(Epi)genetic Modifications in Myogenic Stem Cells: From Novel Insights to Therapeutic Perspectives



Abstract
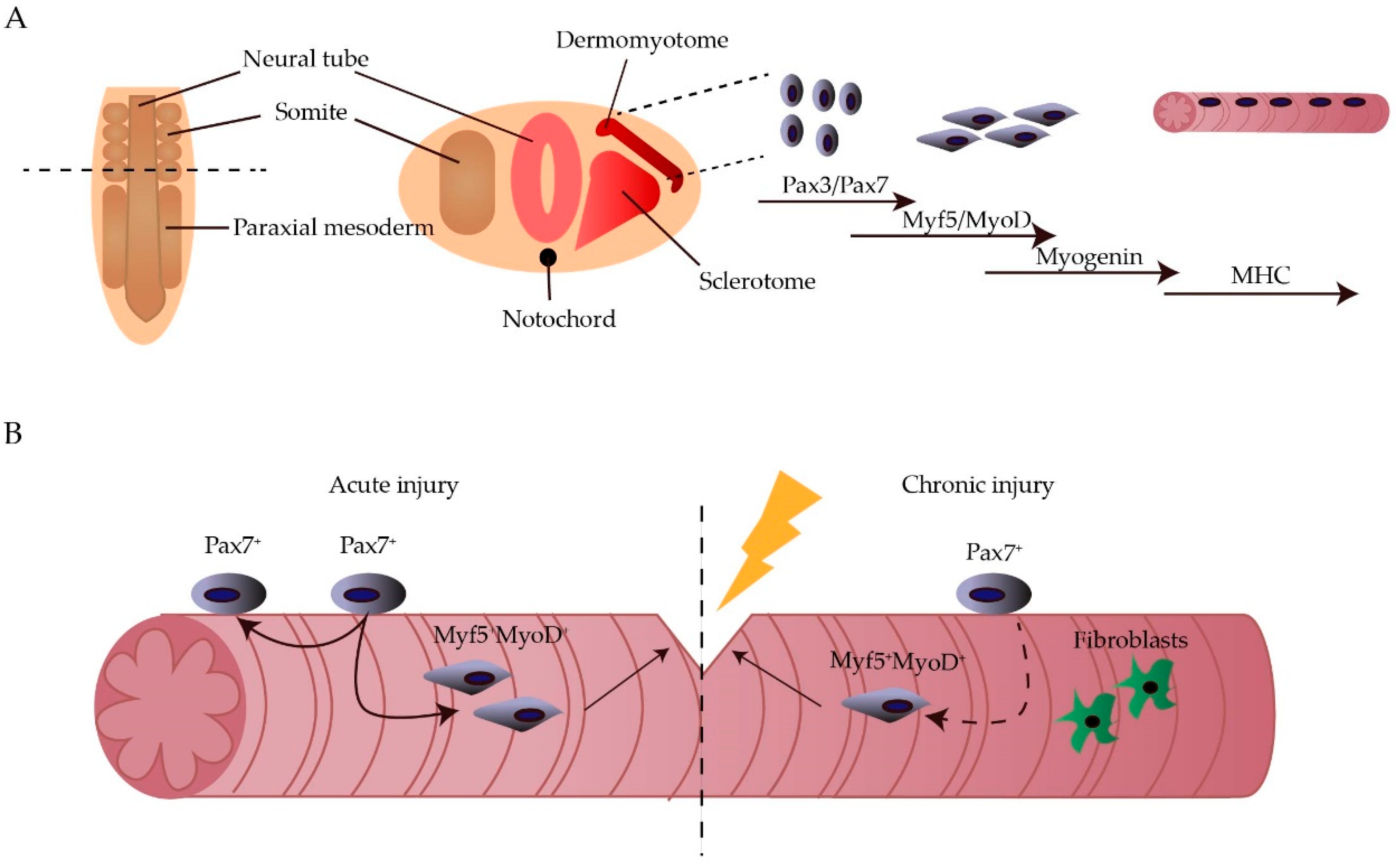
1. Introduction
2. Epigenetics
2.1. Epigenetic Regulation of Myogenesis
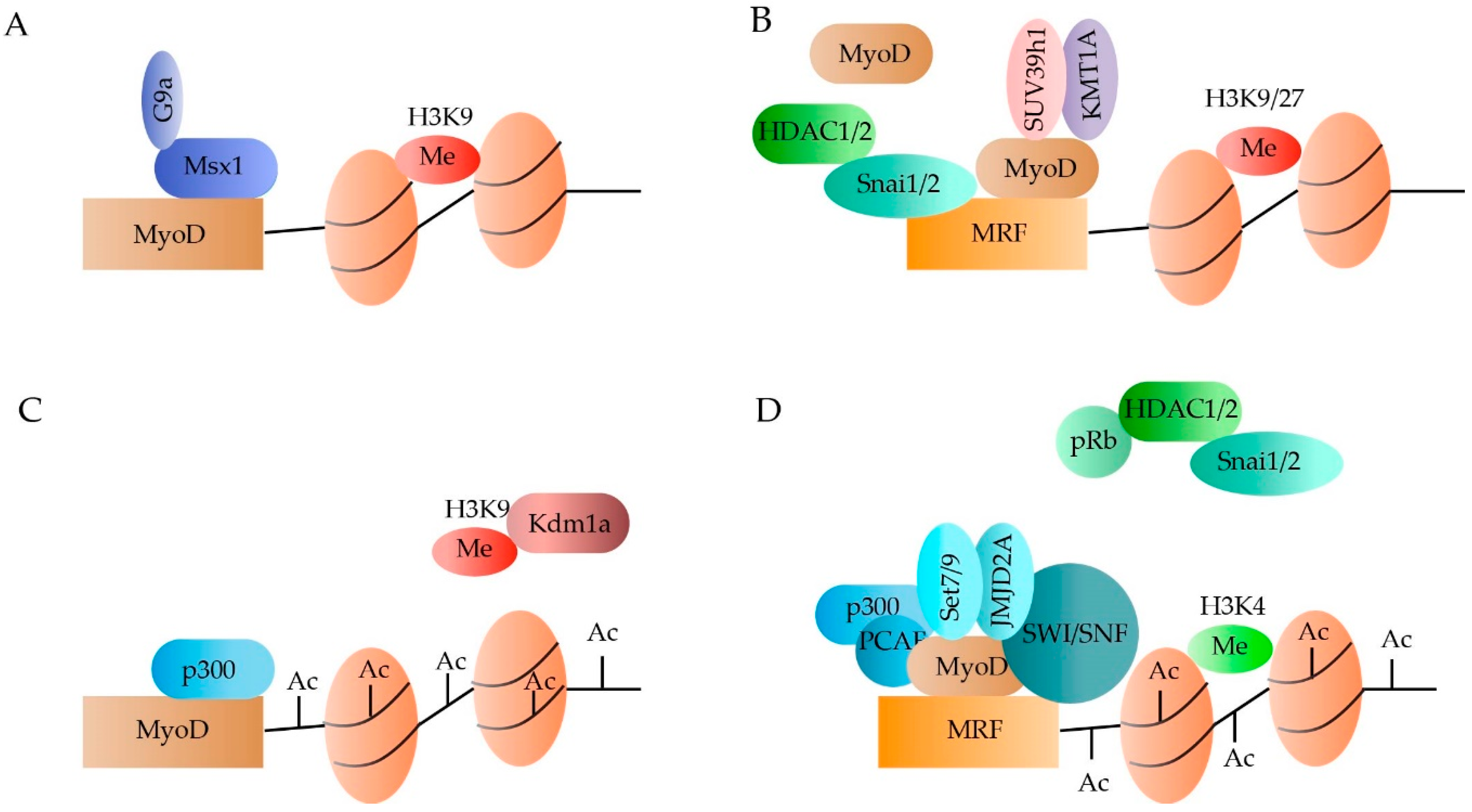
2.1.1. DNA Methylation
2.1.2. Histone Methylation
2.1.3. Histone Acetylation
2.1.4. miRNAs
2.2. Epigenetics to Skew Skeletal Muscle Differentiation
2.2.1. Epigenetic Drugs
2.2.2. miRNA Modulations
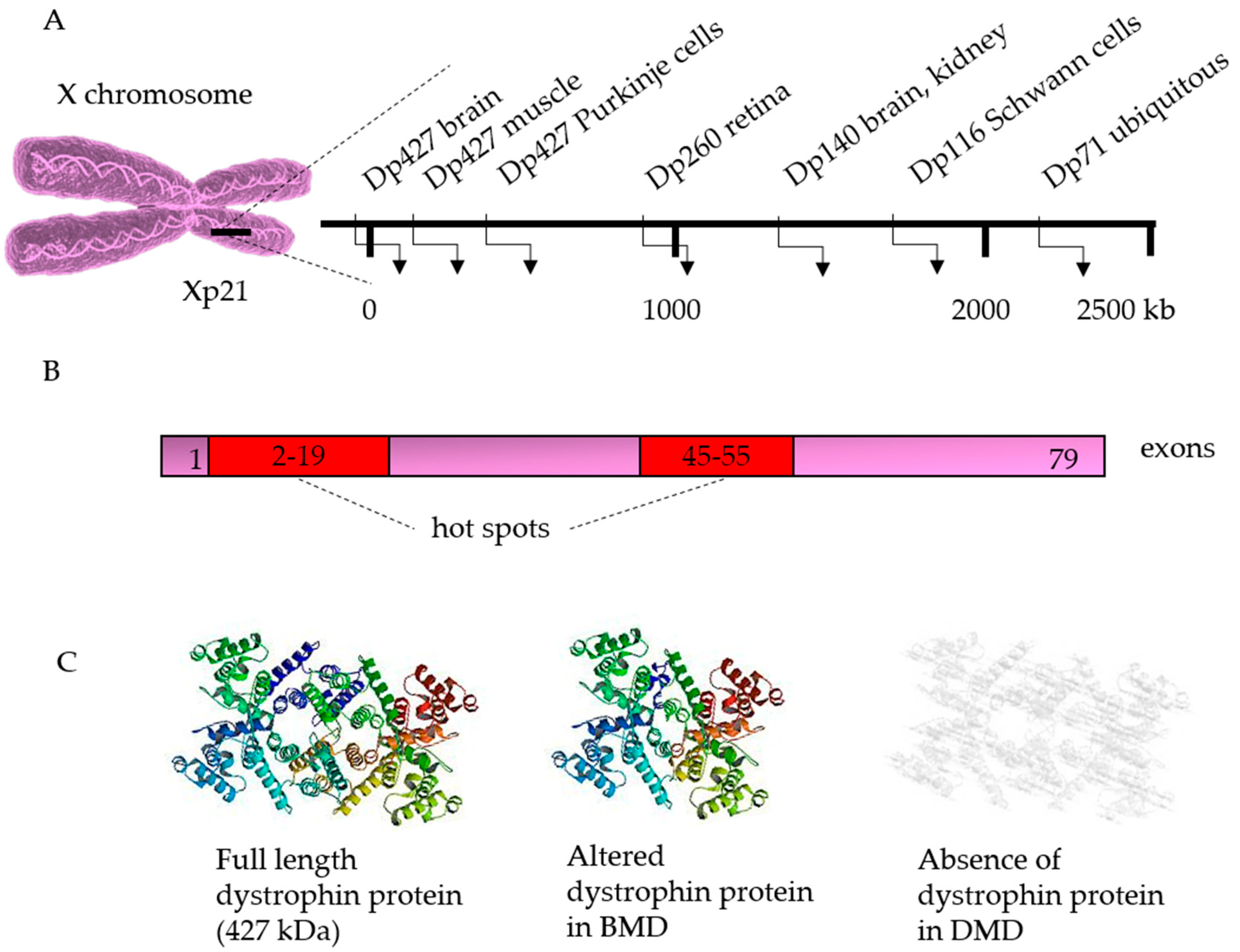
3. Genetics
3.1. Correction of the Disease-Causing Mutation
3.2. Gene Addition
3.2.1. Transcription Factors
3.2.2. Reporter Genes
3.2.3. Suicide Genes
4. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAVS1 | Adeno-Associated Virus Integration-Site 1 |
| Cas | CRISPR-Associated Protein |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DMD | Duchenne Muscular Dystrophy |
| DNMT | DNA Methyltransferases |
| HAT | Histone Acetyltransferases |
| HDAC | Histone Deacetylases |
| HDACi | Histone Deacetylase inhibitors |
| HDR | Homology-Directed Repair |
| HMT | Histone Methyltransferases |
| iPSCs | Induced Pluripotent Stem Cells |
| MAB | Mesoangioblast |
| MD | Muscular Dystrophy |
| miRNA | MicroRNA |
| MRF | Muscle Regulatory Factor |
| Myf5 | Myogenic Factor 5 |
| MyoD | Muscle Determining Factor |
| NHEJ | Non-Homologous End-Joining |
| PAM | Protospacer-Adjacent Motif |
| PAX | Paired Box Gene |
| SCs | Satellite Cells |
| TALEN | Transcription Activator-Like Effector Nucleases |
| ZFN | Zinc-Finger Nuclease |
References
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Yokoyama, S.; Asahara, H. The myogenic transcriptional network. Cell Mol. Life Sci. 2011, 68, 1843–1849. [Google Scholar] [CrossRef]
- Chen, J.C.J.; Goldhamer, D.J. Skeletal muscle stem cells. Reprod. Biol.Endocrin. 2003, 1, 101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Investig. 2010, 120, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli, M.; Cassano, M.; Crippa, S.; Perini, I.; Sampaolesi, M. Cell therapy strategies and improvements for muscular dystrophy. Cell Death Differ. 2010, 17, 1222–1229. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Straub, V.; Campbell, K.P. Muscular dystrophies and the dystrophin-glycoprotein complex. Curr. Opin. Neurol. 1997, 10, 168–175. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Colvin, M.K.; Cripe, L.; Herron, A.R.; Kennedy, A.; Kinnett, K.; et al. Diagnosis and management of duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018, 17, 445–455. [Google Scholar] [CrossRef]
- Fan, Y.; Maley, M.; Beilharz, M.; Grounds, M. Rapid death of injected myoblasts in myoblast transfer therapy. Muscle Nerve. 1996, 19, 853–860. [Google Scholar] [CrossRef]
- Skuk, D.; Goulet, M.; Tremblay, J.P. Use of repeating dispensers to increase the efficiency of the intramuscular myogenic cell injection procedure. Cell Transplant. 2006, 15, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Sampaolesi, M.; Tonlorenzi, R.; Tagliafico, E.; Sacchetti, B.; Perani, L.; Innocenzi, A.; Galvez, B.G.; Messina, G.; Morosetti, R.; et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell. Biol. 2007, 9, 255–267. [Google Scholar] [CrossRef]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of hla-matched donor mesoangioblasts in duchenne muscular dystrophy. EMBO Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Torrente, Y.; Innocenzi, A.; Tonlorenzi, R.; D’Antona, G.; Pellegrino, M.A.; Barresi, R.; Bresolin, N.; De Angelis, M.G.; Campbell, K.P.; et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science 2003, 301, 487–492. [Google Scholar] [CrossRef]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef]
- Barreiro, E.; Tajbakhsh, S. Epigenetic regulation of muscle development. J. Muscle Res. Cell Motil. 2017, 38, 31–35. [Google Scholar] [CrossRef]
- Robinson, D.C.L.; Dilworth, F.J. Epigenetic regulation of adult myogenesis. Curr. Top. Dev. Biol. 2018, 126, 235–284. [Google Scholar] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Aguirre-Arteta, A.M.; Grunewald, I.; Cardoso, M.C.; Leonhardt, H. Expression of an alternative dnmt1 isoform during muscle differentiation. Cell Growth Differ. 2000, 11, 551–559. [Google Scholar]
