mTOR Signaling Pathway in Cancer Targets Photodynamic Therapy In Vitro



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
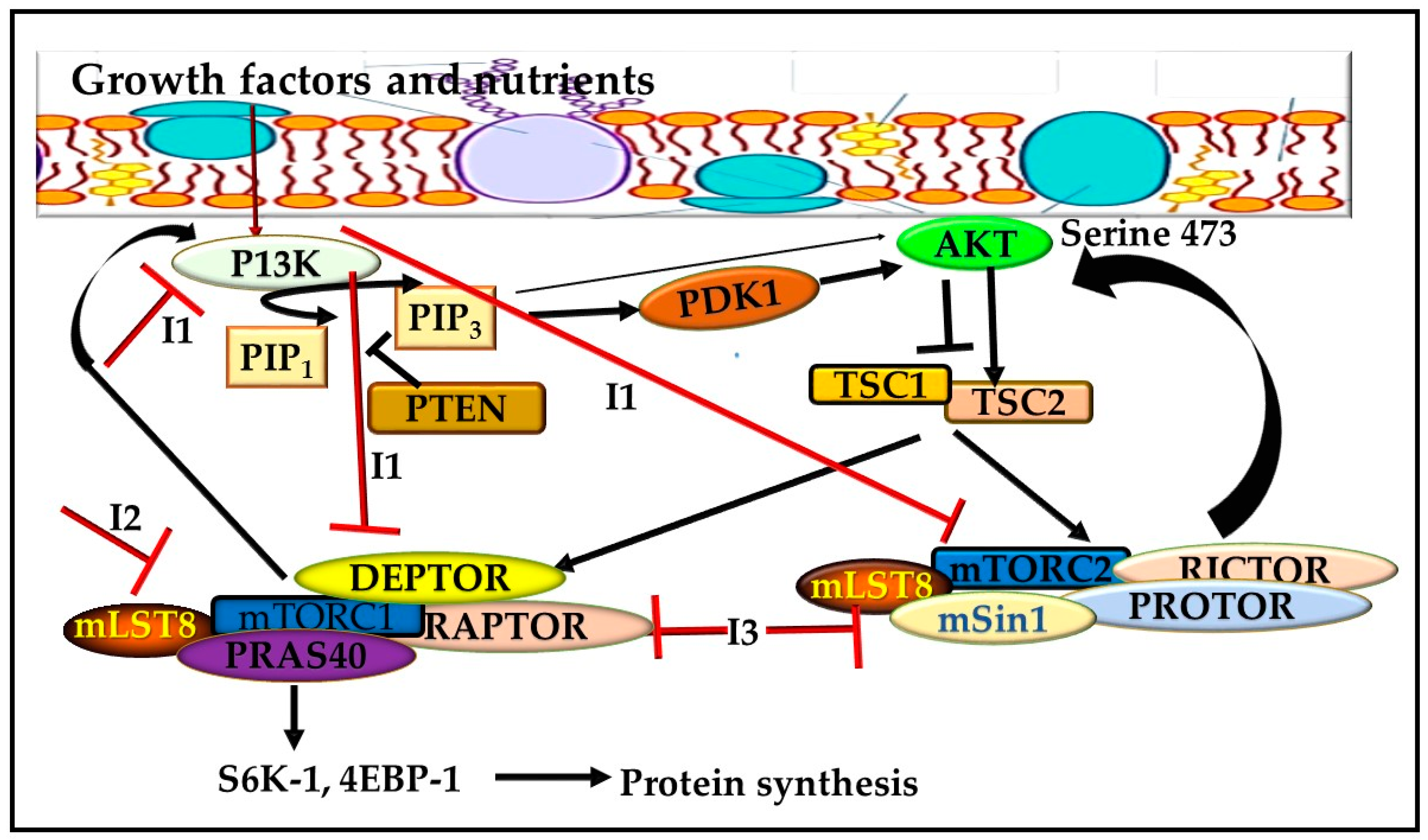
2. The mTOR Pathway
3. The Role of mTOR Inhibitors in Cancer
4. The Role of mTOR Pathway in Cancer Therapy
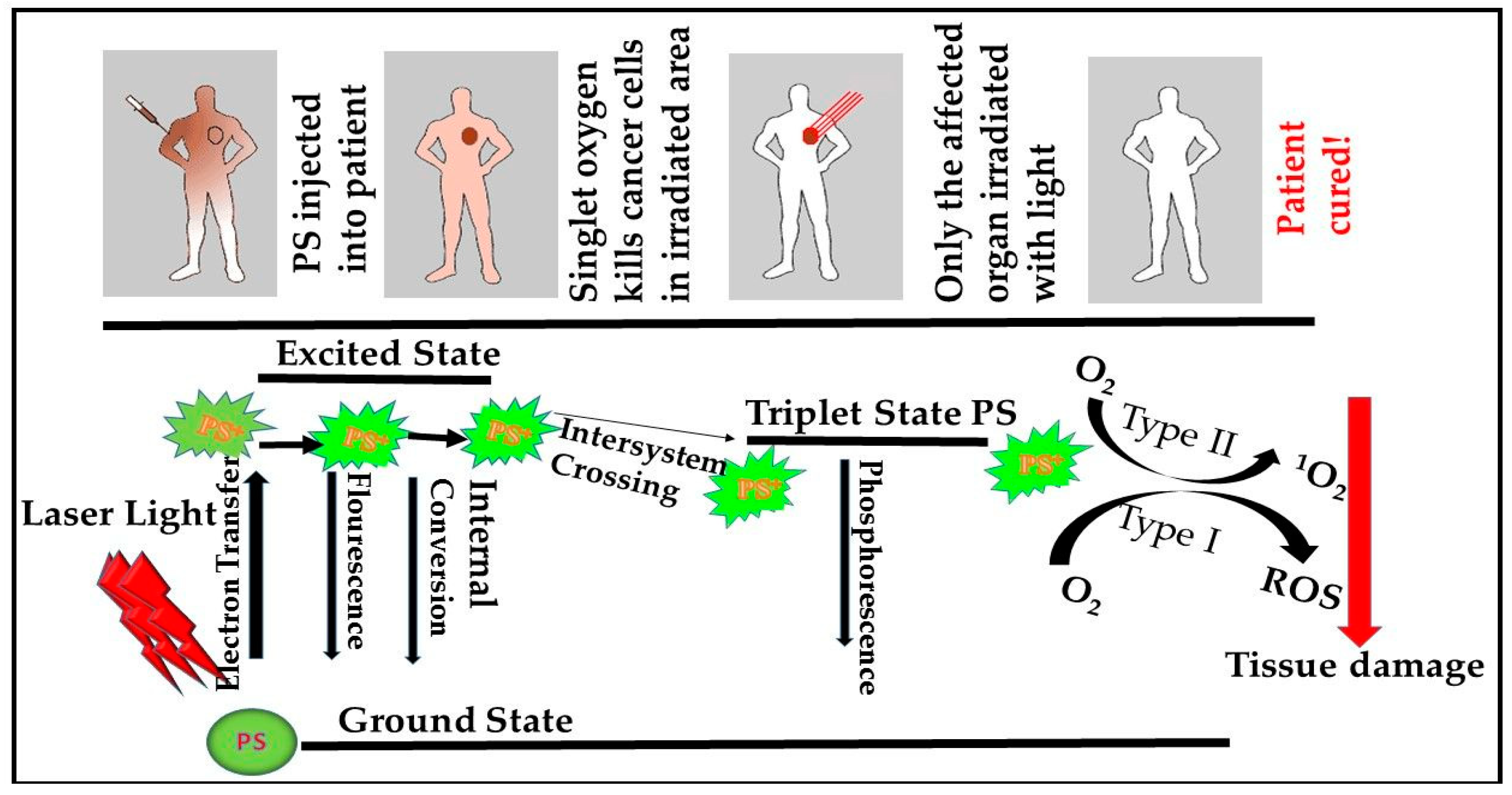
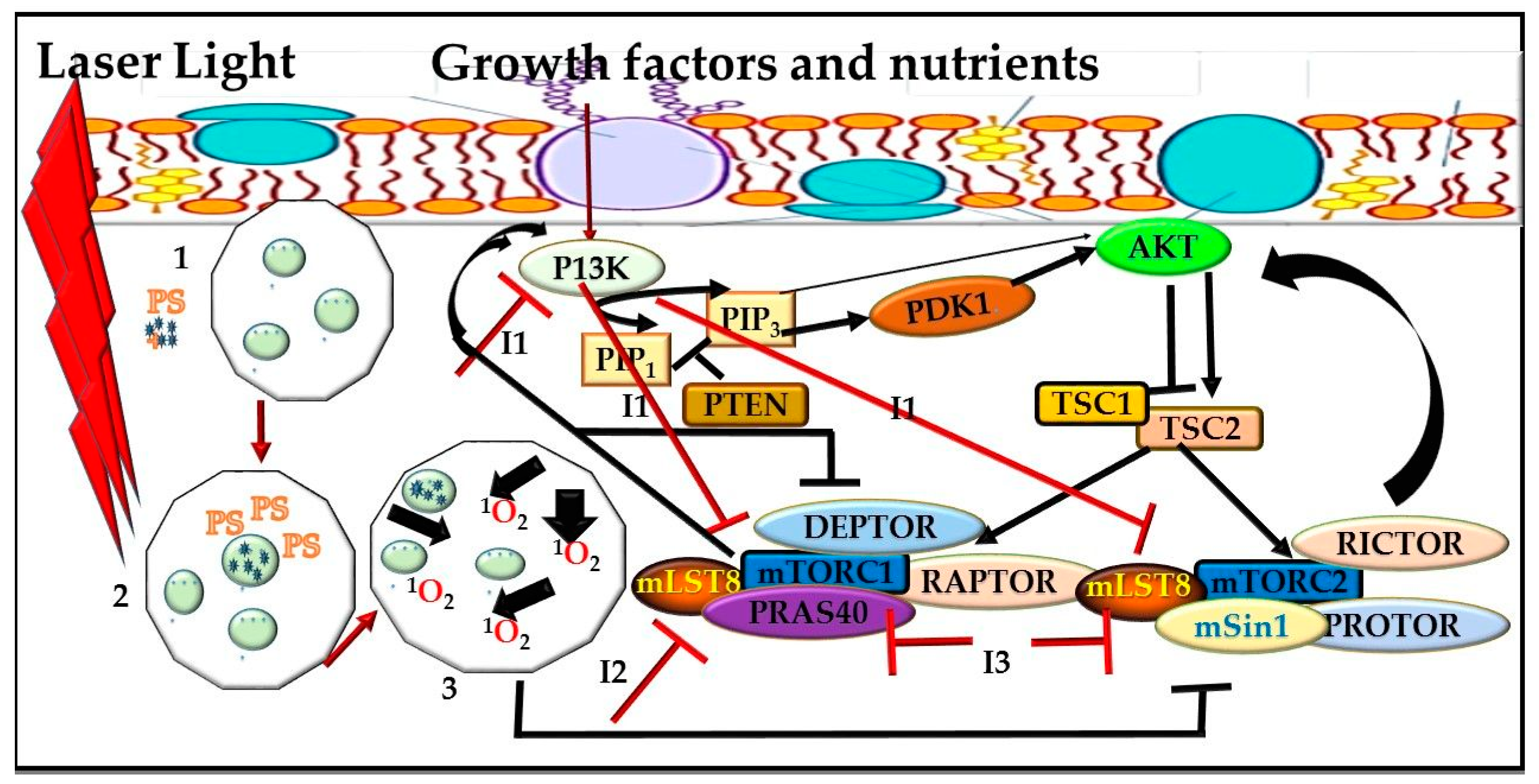
mTOR Signaling Pathway and PDT
5. Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Gomez-Pinillos, A.; Ferrari, A.C. mTOR signaling pathway and mTOR inhibitors in cancer therapy. Hematol. Oncol. N. Am. 2012, 26, 483–505. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Gonzalez-Angulo, A.M. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 121–130. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497. [Google Scholar] [CrossRef]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Evers, B.M. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef]
- Cheng, H.; Walls, M.; Baxi, S.; Yin, M.-J. Targeting the mTOR pathway in tumor malignancy. Curr. Cancer Drug Targets 2013, 13, 267–277. [Google Scholar] [CrossRef]
- Li, T.; Wang, G. Computer-aided targeting of the PI3K/Akt/mTOR pathway: Toxicity reduction and therapeutic opportunities. Int. J. Mol. Sci. 2014, 15, 18856–18891. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Don, A.S.; Zheng, X.F. Recent clinical trials of mTOR-targeted cancer therapies. Rev. Recent Clin. Trials 2011, 6, 24–35. [Google Scholar]
- Volanti, C.; Hendrickx, N.; Van Lint, J.; Matroule, J.-Y.; Agostinis, P.; Piette, J. Distinct transduction mechanisms of cyclooxygenase 2 gene activation in tumour cells after photodynamic therapy. Oncogene 2005, 24, 2981. [Google Scholar] [CrossRef]
- Bozkulak, O.; Wong, S.; Luna, M.; Ferrario, A.; Rucker, N.; Gulsoy, M.; Gomer, C.J. Multiple components of photodynamic therapy can phosphorylate Akt. Photochem. Photobiol. 2007, 83, 1029–1033. [Google Scholar] [CrossRef]
- Koon, H.K.; Chan, P.S.; Wong, R.N.S.; Wu, Z.G.; Lung, M.L.; Chang, C.K.; Mak, N.K. Targeted inhibition of the EGFR pathways enhances Zn-BC-AM PDT-induced apoptosis in well-differentiated nasopharyngeal carcinoma cells. J. Cell. Biochem. 2009, 108, 1356–1363. [Google Scholar] [CrossRef]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. 2009, 27, 2278. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Duan, Y.; Zheng, X.F.S. Targeting the mTOR kinase domain: The second generation of mTOR inhibitors. Drug Discov. Today 2011, 16, 325–331. [Google Scholar] [CrossRef]
