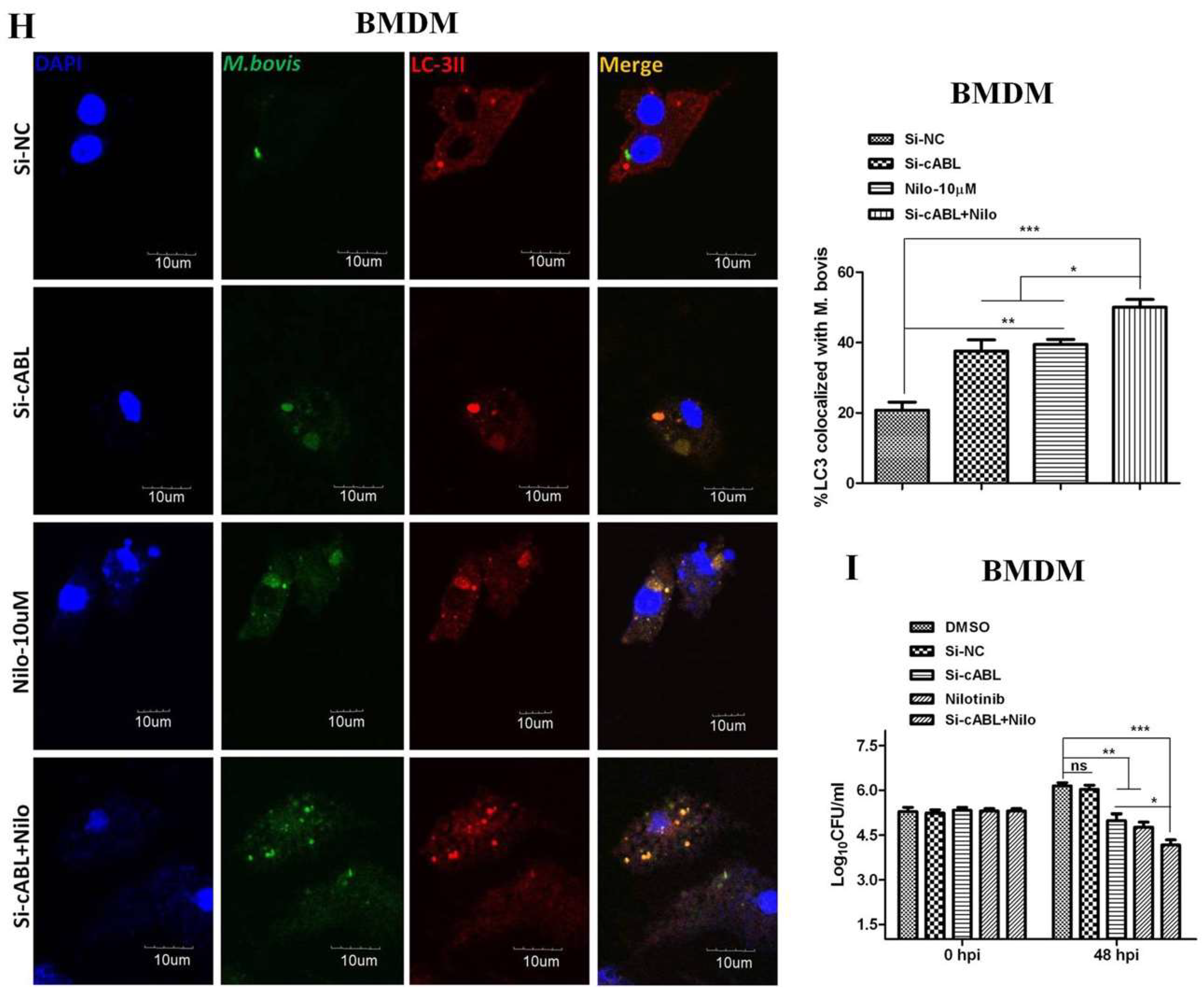
3.2. Nilotinib Induces Autophagy during M. bovis Infection
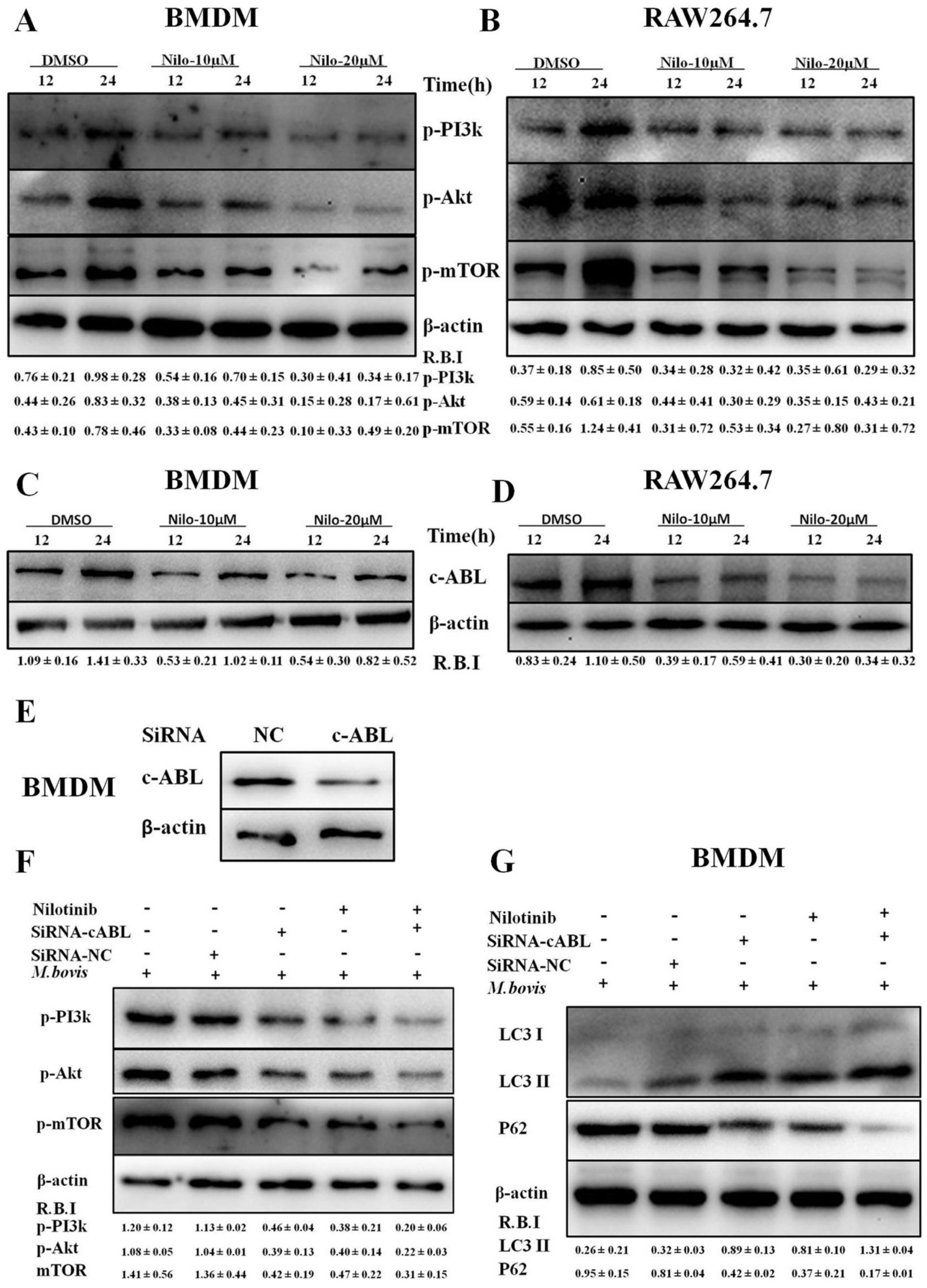
3.3. Nilotinib Attenuates c-ABL Dependent PI3k/Akt/mTOR Signaling Pathway in M. bovis Infected Macrophages
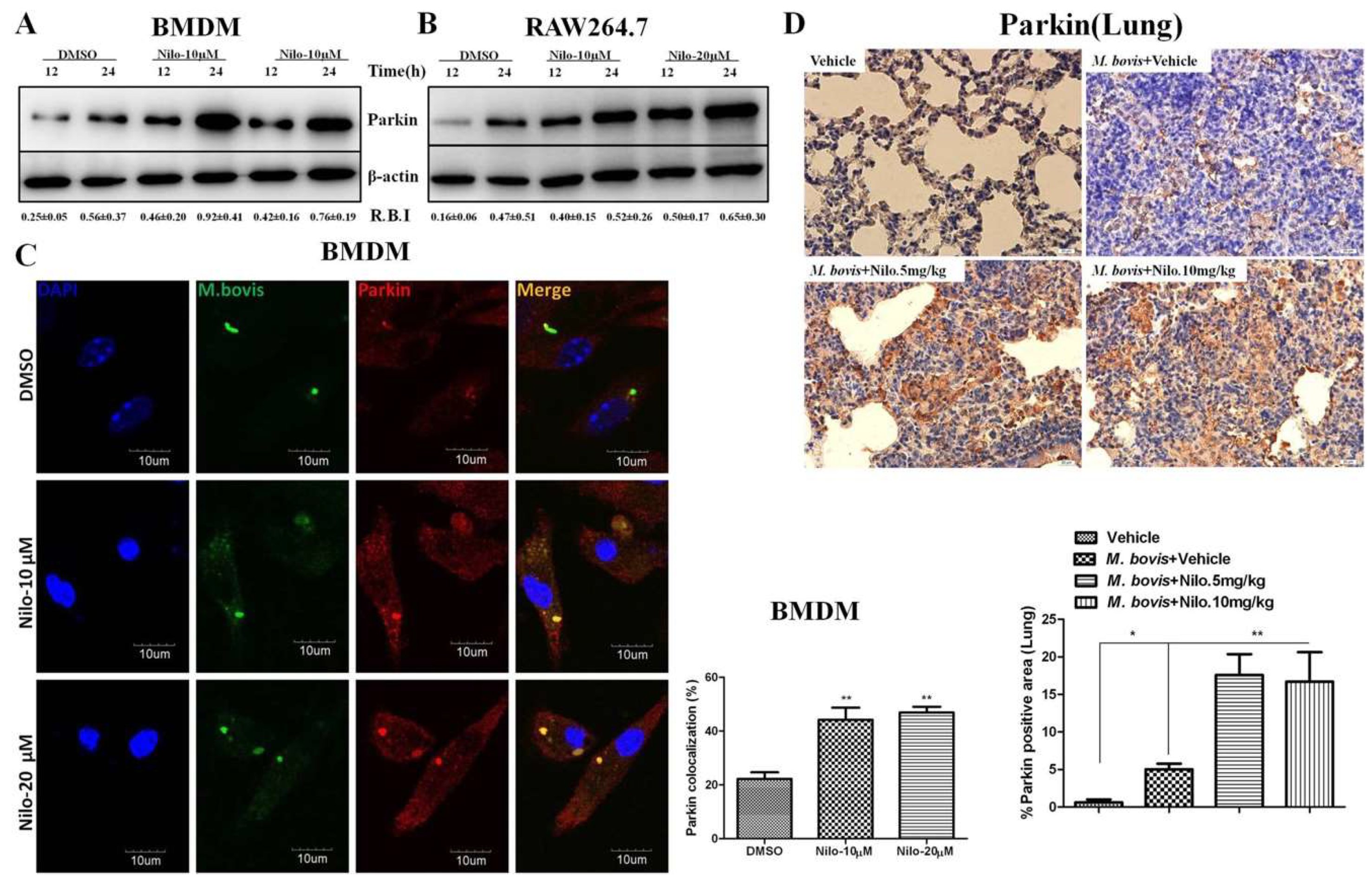
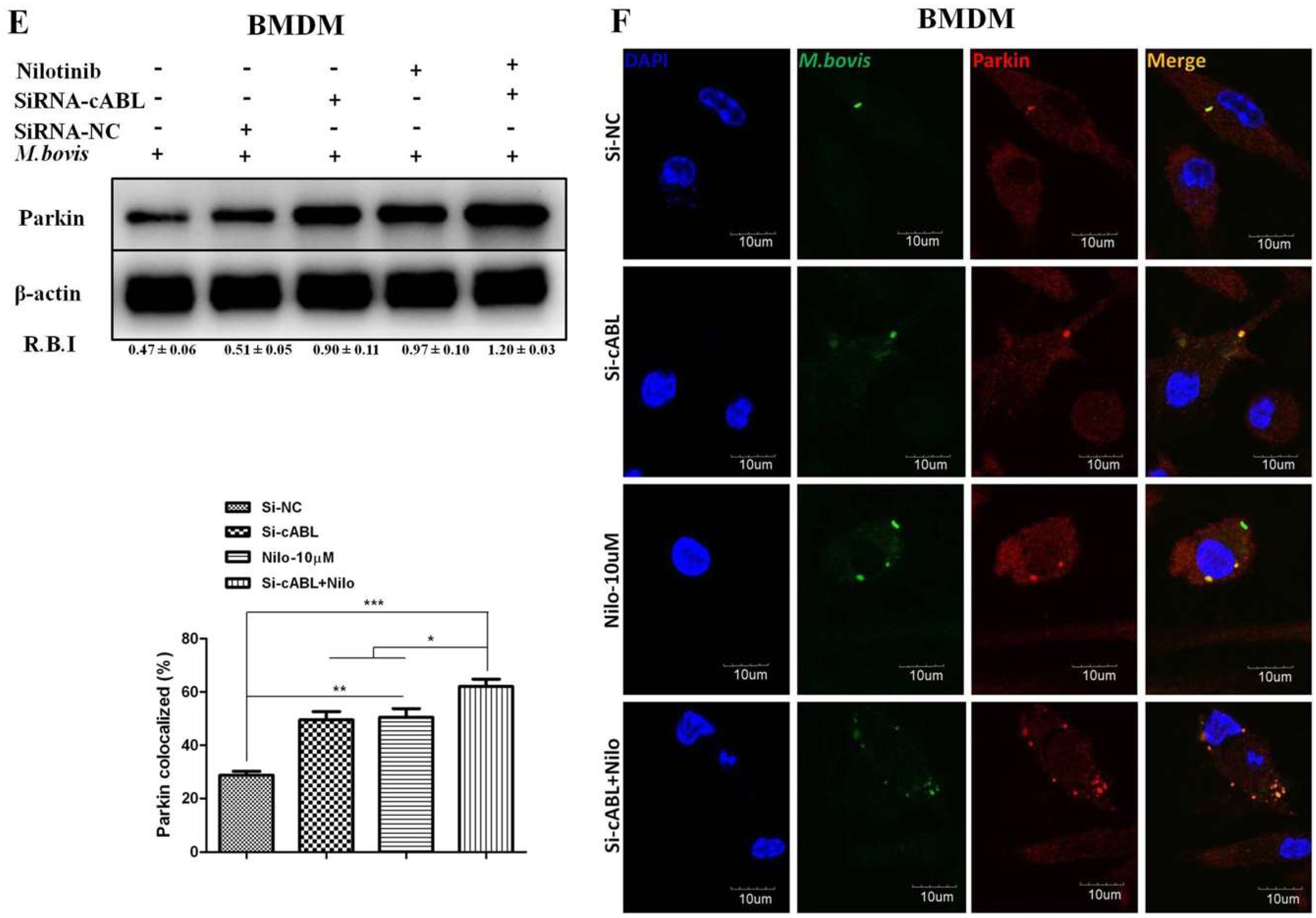
3.4. Nilotinib Increases Parkin Activation During M. bovis Infection
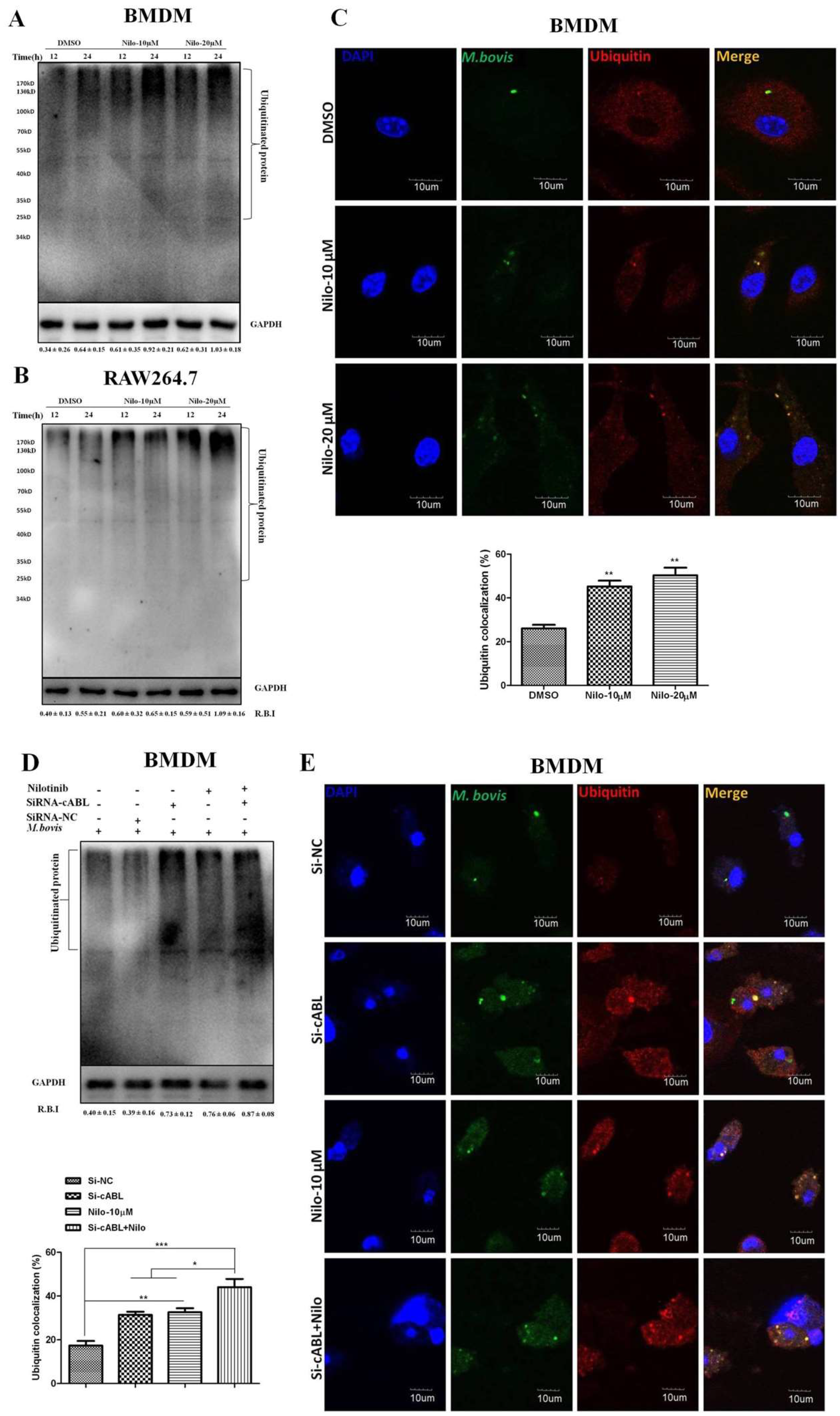
3.5. Nilotinib Promotes Ubiquitination of M. bovis in Infected Macrophages
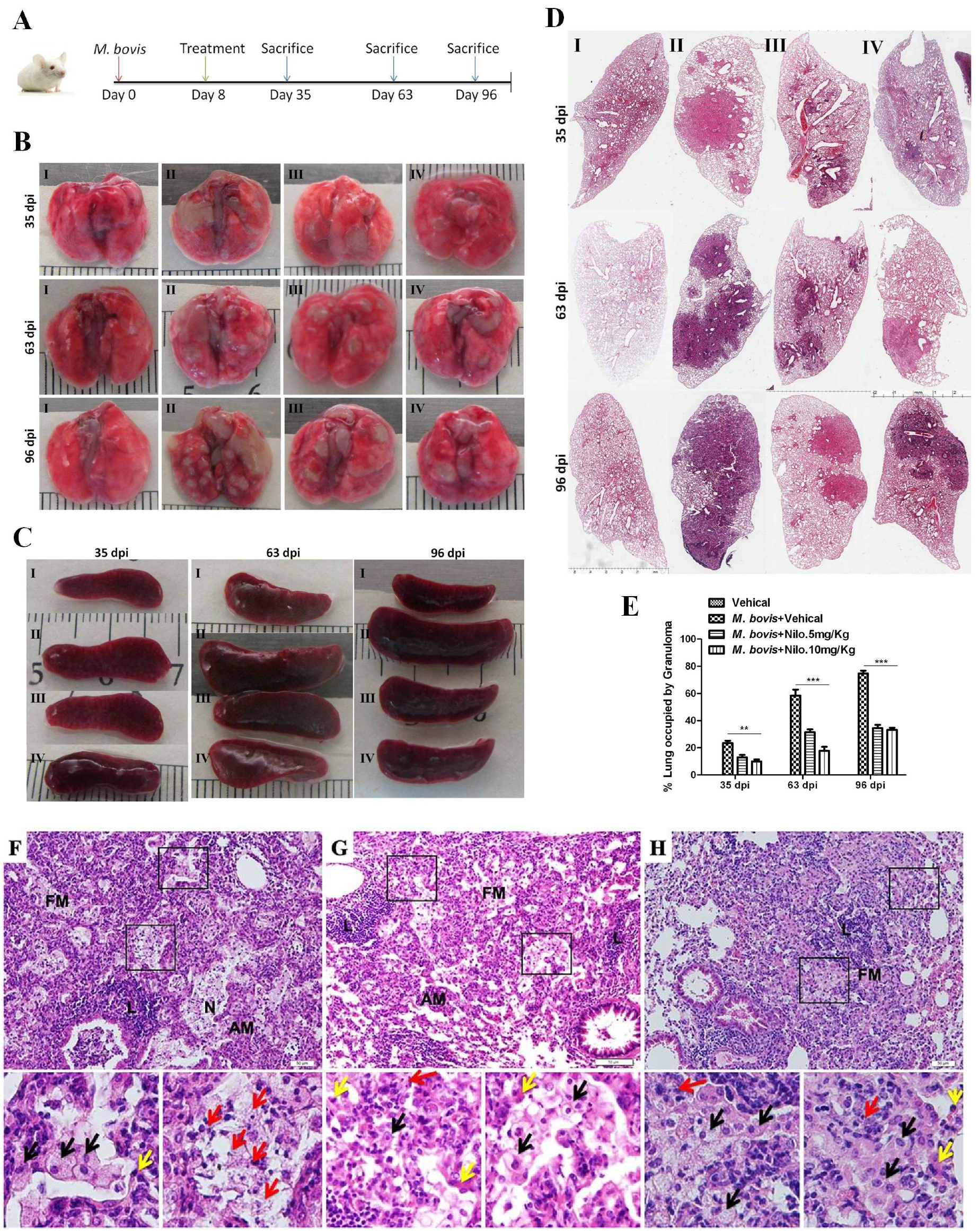
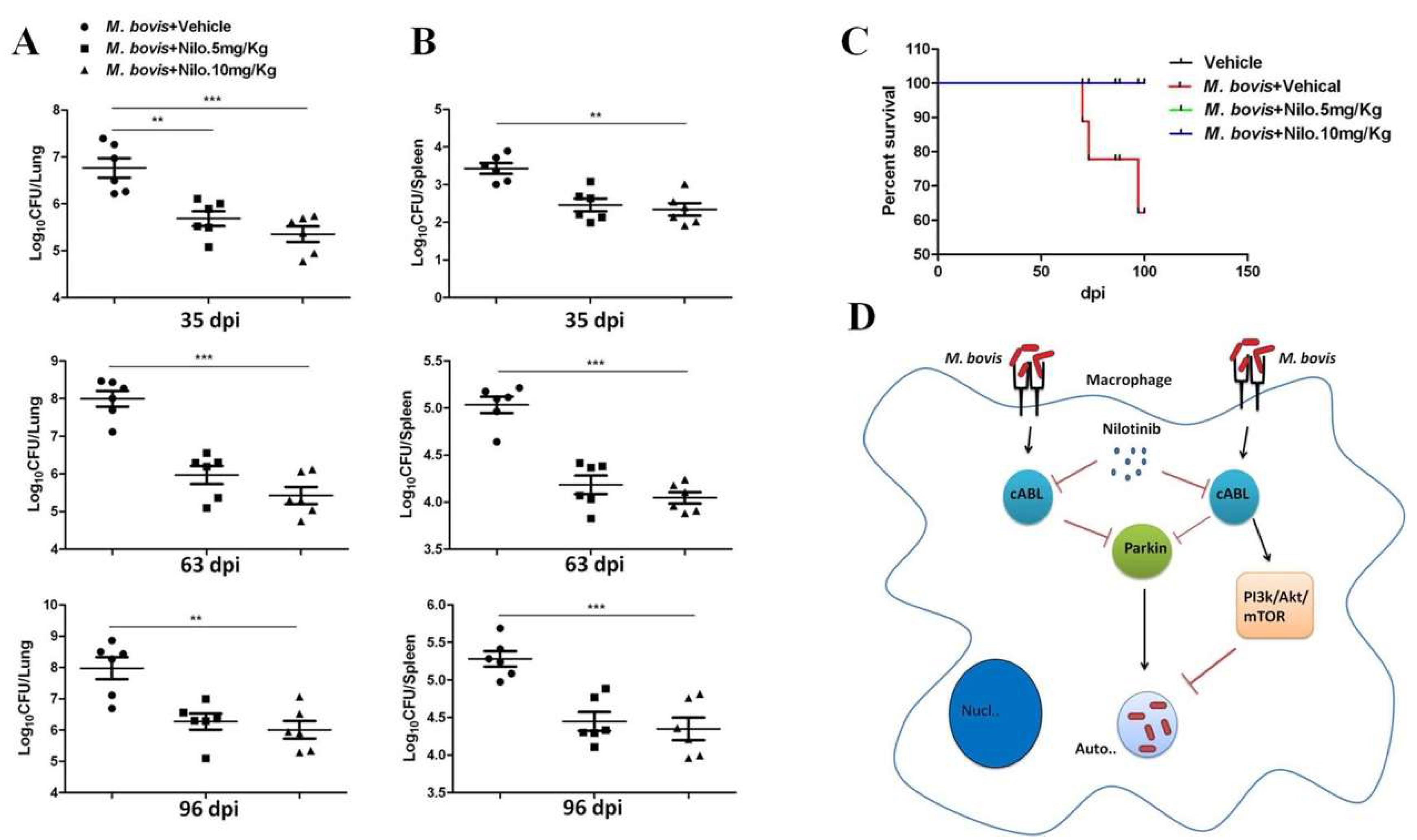
3.6. Nilotinib Reduces the Severity of M. bovis Pathogenesis in Mice
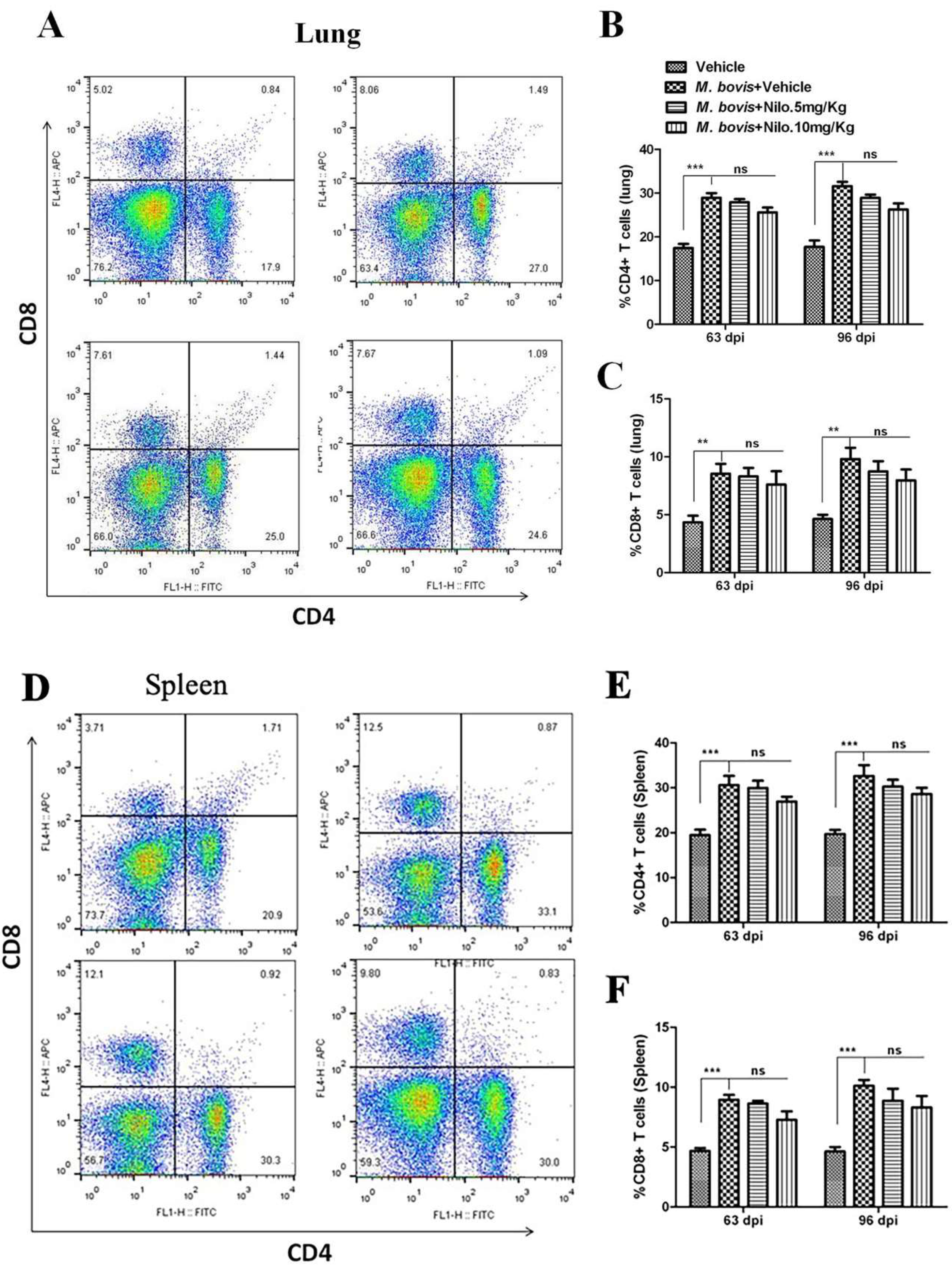
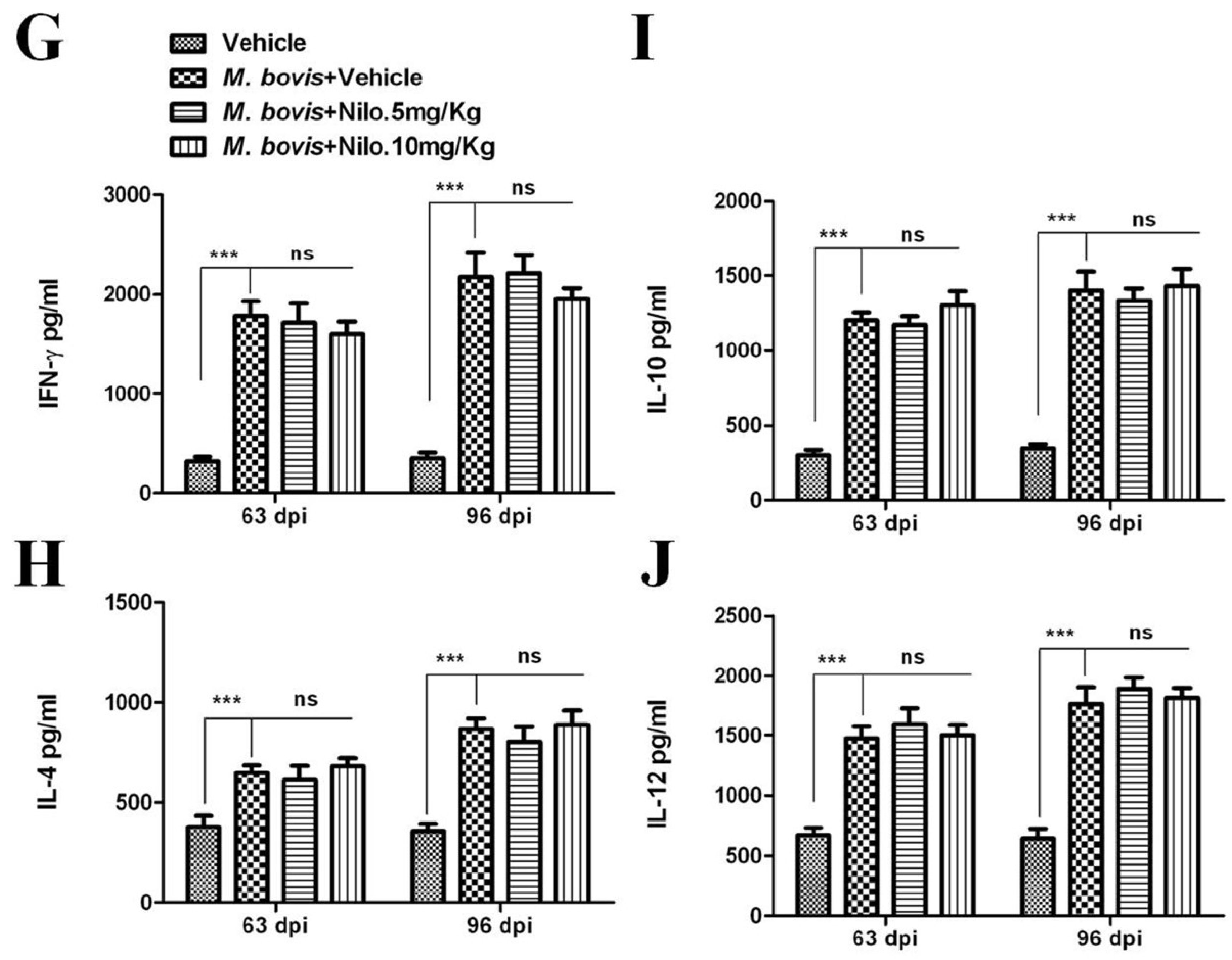
3.7. Nilotinib Reduces M. bovis Burden Irrespective of Modulating T-Cell Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashford, D.A.; Whitney, E.; Raghunathan, P.; Cosivi, O. Epidemiology of selected mycobacteria that infect humans and other animals. J. Revue Scientifique Et Technique De L Office International Des Epizooties 2001, 20, 325–337. [Google Scholar] [CrossRef]
- Grange, J.M. Mycobacterium bovis infection in human beings. J. Tuberc. 2001, 81, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J.E. Epidemiology and economics of paratuberculosis. J. Vet. Clin. North Am. Food Anim. Pract. 2011, 27, 525–535. [Google Scholar] [CrossRef]
- McNees, A.L.; Markesich, D.; Zayyani, N.R.; Graham, D.Y. Mycobacterium paratuberculosis as a cause of Crohn’s disease. J. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1523–1534. [Google Scholar] [CrossRef]
- Dalziel, T.K. Thomas Kennedy Dalziel 1861–1924. Chronic interstitial enteritis. J. Dis. Colon. Rectum. 1989, 32, 1076–1078. [Google Scholar] [CrossRef]
- Kuenstner, J.T.; Naser, S.; Chamberlin, W.; Borody, T.; Graham, D.Y.; McNees, A.; Hermon-Taylor, J.; Hermon-Taylor, A.; Dow, C.T.; Thayer, W.; et al. The Consensus from the Mycobacterium avium ssp. paratuberculosis (MAP) Conference 2017. J. Front. Public Health 2017, 5, 208. [Google Scholar] [CrossRef]