- Carrio, E.; Diez-Villanueva, A.; Lois, S.; Mallona, I.; Cases, I.; Forn, M.; Peinado, M.A.; Suelves, M. Deconstruction of DNA methylation patterns during myogenesis reveals specific epigenetic events in the establishment of the skeletal muscle lineage. Stem Cells 2015, 33, 2025–2036. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, L.; Jost, J.P. In differentiating mouse myoblasts DNA methyltransferase is posttranscriptionally and posttranslationally regulated. Nucleic Acids Res. 1996, 24, 2718–2722. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Mozzetta, C.; Consalvi, S.; Saccone, V.; Forcales, S.V.; Puri, P.L.; Palacios, D. Selective control of pax7 expression by tnf-activated p38alpha/polycomb repressive complex 2 (prc2) signaling during muscle satellite cell differentiation. Cell Cycle 2011, 10, 191–198. [Google Scholar] [CrossRef]
- Palacios, D.; Mozzetta, C.; Consalvi, S.; Caretti, G.; Saccone, V.; Proserpio, V.; Marquez, V.E.; Valente, S.; Mai, A.; Forcales, S.V.; et al. Tnf/p38alpha/polycomb signaling to pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010, 7, 455–469. [Google Scholar] [CrossRef]
- Stojic, L.; Jasencakova, Z.; Prezioso, C.; Stützer, A.; Bodega, B.; Pasini, D.; Klingberg, R.; Mozzetta, C.; Margueron, R.; Puri, P.L.; et al. Chromatin regulated interchange between polycomb repressive complex 2 (prc2)-ezh2 and prc2-ezh1 complexes controls myogenin activation in skeletal muscle cells. Epigenetics Chromatin 2011, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Peng, J.; Jiang, S. The epigenetic regulation of embryonic myogenesis and adult muscle regeneration by histone methylation modification. Biochem. Biophys. Rep. 2016, 6, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Marroncelli, N.; Adamo, S. New insights into the epigenetic control of satellite cells. World J. Stem Cells 2015, 7, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Asp, P.; Blum, R.; Vethantham, V.; Parisi, F.; Micsinai, M.; Cheng, J.; Bowman, C.; Kluger, Y.; Dynlacht, B.D. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc. Natl. Acad. Sci. USA 2011, 108, E149–E158. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Bansal, V.; Grunert, M.; Malecova, B.; Dall’Agnese, A.; Latella, L.; Gatto, S.; Ryan, T.; Schulz, K.; Chen, W.; et al. Muscle-relevant genes marked by stable h3k4me2/3 profiles and enriched myod binding during myogenic differentiation. PLoS ONE 2017, 12, e0179464. [Google Scholar] [CrossRef]
- Lilja, K.C.; Zhang, N.; Magli, A.; Gunduz, V.; Bowman, C.J.; Arpke, R.W.; Darabi, R.; Kyba, M.; Perlingeiro, R.; Dynlacht, B.D. Pax7 remodels the chromatin landscape in skeletal muscle stem cells. PLoS ONE 2017, 12, e0176190. [Google Scholar] [CrossRef]
- Wang, J.; Abate-Shen, C. The msx1 homeoprotein recruits g9a methyltransferase to repressed target genes in myoblast cells. PLoS ONE 2012, 7, e37647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.H.; Judson, R.N.; Liu, D.Y.; Kast, J.; Rossi, F.M. The lysine methyltransferase ehmt2/g9a is dispensable for skeletal muscle development and regeneration. Skelet Muscle 2016, 6, 22. [Google Scholar] [CrossRef]
- Mal, A.K. Histone methyltransferase suv39h1 represses myod-stimulated myogenic differentiation. EMBO J. 2006, 25, 3323–3334. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.; Wolff, D.W.; Jothi, M.; Mal, M.; Mal, A.K. P38alpha mapk disables kmt1a-mediated repression of myogenic differentiation program. Skelet Muscle 2016, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Ling, B.M.; Bharathy, N.; Chung, T.K.; Kok, W.K.; Li, S.; Tan, Y.H.; Rao, V.K.; Gopinadhan, S.; Sartorelli, V.; Walsh, M.J.; et al. Lysine methyltransferase g9a methylates the transcription factor myod and regulates skeletal muscle differentiation. Proc. Natl. Acad. Sci. USA 2012, 109, 841–846. [Google Scholar] [CrossRef]