- van Straten, D.; Mashayekhi, V.; de Bruijn, H.; Oliveira, S.; Robinson, D. Oncologic photodynamic therapy: Basic principles, current clinical status and future directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Ahn, J.-C.; Biswas, R.; Chung, P.-S. Synergistic effect of radachlorin mediated photodynamic therapy on propolis induced apoptosis in AMC-HN-4 cell lines via caspase dependent pathway. Photodiagn. Photodyn. Ther. 2013, 10, 236–243. [Google Scholar] [CrossRef]
- Allison, R.R.; Moghissi, K. Photodynamic therapy (PDT): PDT mechanisms. Clin. Endosc. 2013, 46, 24. [Google Scholar] [CrossRef]
- Baptista, M.S.; Cadet, J.; Di Mascio, P.; Ghogare, A.A.; Greer, A.; Hamblin, M.R.; Lorente, C.; Nunez, S.C.; Ribeiro, M.S.; Thomas, A.H. Type I and type II photosensitized oxidation reactions: Guidelines and mechanistic pathways. Photochem. Photobiol. 2017, 93, 912–919. [Google Scholar] [CrossRef]
- Velloso, N.V.; Muehlmann, L.A.; Longo, J.P.F.; Silva, J.R.; Zancanela, D.C.; Tedesco, A.C.; Azevedo, R.B.D. Aluminum-phthalocyanine chloride-based photodynamic therapy inhibits PI3K/Akt/Mtor pathway in oral squamous cell carcinoma cells in vitro. Chemotherapy 2012, 1, 5. [Google Scholar]
- Fateye, B.; Li, W.; Wang, C.; Chen, B. Combination of Phosphatidylinositol 3-Kinases Pathway Inhibitor and Photodynamic Therapy in Endothelial and Tumor Cells. Photochem. Photobiol. 2012, 88, 1265–1272. [Google Scholar] [CrossRef]
- Fateye, B.; Wan, A.; Yang, X.; Myers, K.; Chen, B. Comparison between endothelial and tumor cells in the response to verteporfin-photodynamic therapy and a PI3K pathway inhibitor. Photodiagn. Photodyn. Ther. 2015, 12, 19–26. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef]
- Weyergang, A.; Berg, K.; Kaalhus, O.; Peng, Q.; Selbo, P.K. Photodynamic therapy targets the mTOR signaling network in vitro and in vivo. Mol. Pharm. 2008, 6, 255–264. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Blouet, C.; Ono, H.; Schwartz, G.J. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab. 2008, 8, 459–467. [Google Scholar] [CrossRef]
- Qian, Y.; Kun-liang, G. Expanding mTOR signaling. Cell Res. 2007, 17, 666–681. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Cantley, L.C.; Engelman, J.A.; Luo, J. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155. [Google Scholar] [CrossRef]
- Russell, R.C.; Fang, C.; Guan, K.-L. An emerging role for TOR signaling in mammalian tissue and stem cell physiology. Development 2011, 138, 3343–3356. [Google Scholar] [CrossRef]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295. [Google Scholar] [CrossRef]
- Verbist, K.C.; Guy, C.S.; Milasta, S.; Liedmann, S.; Kamiński, M.M.; Wang, R.; Green, D.R. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 2016, 532, 389. [Google Scholar] [CrossRef]