- McClean, M.C.; Tobin, D.M. Macrophage form, function, and phenotype in mycobacterial infection: Lessons from tuberculosis and other diseases. J. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [PubMed]
- Mosser, M.D.; Edwards, J.P. Exploring the full spectrum of macrophage activation. J. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Lekes, D.; Szadvari, I.; Krizanova, O.; Lopusna, K.; Rezuchova, I.; Novakova, M.; Novakova, Z.; Parak, T.; Babula, P. Nilotinib induces ER stress and cell death in H9c2 cells. J. Physiol Res. 2016, 65, S505–S514. [Google Scholar]
- Reinwald, M.; Boch, T.; Hofmann, W.K.; Buchheidt, D. Risk of Infectious Complications in Hemato-Oncological Patients Treated with Kinase Inhibitors. J. Biomark Insights 2015, 10, 55–68. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef]
- Yu, H.C.; Lin, C.S.; Tai, W.T.; Liu, C.Y.; Shiau, C.W.; Chen, K.F. Nilotinib Induces Autophagy in Hepatocellular Carcinoma through AMPK Activation. J. Biol. Chem. 2013, 288, 18249–18259. [Google Scholar] [CrossRef]
- Shaker, M.E.; Ghani, A.; Shiha, G.E.; Ibrahim, T.M.; Mehal, W.Z. Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. J. Biochim. Biophys. Acta 2013, 1833, 1992–2003. [Google Scholar] [CrossRef]
- Wyse, R.K.; Brundin, P.; Sherer, T.B. Nilotinib—Differentiating the Hope from the Hype. J. Parkinsons Dis. 2016, 6, 519–522. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Olopade, P.; Selby, S.T.; Pagan, F.; Moussa, C.E. Tyrosine Kinase Inhibition Regulates Early Systemic Immune Changes and Modulates the Neuroimmune Response in alpha-Synucleinopathy. J. Clin. Cell. Immunol. 2014, 5, 259. [Google Scholar] [CrossRef]
- Wilson, L.J.; Weichhart, T. TORching a semaphore for alternative macrophage activation. J. Nat. Immunol. 2018, 19, 512–514. [Google Scholar] [CrossRef]
- Wessler, S.; Backert, S. Abl Family of Tyrosine Kinases and Microbial Pathogenesis. J. Int. Rev. Cell Mol. Biol. 2011, 286, 271–300. [Google Scholar]