- De la Serna, I.L.; Carlson, K.A.; Imbalzano, A.N. Mammalian swi/snf complexes promote myod-mediated muscle differentiation. Nat. Genet. 2001, 27, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Forcales, S.V.; Albini, S.; Giordani, L.; Malecova, B.; Cignolo, L.; Chernov, A.; Coutinho, P.; Saccone, V.; Consalvi, S.; Williams, R.; et al. Signal-dependent incorporation of myod-baf60c into brg1-based swi/snf chromatin-remodelling complex. EMBO J. 2012, 31, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Neppl, R.L.; Huang, Z.P.; Chen, J.; Tang, R.H.; Cao, R.; Zhang, Y.; Jin, S.W.; Wang, D.Z. The histone methyltransferase set7/9 promotes myoblast differentiation and myofibril assembly. J. Cell Biol. 2011, 194, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Verrier, L.; Escaffit, F.; Chailleux, C.; Trouche, D.; Vandromme, M. A new isoform of the histone demethylase jmjd2a/kdm4a is required for skeletal muscle differentiation. PLoS Genet. 2011, 7, e1001390. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, V.D.; Yin, H.; Jahani-Asl, A.; Ming, H.; Kockx, C.E.; van Ijcken, W.F.; Grosveld, F.; Rudnicki, M.A. Snail regulates myod binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol. Cell 2012, 47, 457–468. [Google Scholar] [CrossRef]
- Cho, O.H.; Mallappa, C.; Hernández-Hernández, J.M.; Rivera-Pérez, J.A.; Imbalzano, A.N. Contrasting roles for myod in organizing myogenic promoter structures during embryonic skeletal muscle development. Dev. Dyn. 2015, 244, 43–55. [Google Scholar] [CrossRef]
- Puri, P.L.; Iezzi, S.; Stiegler, P.; Chen, T.T.; Schiltz, R.L.; Muscat, G.E.; Giordano, A.; Kedes, L.; Wang, J.Y.; Sartorelli, V. Class i histone deacetylases sequentially interact with myod and prb during skeletal myogenesis. Mol. Cell 2001, 8, 885–897. [Google Scholar] [CrossRef]
- Fauquier, L.; Azzag, K.; Parra, M.A.M.; Quillien, A.; Boulet, M.; Diouf, S.; Carnac, G.; Waltzer, L.; Gronemeyer, H.; Vandel, L. Cbp and p300 regulate distinct gene networks required for human primary myoblast differentiation and muscle integrity. Sci. Rep. 2018, 8, 12629. [Google Scholar] [CrossRef]
- Sartorelli, V.; Puri, P.L.; Hamamori, Y.; Ogryzko, V.; Chung, G.; Nakatani, Y.; Wang, J.Y.; Kedes, L. Acetylation of myod directed by pcaf is necessary for the execution of the muscle program. Mol. Cell 1999, 4, 725–734. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microrna biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of micrornas in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.F.; Liu, Y.Z.; Du, R.; Wang, B.; Chen, M.T.; Zhang, Y.Y.; Deng, Z.L.; Li, J. Mir-377 induces senescence in human skin fibroblasts by targeting DNA methyltransferase 1. Cell Death Dis. 2017, 8, e2663. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yao, J.; Sun, H.; He, K.; Tong, D.; Song, T.; Huang, C. Microrna-101 suppresses progression of lung cancer through the pten/akt signaling pathway by targeting DNA methyltransferase 3a. Oncol. Lett. 2017, 13, 329–338. [Google Scholar] [CrossRef]
- Song, J.; Jin, E.H.; Kim, D.; Kim, K.Y.; Chun, C.H.; Jin, E.J. Microrna-222 regulates mmp-13 via targeting hdac-4 during osteoarthritis pathogenesis. BBA Clin. 2015, 3, 79–89. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Sampaolesi, M. The mesmirizing complexity of micrornas for striated muscle tissue engineering. Adv. Drug. Deliv. Rev. 2015, 88, 37–52. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microrna-1 and microrna-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Lee, Y.S.; Sivaprasad, U.; Malhotra, A.; Dutta, A. Muscle-specific microrna mir-206 promotes muscle differentiation. J. Cell Biol. 2006, 174, 677–687. [Google Scholar] [CrossRef]
- Naguibneva, I.; Ameyar-Zazoua, M.; Polesskaya, A.; Ait-Si-Ali, S.; Groisman, R.; Souidi, M.; Cuvellier, S.; Harel-Bellan, A. The microrna mir-181 targets the homeobox protein hox-a11 during mammalian myoblast differentiation. Nat. Cell Biol. 2006, 8, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, Y.; Yang, G.; Chen, X.; Zhang, Y.; Cao, G.; Wang, J.; Sun, Y.; Zhang, P.; Fan, M.; et al. Transforming growth factor-beta-regulated mir-24 promotes skeletal muscle differentiation. Nucleic Acids Res. 2008, 36, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Crist, C.G.; Montarras, D.; Pallafacchina, G.; Rocancourt, D.; Cumano, A.; Conway, S.J.; Buckingham, M. Muscle stem cell behavior is modified by microrna-27 regulation of pax3 expression. Proc. Natl. Acad. Sci. USA 2009, 106, 13383–13387. [Google Scholar] [CrossRef] [PubMed]