- Vadlakonda, L.; Dash, A.; Pasupuleti, M.; Kotha, A.K.; Reddanna, P. The paradox of Akt-mTOR interactions. Front. Oncol. 2013, 3, 165. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. The pharmacology of mTOR inhibition. Sci. Signal. 2009, 2, pe24. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5, F1000. [Google Scholar] [CrossRef]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22, 989), a new antifungal antibiotic. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Singh, K.; Sun, S.; Vezina, C. Rapamycin (AY-22, 989), A New Antifungal Antibiotic. J. Antibiot. 1979, 32, 630–645. [Google Scholar] [CrossRef]
- Tsang, C.K.; Qi, H.; Liu, L.F.; Zheng, X.F.S. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov. Today 2007, 12, 112–124. [Google Scholar] [CrossRef]
- Shafer, A.; Zhou, C.; Gehrig, P.A.; Boggess, J.F.; Bae-Jump, V.L. Rapamycin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and induction of apoptosis. Int. J. Cancer 2010, 126, 1144–1154. [Google Scholar] [CrossRef]
- Kunz, J.; Henriquez, R.; Schneider, U.; Deuter-Reinhard, M.; Movva, N.R.; Hall, M.N. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell 1993, 73, 585–596. [Google Scholar] [CrossRef]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.-i.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Aylett, C.H.S.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mTOR complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef]
- Yuan, H.-X.; Guan, K.-L. Structural insights of mTOR complex 1. Cell Res. 2016, 26, 267. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Sabatini, D.M. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009, 5, 725–726. [Google Scholar] [CrossRef]
- Calimeri, T.; Ferreri, A.J.M. m-TOR inhibitors and their potential role in haematological malignancies. Br. J. Haematol. 2017, 177, 684–702. [Google Scholar] [CrossRef]
- Pinto-Leite, R.; Arantes-Rodrigues, R.; Sousa, N.; Oliveira, P.A.; Santos, L. mTOR inhibitors in urinary bladder cancer. Tumour Biol. 2016, 37, 11541–11551. [Google Scholar] [CrossRef]
- Liu, L.; Luo, Y.; Chen, L.; Shen, T.; Xu, B.; Chen, W.; Zhou, H.; Han, X.; Huang, S. Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J. Biol. Chem. 2010, 285, 38362–38373. [Google Scholar] [CrossRef]
- Alvarado, Y.; Mita, M.M.; Vemulapalli, S.; Mahalingam, D.; Mita, A.C. Clinical activity of mammalian target of rapamycin inhibitors in solid tumors. Target. Oncol. 2011, 6, 69–94. [Google Scholar] [CrossRef]