- Bruns, H.; Stegelmann, F.; Fabri, M.; Dohner, K.; van Zandbergen, G.; Wagner, M.; Skinner, M.; Modlin, R.L.; Stenger, S. Abelson Tyrosine Kinase Controls Phagosomal Acidification Required for Killing of Mycobacterium tuberculosis in Human Macrophages. J. Immunol. 2012, 189, 4069–4078. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Fauvet, B.; Gysbers, A.; Dikiy, I.; Oueslati, A.; Georgeon, S.; Lamontanara, A.J.; Bisquertt, A.; Eliezer, D.; Masliah, E.; et al. c-Abl phosphorylates alpha-synuclein and regulates its degradation: Implication for alpha-synuclein clearance and contribution to the pathogenesis of Parkinson’s disease. J. Hum. Mol. Genet. 2014, 23, 2858–2879. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of -synuclein in Parkinsons disease models. J. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. J. Nat. 2013, 501, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.B.; Lamark, T.; Overvatn, A.; Harneshaug, I.; Johansen, T.; Bjorkoey, G. A reporter cell system to monitor autophagy based on p62/SQSTM1. J. Autophagy 2010, 6, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. J. Mol. Aspects Med. 2006, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. J. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Subbian, S. Harnessing the mTOR Pathway for Tuberculosis Treatment. J. Front. Microbiol. 2018, 9, 70. [Google Scholar] [CrossRef]
- Airiau, K.; Mahon, F.X.; Josselin, M.; Jeanneteau, M.; Belloc, F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. J. Cell Death Dis. 2013, 4, e827. [Google Scholar] [CrossRef]
- Ly, C.; Arechiga, A.F.; Melo, J.V.; Walsh, C.M.; Ong, S.T. Bcr-Abl kinase modulates the translation regulators ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia cells via the mammalian target of rapamycin. J. Cancer Res. 2003, 63, 5716–5722. [Google Scholar]
- Chandra, P.; Rajmani, R.S.; Verma, G.; Bhavesh, N.S.; Kumar, D. Targeting Drug-Sensitive and -Resistant Strains of Mycobacterium tuberculosis by Inhibition of Src Family Kinases Lowers Disease Burden and Pathology. J. Msphere 2016, 1, e00043-15. [Google Scholar] [CrossRef]