- Crippa, S.; Cassano, M.; Messina, G.; Galli, D.; Galvez, B.G.; Curk, T.; Altomare, C.; Ronzoni, F.; Toelen, J.; Gijsbers, R.; et al. Mir669a and mir669q prevent skeletal muscle differentiation in postnatal cardiac progenitors. J. Cell Biol. 2011, 193, 1197–1212. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli, M.; Crippa, S.; Montecchiani, C.; Camps, J.; Cornaglia, A.I.; Boldrin, L.; Morgan, J.; Calligaro, A.; Casasco, A.; Orlacchio, A.; et al. Long-term mir-669a therapy alleviates chronic dilated cardiomyopathy in dystrophic mice. J. Am. Heart Assoc. 2013, 2, e000284. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Sandona, M.; Saccone, V. Epigenetic reprogramming of muscle progenitors: Inspiration for clinical therapies. Stem Cells Int. 2016, 2016, 6093601. [Google Scholar] [CrossRef]
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; Del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol. Med 2013, 19, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Minetti, G.C.; Colussi, C.; Adami, R.; Serra, C.; Mozzetta, C.; Parente, V.; Fortuni, S.; Straino, S.; Sampaolesi, M.; Di Padova, M.; et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 2006, 12, 1147–1150. [Google Scholar] [CrossRef]
- Furlan, A.; Monzani, V.; Reznikov, L.L.; Leoni, F.; Fossati, G.; Modena, D.; Mascagni, P.; Dinarello, C.A. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor itf2357 (givinostat). Mol. Med. 2011, 17, 353–362. [Google Scholar] [CrossRef]
- Bettica, P.; Petrini, S.; D’Oria, V.; D’Amico, A.; Catteruccia, M.; Pane, M.; Sivo, S.; Magri, F.; Brajkovic, S.; Messina, S.; et al. Histological effects of givinostat in boys with duchenne muscular dystrophy. Neuromuscul Disord 2016, 26, 643–649. [Google Scholar] [CrossRef]
- Iezzi, S.; Di Padova, M.; Serra, C.; Caretti, G.; Simone, C.; Maklan, E.; Minetti, G.; Zhao, P.; Hoffman, E.P.; Puri, P.L.; et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev. Cell 2004, 6, 673–684. [Google Scholar] [CrossRef]
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandoná, M.; Ryan, T.; Rojas-Muñoz, A.; Madaro, L.; Fasanaro, P.; et al. Hdac-regulated myomirs control baf60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes. Dev. 2014, 28, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Albini, S.; Coutinho, P.; Malecova, B.; Giordani, L.; Savchenko, A.; Forcales, S.V.; Puri, P.L. Epigenetic reprogramming of human embryonic stem cells into skeletal muscle cells and generation of contractile myospheres. Cell Rep. 2013, 3, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Foote, M.; Chen, J. Effects of histone deacetylase inhibitor valproic acid on skeletal myocyte development. Sci. Rep. 2014, 4, 7207. [Google Scholar] [CrossRef] [PubMed]
- Bansal, V.; De, D.; An, J.; Kang, T.M.; Jeong, H.J.; Kang, J.S.; Kim, K.K. Chemical induced conversion of mouse fibroblasts and human adipose-derived stem cells into skeletal muscle-like cells. Biomaterials 2019, 193, 30–46. [Google Scholar] [CrossRef] [PubMed]
- Iezzi, S.; Cossu, G.; Nervi, C.; Sartorelli, V.; Puri, P.L. Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc. Natl. Acad. Sci. USA 2002, 99, 7757–7762. [Google Scholar] [CrossRef]
- Mal, A.; Sturniolo, M.; Schiltz, R.L.; Ghosh, M.K.; Harter, M.L. A role for histone deacetylase hdac1 in modulating the transcriptional activity of myod: Inhibition of the myogenic program. EMBO J. 2001, 20, 1739–1753. [Google Scholar] [CrossRef]
- Johnston, L.A.; Tapscott, S.J.; Eisen, H. Sodium butyrate inhibits myogenesis by interfering with the transcriptional activation function of myod and myogenin. Mol. Cell Biol. 1992, 12, 5123–5130. [Google Scholar] [CrossRef]