- Gulhati, P.; Cai, Q.; Li, J.; Liu, J.; Rychahou, P.G.; Qiu, S.; Lee, E.Y.; Silva, S.R.; Bowen, K.A.; Gao, T. Targeted inhibition of mammalian target of rapamycin signaling inhibits tumorigenesis of colorectal cancer. Clin. Cancer Res. 2009, 15, 7207–7216. [Google Scholar] [CrossRef]
- Roulin, D.; Cerantola, Y.; Dormond-Meuwly, A.; Demartines, N.; Dormond, O. Targeting mTORC2 inhibits colon cancer cell proliferation in vitro and tumor formation in vivo. Mol. Cancer 2010, 9, 57. [Google Scholar] [CrossRef]
- Wu, W.K.K.; Lee, C.W.; Cho, C.H.; Chan, F.K.L.; Yu, J.; Sung, J.J.Y. RNA interference targeting raptor inhibits proliferation of gastric cancer cells. Exp. Cell Res. 2011, 317, 1353–1358. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nature Reviews Immunology 2009, 9, 324. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.; Fantus, D.; Solari, M.; Thomson, A.W. New perspectives on mTOR inhibitors (rapamycin, rapalogs and TORKinibs) in transplantation. Br. J. Clin. Pharmacol. 2016, 82, 1158–1170. [Google Scholar] [CrossRef]
- Habib, S.L.; Al-Obaidi, N.Y.; Nowacki, M.; Pietkun, K.; Zegarska, B.; Kloskowski, T.; Zegarski, W.; Drewa, T.; Medina, E.A.; Zhao, Z. Is mTOR inhibitor good enough for treatment all tumors in TSC patients? J. Cancer 2016, 7, 1621. [Google Scholar] [CrossRef]
- Sasongko, T.H.; Ismail, N.F.D.; Zabidi-Hussin, Z. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef]
- Chiarini, F.; Evangelisti, C.; McCubrey, J.A.; Martelli, A.M. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol. Sci. 2015, 36, 124–135. [Google Scholar] [CrossRef]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris Iii, H.A.; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F. Everolimus in postmenopausal hormone-receptor–positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; De Vries, E.G.E. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Hess, G.; Herbrecht, R.; Romaguera, J.; Verhoef, G.; Crump, M.; Gisselbrecht, C.; Laurell, A.; Offner, F.; Strahs, A.; Berkenblit, A. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2009, 27, 3822–3829. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.-H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef]
- Masri, J.; Bernath, A.; Martin, J.; Jo, O.D.; Vartanian, R.; Funk, A.; Gera, J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007, 67, 11712–11720. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; Marsh, C.A.; Dorsam, R.T.; Mikelis, C.M.; Masedunskas, A.; Amornphimoltham, P.; Nathan, C.A.; Singh, B.; Weigert, R.; Molinolo, A.A. Decreased lymphangiogenesis and lymph node metastasis by mTOR inhibition in head and neck cancer. Cancer Res. 2011, 71, 7103–7112. [Google Scholar] [CrossRef]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Hörsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Burris Iii, H.A.; Lebrun, F.; Rugo, H.S.; Beck, J.T.; Piccart, M.; Neven, P.; Baselga, J.; Petrakova, K.; Hortobagyi, G.N.; Komorowski, A. Health-related quality of life of patients with advanced breast cancer treated with everolimus plus exemestane versus placebo plus exemestane in the phase 3, randomized, controlled, BOLERO-2 trial. Cancer 2013, 119, 1908–1915. [Google Scholar] [CrossRef]
- André, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. The lancet oncology 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Dalenc, F.; Campone, M.; O’Regan, R.M.; Tjan-Heijnen, V.C.; Gligorov, J.; Llombart, A.; Jhangiani, H.; Mirshahidi, H.R.; Tan-Chiu, E. A phase 2 study of everolimus combined with trastuzumab and paclitaxel in patients with HER2-overexpressing advanced breast cancer that progressed during prior trastuzumab and taxane therapy. Breast Cancer Res. Treat. 2013, 141, 437–446. [Google Scholar] [CrossRef]
- Temkin, S.M.; Fleming, G. Current treatment of metastatic endometrial cancer. Cancer Control. 2009, 16, 38–45. [Google Scholar] [CrossRef]
- Tinker, A.V.; Ellard, S.; Welch, S.; Moens, F.; Allo, G.; Tsao, M.S.; Squire, J.; Tu, D.; Eisenhauer, E.A.; MacKay, H. Phase II study of temsirolimus (CCI-779) in women with recurrent, unresectable, locally advanced or metastatic carcinoma of the cervix. A trial of the NCIC Clinical Trials Group (NCIC CTG IND 199). Gynecol. Oncol. 2013, 130, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Takatori, E.; Shoji, T.; Miura, Y.; Takada, A.; Takeuchi, S.; Sugiyama, T. Effective use of everolimus as salvage chemotherapy for ovarian clear cell carcinoma: A case report. Onco Targets Ther. 2014, 7, 165. [Google Scholar]
- Behbakht, K.; Sill, M.W.; Darcy, K.M.; Rubin, S.C.; Mannel, R.S.; Waggoner, S.; Schilder, R.J.; Cai, K.Q.; Godwin, A.K.; Alpaugh, R.K. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: A Gynecologic Oncology Group study. Gynecol. Oncol. 2011, 123, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; McMeekin, S.; Schwartz, P.; Kostka, J.; Sessa, C.; Gehrig, P.; Holloway, R.; Braly, P.; Matei, D.; Einstein, M. A phase II trial of the mTOR inhibitor AP23573 as a single agent in advanced endometrial cancer. J. Clin. Oncol. 2007, 25, 5516. [Google Scholar]