- Yue, R.C.; Liu, C.F.; Barrow, P.; Liu, F.; Cui, Y.Y.; Yang, L.F.; Zhao, D.M.; Zhou, X.M. The isolation and molecular characterization of Mycobacterium avium subsp paratuberculosis in Shandong province, China. J. Gut Pathog. 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Boute, M.; Carreras, F.; Rossignol, C.; Doz, E.; Winter, N.; Epardaud, M. The C3HeB/FeJ mouse model recapitulates the hallmark of bovine tuberculosis lung lesions following Mycobacterium bovis aerogenous infection. J. Vet. Res. 2017, 48, 73. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.K.; Bohn, E.; Judd, B.A.; Gil, M.P.; Schreiber, R.D. ABIN-3: A molecular basis for species divergence in interleukin-10-induced anti-inflammatory actions. J. Mol. Cell. Biol. 2007, 27, 4603–4616. [Google Scholar] [CrossRef]
- Srinivasan, L.; Gurses, S.A.; Hurley, B.E.; Miller, J.L.; Karakousis, P.C.; Briken, V. Identification of a Transcription Factor That Regulates Host Cell Exit and Virulence of Mycobacterium tuberculosis. J. PLoS Pathog. 2016, 12, e1005652. [Google Scholar] [CrossRef] [PubMed]
- Pooley, H.B.; de Silva, K.; Purdie, A.C.; Begg, D.J.; Whittington, R.J.; Plain, K.M. A Rapid Method for Quantifying Viable Mycobacterium avium subsp. paratuberculosis in Cellular Infection Assays. J. Appl. Environ. Microbiol. 2016, 82, 5553–5562. [Google Scholar] [CrossRef] [PubMed]
- Bai, W.; Liu, H.; Ge, B.; Kaufmann, S.H.E. TLR3 regulates mycobacterial RNA-induced IL-10 production through the PI3K/AKT signaling pathway. Eur. J. Clin. Investig. 2014, 44, 27. [Google Scholar] [CrossRef] [PubMed]
- Shaker, M.E. Nilotinib interferes with the signalling pathways implicated in acetaminophen hepatotoxicity. J. Basic Clin. Pharmacol. Toxicol. 2014, 114, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Zhao, D.; Shah, S.Z.A.; Wang, J.; Yue, R.; Liao, Y.; Sabir, N.; Yang, L.; Zhou, X. MicroRNA 27a-3p Regulates Antimicrobial Responses of Murine Macrophages Infected by Mycobacterium avium subspecies paratuberculosis by Targeting Interleukin-10 and TGF-beta-Activated Protein Kinase 1 Binding Protein 2. J. Front. Immunol. 