- Kazama, T.; Fujie, M.; Endo, T.; Kano, K. Mature adipocyte-derived dedifferentiated fat cells can transdifferentiate into skeletal myocytes in vitro. Biochem. Biophys. Res. Commun. 2008, 377, 780–785. [Google Scholar] [CrossRef]
- Kaur, K.; Yang, J.; Eisenberg, C.A.; Eisenberg, L.M. 5-azacytidine promotes the transdifferentiation of cardiac cells to skeletal myocytes. Cell. Reprogram. 2014, 16, 324–330. [Google Scholar] [CrossRef]
- Taylor, S.M.; Jones, P.A. Multiple new phenotypes induced in 10t1/2 and 3t3 cells treated with 5-azacytidine. Cell 1979, 17, 771–779. [Google Scholar] [CrossRef]
- Constantinides, P.G.; Jones, P.A.; Gevers, W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment. Nature 1977, 267, 364–366. [Google Scholar] [CrossRef]
- Senesi, P.; Luzi, L.; Montesano, A.; Terruzzi, I. DNA demethylation enhances myoblasts hypertrophy during the late phase of myogenesis activating the igf-i pathway. Endocrine 2014, 47, 244–254. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Scarpa, S.; Lucarelli, M.; Palitti, F.; Carotti, D.; Strom, R. Simultaneous myogenin expression and overall DNA hypomethylation promote in vitro myoblast differentiation. Cell Growth Differ. 1996, 7, 1051–1058. [Google Scholar]
- Choi, S.C.; Yoon, J.; Shim, W.J.; Ro, Y.M.; Lim, D.S. 5-azacytidine induces cardiac differentiation of p19 embryonic stem cells. Exp. Mol. Med. 2004, 36, 515–523. [Google Scholar] [CrossRef]
- Qian, Q.; Qian, H.; Zhang, X.; Zhu, W.; Yan, Y.; Ye, S.; Peng, X.; Li, W.; Xu, Z.; Sun, L.; et al. 5-azacytidine induces cardiac differentiation of human umbilical cord-derived mesenchymal stem cells by activating extracellular regulated kinase. Stem Cells Dev. 2012, 21, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Wakitani, S.; Saito, T.; Caplan, A.I. Myogenic cells derived from rat bone marrow mesenchymal stem cells exposed to 5-azacytidine. Muscle Nerve. 1995, 18, 1417–1426. [Google Scholar] [CrossRef]
- Liu, N.; Williams, A.H.; Maxeiner, J.M.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Microrna-206 promotes skeletal muscle regeneration and delays progression of duchenne muscular dystrophy in mice. J. Clin. Investig. 2012, 122, 2054–2065. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. Microrna-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, M.; Heo, H.R.; Ha, K.S.; Han, E.T.; Park, W.S.; Yang, S.R.; Hong, S.H. Inhibition of microrna-221 and 222 enhances hematopoietic differentiation from human pluripotent stem cells via c-kit upregulation. Mol. Cells 2018, 41, 971–978. [Google Scholar] [PubMed]
- Judson, R.L.; Babiarz, J.E.; Venere, M.; Blelloch, R. Embryonic stem cell-specific micrornas promote induced pluripotency. Nat. Biotechnol. 2009, 27, 459–461. [Google Scholar] [CrossRef]
- Ying, S.Y.; Fang, W.; Lin, S.L. The mir-302-mediated induction of pluripotent stem cells (ipsc): Multiple synergistic reprogramming mechanisms. Methods Mol. Biol. 2018, 1733, 283–304. [Google Scholar]
- Yoshida, S.; Miyagawa, S.; Fukushima, S.; Kawamura, T.; Kashiyama, N.; Ohashi, F.; Toyofuku, T.; Toda, K.; Sawa, Y. Maturation of human induced pluripotent stem cell-derived cardiomyocytes by soluble factors from human mesenchymal stem cells. Mol. Ther. 2018, 26, 2681–2695. [Google Scholar] [CrossRef]
- Iwasaki, H.; Imamura, T.; Morino, K.; Shimosato, T.; Tawa, M.; Ugi, S.; Sakurai, H.; Maegawa, H.; Okamura, T. Microrna-494 plays a role in fiber type-specific skeletal myogenesis in human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2015, 468, 208–213. [Google Scholar] [CrossRef]
- Quattrocelli, M.; Swinnen, M.; Giacomazzi, G.; Camps, J.; Barthelemy, I.; Ceccarelli, G.; Caluwe, E.; Grosemans, H.; Thorrez, L.; Pelizzo, G.; et al. Mesodermal ipsc-derived progenitor cells functionally regenerate cardiac and skeletal muscle. J. Clin. Investig. 2015, 125, 4463–4482. [Google Scholar] [CrossRef]
- Kondo, H.; Kim, H.W.; Wang, L.; Okada, M.; Paul, C.; Millard, R.W.; Wang, Y. Blockade of senescence-associated microrna-195 in aged skeletal muscle cells facilitates reprogramming to produce induced pluripotent stem cells. Aging Cell 2016, 15, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; Gersbach, C.A. Enabling functional genomics with genome engineering. Genome Res. 2015, 25, 1442–1455. [Google Scholar] [CrossRef]