- Mackay, H.; Welch, S.; Tsao, M.S.; Biagi, J.J.; Elit, L.; Ghatage, P.; Martin, L.A.; Tonkin, K.S.; Ellard, S.; Lau, S.K. Phase II study of oral ridaforolimus in patients with metastatic and/or locally advanced recurrent endometrial cancer: NCIC CTG IND 192. J. Clin. Oncol. 2011, 29, 5013. [Google Scholar] [CrossRef]
- Vignot, S.; Faivre, S.; Aguirre, D.; Raymond, E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann. Oncol. 2005, 16, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Duran, I.; Siu, L.L.; Oza, A.M.; Chung, T.B.; Sturgeon, J.; Townsley, C.A.; Pond, G.R.; Seymour, L.; Niroumand, M. Characterisation of the lung toxicity of the cell cycle inhibitor temsirolimus. Eur. J. Cancer 2006, 42, 1875–1880. [Google Scholar] [CrossRef]
- Leary, A.; Auclin, E.; Pautier, P.; Lhommé, C. The PI3K/Akt/mTOR pathway in ovarian cancer: Biological rationale and therapeutic opportunities. In Ovarian Cancer-A Clinical and Translational Update; Díaz-Padilla, I., Ed.; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef]
- Cho, D.C.; Cohen, M.B.; Panka, D.J.; Collins, M.; Ghebremichael, M.; Atkins, M.B.; Signoretti, S.; Mier, J.W. The efficacy of the novel dual PI3-kinase/mTOR inhibitor NVP-BEZ235 compared with rapamycin in renal cell carcinoma. Clin. Cancer Res. 2010, 16, 3628–3638. [Google Scholar] [CrossRef]
- Blaser, B.; Waselle, L.; Dormond-Meuwly, A.; Dufour, M.; Roulin, D.; Demartines, N.; Dormond, O. Antitumor activities of ATP-competitive inhibitors of mTOR in colon cancer cells. BMC Cancer 2012, 12, 86. [Google Scholar] [CrossRef]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Hennessy, B.T.; Slingerland, J.M. Next-generation mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J. Clin. Investig. 2011, 121, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272. [Google Scholar] [CrossRef] [PubMed]
- Steuer-Vogt, M.K.; Bonkowsky, V.; Ambrosch, P.; Scholz, M.; Neiβ, A.; Strutz, J.; Hennig, M.; Lenarz, T.; Arnold, W. The effect of an adjuvant mistletoe treatment programme in resected head and neck cancer patients: A randomised controlled clinical trial. Eur. J. Cancer 2001, 37, 23–31. [Google Scholar] [CrossRef]
- Huang, S.; Houghton, P.J. Inhibitors of mammalian target of rapamycin as novel antitumor agents: From bench to clinic. Curr. Opin. Investig. Drugs 2002, 3, 295–304. [Google Scholar]
- Dancey, J.E. Clinical development of mammalian target of rapamycin inhibitors. Hematol./Oncol. Clin. 2002, 16, 1101–1114. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Hardt, M.; Chantaravisoot, N.; Tamanoi, F. Activating mutations of TOR (target of rapamycin). Genes Cells 2011, 16, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Chiang, G.G.; Abraham, R.T. Targeting the mTOR signaling network in cancer. Trends Mol. Med. 2007, 13, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Salazar, R.; Halperín, I.; Abad, A.; Yao, J.C. Innovations therapy: Mammalian target of rapamycin (mTOR) inhibitors for the treatment of neuroendocrine tumors. Cancer Metastasis Rev. 2011, 30, 27–34. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Song, J.; Wang, X.; Weiss, H.L.; Townsend, C.M., Jr.; Gao, T.; Evers, B.M. mTORC1 inhibition increases neurotensin secretion and gene expression through activation of the MEK/ERK/c-Jun pathway in the human endocrine cell line BON. Am. J. Phys.-Cell Phys. 2011, 301, C213–C226. [Google Scholar]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef]
- Balgi, A.D.; Diering, G.H.; Donohue, E.; Lam, K.K.Y.; Fonseca, B.D.; Zimmerman, C.; Numata, M.; Roberge, M. Regulation of mTORC1 signaling by pH. PLoS ONE 2011, 6, e21549. [Google Scholar] [CrossRef] [PubMed]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Lamming, D.W. The mechanistic target of rapamycin: The grand conducTOR of metabolism and aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Gulhati, P.; Bowen, K.A.; Liu, J.; Stevens, P.D.; Rychahou, P.G.; Chen, M.; Lee, E.Y.; Weiss, H.L.; O’Connor, K.L.; Gao, T. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011, 71, 3246–3256. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, S. mTOR signaling in cancer cell motility and tumor metastasis. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 1–16. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954. [Google Scholar] [CrossRef]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef]
- Inoki, K.; Corradetti, M.N.; Guan, K.-L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19. [Google Scholar] [CrossRef]
- Mabuchi, S.; Kawase, C.; Altomare, D.A.; Morishige, K.; Sawada, K.; Hayashi, M.; Tsujimoto, M.; Yamoto, M.; Klein-Szanto, A.J.; Schilder, R.J. mTOR is a promising therapeutic target both in cisplatin-sensitive and cisplatin-resistant clear cell carcinoma of the ovary. Clin. Cancer Res. 2009, 15, 5404–5413. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.-L.; Lai, P.-S.; Lin, F.-H.; Wu, S.Y.-H.; Shieh, M.-J. Dual chemotherapy and photodynamic therapy in an HT-29 human colon cancer xenograft model using SN-38-loaded chlorin-core star block copolymer micelles. Biomaterials 2009, 30, 3614–3625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, L.; Yin, Q.; Zhang, Z.; Feng, L.; Li, Y. Transferrin-conjugated polyphosphoester hybrid micelle loading paclitaxel for brain-targeting delivery: Synthesis, preparation and in vivo evaluation. J. Control. Release 