2017, 8, 1915. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Zhao, D.M.; Taglialatela, G.; Khan, S.H.; Hussain, T.; Dong, H.D.; Lai, M.Y.; Zhou, X.M.; Yang, L.F. Early Minocycline and Late FK506 Treatment Improves Survival and Alleviates Neuroinflammation, Neurodegeneration, and Behavioral Deficits in Prion-Infected Hamsters. J. Neurotherap. 2017, 14, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Mahadik, K.; Prakhar, P.; Rajmani, R.S.; Singh, A.; Balaji, K.N. c-Abl-TWIST1 Epigenetically Dysregulate Inflammatory Responses during Mycobacterial Infection by Co-Regulating Bone Morphogenesis Protein and miR27a. J. Front. Immunol. 2018, 9, 85. [Google Scholar] [CrossRef]
- Wang, J.; Hussain, T.; Yue, R.; Liao, Y.; Li, Q.; Yao, J.; Song, Y.; Sun, X.; Wang, N.; Xu, L.; et al. MicroRNA-199a Inhibits Cellular Autophagy and Downregulates IFN-beta Expression by Targeting TBK1 in Mycobacterium bovis Infected Cells. J. Front. Cell Infect. Microbiol. 2018, 8, 238. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. J. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef]
- Serbina, N.V.; Liu, C.C.; Scanga, C.A.; Flynn, L.J. CD8+ CTL from lungs of Mycobacterium tuberculosis-infected mice express perforin in vivo and lyse infected macrophages. J. Immunol. 2000, 165, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Yu, Y.; Schmitt, A.; Rojewski, M.T.; Chen, B.; Greiner, J.; Gotz, M.; Bunjes, D.; Schmitt, M. Effects of nilotinib on regulatory T cells: The dose matters. J. Mol. Cancer 2010, 9, 22. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. J. Cell Signal 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, K.; Shi, X.; Wang, C.; Wang, F.; Fan, J.; Shen, F.; Xu, J.; Bao, W.; Liu, M.; et al. Critical role of bacterial isochorismatase in the autophagic process induced by Acinetobacter baumannii in mammalian cells. FASEB J. 2016, 30, 3563–3577. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.; Mestan, J.; Cowan-Jacob, S.; Ray, A.; Griffin, J.D. AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL. Br. J. Cancer 2006, 94, 1765–1769. [Google Scholar] [CrossRef]