- Turan, S.; Farruggio, A.P.; Srifa, W.; Day, J.W.; Calos, M.P. Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy. Mol. Ther. 2016, 24, 685–696. [Google Scholar] [CrossRef]
- Ikink, G.J.; Boer, M.; Bakker, E.R.M.; Vendel-Zwaagstra, A.; Klijn, C.; Ten Hoeve, J.; Jonkers, J.; Wessels, L.F.; Hilkens, J. Insertional mutagenesis in a her2-positive breast cancer model reveals eras as a driver of cancer and therapy resistance. Oncogene 2018, 37, 1594–1609. [Google Scholar] [CrossRef] [PubMed]
- Bii, V.M.; Collins, C.P.; Hocum, J.D.; Trobridge, G.D. Replication-incompetent gammaretroviral and lentiviral vector-based insertional mutagenesis screens identify prostate cancer progression genes. Oncotarget 2018, 9, 15451–15463. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of scid-x1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., 3rd. Zfn, talen, and crispr/cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Merkert, S.; Martin, U. Targeted gene editing in human pluripotent stem cells using site-specific nucleases. Adv. Biochem. Eng. Biotechnol. 2018, 163, 169–186. [Google Scholar] [PubMed]
- Guha, T.K.; Wai, A.; Hausner, G. Programmable genome editing tools and their regulation for efficient genome engineering. Comput. Struct. Biotechnol. J. 2017, 15, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by rna-guided crispr cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Park, A.; Hong, P.; Won, S.T.; Thibault, P.A.; Vigant, F.; Oguntuyo, K.Y.; Taft, J.D.; Lee, B. Sendai virus, an rna virus with no risk of genomic integration, delivers crispr/cas9 for efficient gene editing. Mol. Ther. Methods Clin. Dev. 2016, 3, 16057. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation spectrum of the dystrophin gene in 442 duchenne/becker muscular dystrophy cases from one japanese referral center. J. Hum. Genet. 2010, 55, 379–388. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Filareto, A.; Parker, S.; Darabi, R.; Borges, L.; Iacovino, M.; Schaaf, T.; Mayerhofer, T.; Chamberlain, J.S.; Ervasti, J.M.; McIvor, R.S.; et al. An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells. Nat. Commun. 2013, 4, 1549. [Google Scholar] [CrossRef]
- Kazuki, Y.; Hiratsuka, M.; Takiguchi, M.; Osaki, M.; Kajitani, N.; Hoshiya, H.; Hiramatsu, K.; Yoshino, T.; Kazuki, K.; Ishihara, C.; et al. Complete genetic correction of ips cells from duchenne muscular dystrophy. Mol. Ther. 2010, 18, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Keller, B.; Makalou, N.; Sutton, R.E. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 2001, 12, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, C.; Bassel-Duby, R.; Olson, E.N. Myoediting: Toward prevention of muscular dystrophy by therapeutic genome editing. Physiol. Rev. 2018, 98, 1205–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wu, F.; Mosenson, J.; Zhang, H.; He, T.C.; Wu, W.S. Crispr/cas9-mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids 2017, 7, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Perez-Pinera, P.; Brown, M.T.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Correction of dystrophin expression in cells from duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol. Ther. 2015, 23, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, V.; Kyrychenko, S.; Tiburcy, M.; Shelton, J.M.; Long, C.; Schneider, J.W.; Zimmermann, W.H.; Bassel-Duby, R.; Olson, E.N. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Amoasii, L.; Hildyard, J.C.W.; Li, H.; Sanchez-Ortiz, E.; Mireault, A.; Caballero, D.; Harron, R.; Stathopoulou, T.-R.; Massey, C.; Shelton, J.M.; et al. Gene editing restores dystrophin expression in a canine model of duchenne muscular dystrophy. Science 2018, 362, 86–91. [Google Scholar] [CrossRef]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Bhattacharyya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by talen and crispr-cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Li, H.; Tiburcy, M.; Rodriguez-Caycedo, C.; Kyrychenko, V.; Zhou, H.; Zhang, Y.; Min, Y.L.; Shelton, J.M.; Mammen, P.P.A.; et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv. 2018, 4, eaap9004. [Google Scholar] [CrossRef]