2012, 159, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; De Jesus, J. Oncogenic EGFR signaling activates an mTORC2–NF-κB pathway that promotes chemotherapy resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.R.; Karim, S.A.; Sano, M.; Gay, D.M.; Jacob, W.; Yu, J.; Mizukami, Y.; Gopinathan, A.; Jodrell, D.I.; Evans, T.R.J. mTORC2 signaling drives the development and progression of pancreatic cancer. Cancer Res. 2016, 76, 6911–6923. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Jänne, P.A. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012, 2, 214–226. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Carvalho, A.P.; Duarte, C.B. Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2004, 1704, 59–86. [Google Scholar] [CrossRef]
- Kraus, D.; Palasuberniam, P.; Chen, B. Targeting phosphatidylinositol 3-kinase signaling pathway for therapeutic enhancement of vascular-targeted photodynamic therapy. Mol. Cancer Ther. 2017, 16, 2422–2431. [Google Scholar] [CrossRef] [PubMed]
- Sasore, T.; Kennedy, B. Deciphering Combinations of PI3K/AKT/mTOR Pathway Drugs Augmenting Anti-Angiogenic Efficacy In Vivo. PLoS ONE 2014, 9, e105280. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Wang, Q.; Zouboulis, C.C.; Liu, Y.; Ma, Y.; Ma, L.; Ying, J.; Zhang, C.; Xiang, L. ALA-PDT suppressing the cell growth and reducing the lipogenesis in human SZ95 sebocytes by mTOR signaling pathway in vitro. Photodiagn. Photodyn. Ther. 2017, 18, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Brodin, N.P.; Guha, C.; Tomé, W.A. Photodynamic therapy and its role in combined modality anticancer treatment. Technol. Cancer Res. Treat. 2015, 14, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, K.; Kotulska, K.; Jozwiak, S. Management of side effects of mTOR inhibitors in tuberous sclerosis patients. Pharmacol. Rep. 2016, 68, 536–542. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayuk, S.M.; Abrahamse, H. mTOR Signaling Pathway in Cancer Targets Photodynamic Therapy In Vitro. Cells 2019, 8, 431. https://doi.org/10.3390/cells8050431
Ayuk SM, Abrahamse H. mTOR Signaling Pathway in Cancer Targets Photodynamic Therapy In Vitro. Cells. 2019; 8(5):431. https://doi.org/10.3390/cells8050431
Chicago/Turabian StyleAyuk, Sandra M., and Heidi Abrahamse. 2019. "mTOR Signaling Pathway in Cancer Targets Photodynamic Therapy In Vitro" Cells 8, no. 5: 431. https://doi.org/10.3390/cells8050431
APA StyleAyuk, S. M., & Abrahamse, H. (2019). mTOR Signaling Pathway in Cancer Targets Photodynamic Therapy In Vitro. Cells, 8(5), 431. https://doi.org/10.3390/cells8050431