- Garcia, M.; Cooper, A.; Shi, W.; Bornmann, W.; Carrion, R.; Kalman, D.; Nabel, G.J. Productive Replication of Ebola Virus Is Regulated by the c-Abl1 Tyrosine Kinase. J. Sci. Transl. Med. 2012, 4, 123ra24. [Google Scholar] [CrossRef] [PubMed]
- Napier, R.J.; Rafi, W.; Cheruvu, M.; Powell, K.R.; Zaunbrecher, M.A.; Bornmann, W.; Salgame, P.; Shinnick, T.M.; Kalman, D. Imatinib-sensitive tyrosine kinases regulate mycobacterial pathogenesis and represent therapeutic targets against tuberculosis. J. Cell Host Microbe 2011, 10, 475–485. [Google Scholar] [CrossRef]
- Imam, S.Z.; Zhou, Q.; Yamamoto, A.; Valente, A.J.; Ali, S.F.; Bains, M.; Roberts, J.L.; Kahle, P.J.; Clark, R.A.; Li, S.L. Novel Regulation of Parkin Function through c-Abl-Mediated Tyrosine Phosphorylation: Implications for Parkinson’s Disease. J. Neurosci. 2011, 31, 157–163. [Google Scholar] [CrossRef]
- Sia, J.K.; Bizzell, E.; Madan-Lala, R.; Rengarajan, J. Engaging the CD40-CD40L pathway augments T-helper cell responses and improves control of Mycobacterium tuberculosis infection. J. PLoS Pathog. 2017, 13, e1006530. [Google Scholar] [CrossRef]
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Autophagy in Human Health and Disease REPLY. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.C.; Yao, Z.Y.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. J. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Steiger, J.; Stephan, A.; Inkeles, M.S.; Realegeno, S.; Bruns, H.; Kroll, P.; Kroner, J.d.; Sommer, A.; Batinica, M.; Pitzler, L.; et al. Imatinib Triggers Phagolysosome Acidification and Antimicrobial Activity against Mycobacterium bovis Bacille Calmette-Guerin in Glucocorticoid-Treated Human Macrophages. J. Immunol. 2016, 197, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J. M-tuberculosis and M-leprae translocate from the phagolysosome to the cytosol in myeloid cells. J. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Barczak, A.K.; Silvis, M.R.; Luo, S.S.; Sogi, K.; Vokes, M.; Bray, M.A.; Carpenter, A.E.; Moore, C.B.; Siddiqi, N.; et al. Identification of Host-Targeted Small Molecules That Restrict Intracellular Mycobacterium tuberculosis Growth. J. PLoS Pathog. 2014, 10, e1003946. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, M.A.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. J. Neuron. 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F.R.G.; Lepelley, A.; Seeley, J.J.; Hayden, M.S.; Ghosh, S. An Essential Role for ECSIT in Mitochondrial Complex I Assembly and Mitophagy in Macrophages. J. Cell Rep. 2018, 22, 2654–2666. [Google Scholar] [CrossRef]
- Franco, L.H.; Nair, V.R.; Scharn, C.R.; Xavier, R.J.; Torrealba, J.R.; Shiloh, M.U.; Levine, B. The Ubiquitin Ligase Smurf1 Functions in Selective Autophagy of Mycobacterium tuberculosis and Anti-tuberculous Host Defense. J. Cell Host Microbe 2017, 22, 421–423. [Google Scholar] [CrossRef]
- Pei, G.; Buijze, H.; Liu, H.P.; Moura-Alves, P.; Goosmann, C.; Brinkmann, V.; Kawabe, H.; Dorhoi, A.; Kaufmann, S.H.E. The E3 ubiquitin ligase NEDD4 enhances killing of membrane-perturbing intracellular bacteria by promoting autophagy. J. Autophagy 2017, 13, 2041–2055. [Google Scholar] [CrossRef]
- Ishiyama, A.; Iwatsuki, M.; Hokari, R.; Sawa, M.; Omura, S.; Otoguro, K. Antimalarial activity of kinase inhibitor, nilotinib, in vitro and in vivo. J. Antibiot. 2015, 68, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Grangeasse, C.; Nessler, S.; Mijakovic, I. Bacterial tyrosine kinases: Evolution, biological function and structural insights. J. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2640–2655. [Google Scholar] [CrossRef] [PubMed]
- Bach, H.; Wong, D.; Av-Gay, Y. Mycobacterium tuberculosis PtkA is a novel protein tyrosine kinase whose substrate is PtpA. Biochem. J. 2009, 420, 155–160. [Google Scholar] [CrossRef]