- Young, C.S.; Hicks, M.R.; Ermolova, N.V.; Nakano, H.; Jan, M.; Younesi, S.; Karumbayaram, S.; Kumagai-Cresse, C.; Wang, D.; Zack, J.A.; et al. A single crispr-cas9 deletion strategy that targets the majority of dmd patients restores dystrophin function in hipsc-derived muscle cells. Cell Stem Cell 2016, 18, 533–540. [Google Scholar] [CrossRef]
- Iyombe-Engembe, J.-P.; Ouellet, D.L.; Barbeau, X.; Rousseau, J.; Chapdelaine, P.; Lagüe, P.; Tremblay, J.P. Efficient restoration of the dystrophin gene reading frame and protein structure in dmd myoblasts using the cindel method. Mol. Ther. Nucleic Acids 2016, 5, e283. [Google Scholar] [CrossRef]
- Popplewell, L.; Koo, T.; Leclerc, X.; Duclert, A.; Mamchaoui, K.; Gouble, A.; Mouly, V.; Voit, T.; Paques, F.; Cedrone, F.; et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum. Gene Ther. 2013, 24, 692–701. [Google Scholar] [CrossRef]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Corrigendum: Muscle-specific crispr/cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for duchenne muscular dystrophy. Nat. Commun. 2017, 8, 16007. [Google Scholar] [CrossRef]
- El Refaey, M.; Xu, L.; Gao, Y.; Canan, B.D.; Adesanya, T.M.A.; Warner, S.C.; Akagi, K.; Symer, D.E.; Mohler, P.J.; Ma, J.; et al. In vivo genome editing restores dystrophin expression and cardiac function in dystrophic mice. Circ. Res. 2017, 121, 923–929. [Google Scholar] [CrossRef]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.W.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. Crispr-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging issues in aav-mediated in vivo gene therapy. Mol. Ther. Meth. Clin. Dev. 2017, 8, 87–104. [Google Scholar] [CrossRef]
- Maggio, I.; Stefanucci, L.; Janssen, J.M.; Liu, J.; Chen, X.; Mouly, V.; Goncalves, M.A. Selection-free gene repair after adenoviral vector transduction of designer nucleases: Rescue of dystrophin synthesis in dmd muscle cell populations. Nucleic Acids Res. 2016, 44, 1449–1470. [Google Scholar] [CrossRef]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Goncalves, M.A. Adenoviral vectors encoding crispr/cas9 multiplexes rescue dystrophin synthesis in unselected populations of dmd muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, C.; Li, H.; McAnally, J.R.; Baskin, K.K.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Crispr-cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017, 3, e1602814. [Google Scholar] [CrossRef]
- Lattanzi, A.; Duguez, S.; Moiani, A.; Izmiryan, A.; Barbon, E.; Martin, S.; Mamchaoui, K.; Mouly, V.; Bernardi, F.; Mavilio, F.; et al. Correction of the exon 2 duplication in dmd myoblasts by a single crispr/cas9 system. Mol. Ther. Nucleic Acids 2017, 7, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Charville, G.W.; Cheung, T.H.; Yoo, B.; Santos, P.J.; Lee, G.K.; Shrager, J.B.; Rando, T.A. Ex vivo expansion and in vivo self-renewal of human muscle stem cells. Stem Cell Rep. 2015, 5, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Regan, S.N.; Xia, Y.; Oostrom, L.A.; Cowan, C.A.; Musunuru, K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing talens with crisprs. Cell Stem Cell 2013, 12, 393–394. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Hoshiya, H.; D’Antona, G.; Gerli, M.F.; Messina, G.; Antonini, S.; Tonlorenzi, R.; Benedetti, S.; Berghella, L.; Torrente, Y.; et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011, 3, 96ra78. [Google Scholar] [CrossRef]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient crispr/cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived ips and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef]
- Papapetrou, E.P.; Schambach, A. Gene insertion into genomic safe harbors for human gene therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef]