- Napier, R.J.; Norris, B.A.; Swimm, A.; Giver, C.R.; Harris, W.A.; Laval, J.; Napier, B.A.; Patel, G.; Crump, R.; Peng, Z.; et al. Low doses of imatinib induce myelopoiesis and enhance host anti-microbial immunity. J. PLoS Pathog. 2015, 11, e1004770. [Google Scholar] [CrossRef]
- Varda-Bloom, N.; Somech, R.; Vokhek, Y.; Davidson, J.; Lev, A.; Yerushalmi, R.; Shimoni, A.; Nagler, A. Assessment of the Effect of Nilotinib (Tasigna) Maintenance Therapy After Allogeneic Stem Cell Transplantation in Patients with Advanced CML and Ph plus ALL On Immune Reconstitution and Lymphocyte Function. J. Blood 2012, 120, 21. [Google Scholar] [CrossRef][Green Version]
- Wehrstedt, S.; Kubis, J.; Zimmermann, A.; Bruns, H.; Mayer, D.; Grieshober, M.; Stenger, S. The tyrosine kinase inhibitor dasatinib reduces the growth of intracellular Mycobacterium tuberculosis despite impairing T-cell function. J. Eur. J. Immunol. 2018, 48, 1892–1903. [Google Scholar] [CrossRef] [PubMed]













© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, T.; Zhao, D.; Shah, S.Z.A.; Sabir, N.; Wang, J.; Liao, Y.; Song, Y.; Dong, H.; Hussain Mangi, M.; Ni, J.; et al. Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy. Cells 2019, 8, 506. https://doi.org/10.3390/cells8050506
Hussain T, Zhao D, Shah SZA, Sabir N, Wang J, Liao Y, Song Y, Dong H, Hussain Mangi M, Ni J, et al. Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy. Cells. 2019; 8(5):506. https://doi.org/10.3390/cells8050506
Chicago/Turabian StyleHussain, Tariq, Deming Zhao, Syed Zahid Ali Shah, Naveed Sabir, Jie Wang, Yi Liao, Yinjuan Song, Haodi Dong, Mazhar Hussain Mangi, Jiamin Ni, and et al. 2019. "Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy" Cells 8, no. 5: 506. https://doi.org/10.3390/cells8050506
APA StyleHussain, T., Zhao, D., Shah, S. Z. A., Sabir, N., Wang, J., Liao, Y., Song, Y., Dong, H., Hussain Mangi, M., Ni, J., Yang, L., & Zhou, X. (2019). Nilotinib: A Tyrosine Kinase Inhibitor Mediates Resistance to Intracellular Mycobacterium Via Regulating Autophagy. Cells, 8(5), 506. https://doi.org/10.3390/cells8050506