- Oceguera-Yanez, F.; Kim, S.I.; Matsumoto, T.; Tan, G.W.; Xiang, L.; Hatani, T.; Kondo, T.; Ikeya, M.; Yoshida, Y.; Inoue, H.; et al. Engineering the aavs1 locus for consistent and scalable transgene expression in human ipscs and their differentiated derivatives. Methods 2016, 101, 43–55. [Google Scholar] [CrossRef]
- Wallen, M.C.; Gaj, T.; Barbas, C.F., 3rd. Redesigning recombinase specificity for safe harbor sites in the human genome. PLoS ONE 2015, 10, e0139123. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kodaka, Y.; Rabu, G.; Asakura, A. Skeletal muscle cell induction from pluripotent stem cells. Stem Cells Int. 2017, 2017, 1376151. [Google Scholar] [CrossRef]
- Tanaka, A.; Woltjen, K.; Miyake, K.; Hotta, A.; Ikeya, M.; Yamamoto, T.; Nishino, T.; Shoji, E.; Sehara-Fujisawa, A.; Manabe, Y.; et al. Efficient and reproducible myogenic differentiation from human ips cells: Prospects for modeling miyoshi myopathy in vitro. PLoS ONE 2013, 8, e61540. [Google Scholar] [CrossRef]
- Maffioletti, S.M.; Gerli, M.F.; Ragazzi, M.; Dastidar, S.; Benedetti, S.; Loperfido, M.; VandenDriessche, T.; Chuah, M.K.; Tedesco, F.S. Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human es and patient-specific ips cells. Nat. Protoc. 2015, 10, 941–958. [Google Scholar] [CrossRef]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C. Human es- and ips-derived myogenic progenitors restore dystrophin and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef]
- Shoji, E.; Woltjen, K.; Sakurai, H. Directed myogenic differentiation of human induced pluripotent stem cells. Meth. Mol. Biol. 2016, 1353, 89–99. [Google Scholar]
- Cadiñanos, J.; Bradley, A. Generation of an inducible and optimized piggybac transposon system. Nucleic Acids Res. 2007, 35, e87. [Google Scholar] [CrossRef]
- Wolfs, E.; Verfaillie, C.M.; Van Laere, K.; Deroose, C.M. Radiolabeling strategies for radionuclide imaging of stem cells. Stem Cell Rev. 2015, 11, 254–274. [Google Scholar] [CrossRef]
- Kim, M.H.; Lee, Y.J.; Kang, J.H. Stem cell monitoring with a direct or indirect labeling method. Nucl. Med. Mol. Imaging 2016, 50, 275–283. [Google Scholar] [CrossRef]
- Farruggio, A.P.; Bhakta, M.S.; du Bois, H.; Ma, J.; Michele, P.C. Genomic integration of the full-length dystrophin coding sequence in duchenne muscular dystrophy induced pluripotent stem cells. Biotechnol. J. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Holvoet, B.; Quattrocelli, M.; Belderbos, S.; Pollaris, L.; Wolfs, E.; Gheysens, O.; Gijsbers, R.; Vanoirbeek, J.; Verfaillie, C.M.; Sampaolesi, M.; et al. Sodium iodide symporter pet and bli noninvasively reveal mesoangioblast survival in dystrophic mice. Stem Cell Rep. 2015, 5, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Kristin, S.; Radojewski, P.; Tobias, W.; Georg, D. In vivo bioluminescence imaging—A suitable method to track mesenchymal stromal cells in a skeletal muscle trauma. Open Orthop. J. 2015, 9, 262–269. [Google Scholar]
- Wolfs, E.; Holvoet, B.; Ordovas, L.; Breuls, N.; Helsen, N.; Schonberger, M.; Raitano, S.; Struys, T.; Vanbilloen, B.; Casteels, C.; et al. Molecular imaging of human embryonic stem cells stably expressing human pet reporter genes after zinc finger nuclease-mediated genome editing. J. Nucl. Med. 2017, 58, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Tang, C.; Cao, F.; Xie, X.; van der Bogt, K.; Hwang, A.; Connolly, A.J.; Robbins, R.C.; Wu, J.C. Effects of cell number on teratoma formation by human embryonic stem cells. Cell Cycle 2009, 8, 2608–2612. [Google Scholar] [CrossRef]
- Giacomazzi, G.; Holvoet, B.; Trenson, S.; Caluwe, E.; Kravic, B.; Grosemans, H.; Cortes-Calabuig, A.; Deroose, C.M.; Huylebroeck, D.; Hashemolhosseini, S.; et al. Micrornas promote skeletal muscle differentiation of mesodermal ipsc-derived progenitors. Nat. Commun. 2017, 8, 1249. [Google Scholar] [CrossRef] [PubMed]
- Neyrinck, K.; Breuls, N.; Holvoet, B.; Oosterlinck, W.; Wolfs, E.; Vanbilloen, H.; Gheysens, O.; Duelen, R.; Gsell, W.; Lambrichts, I.; et al. The human somatostatin receptor type 2 as an imaging and suicide reporter gene for pluripotent stem cell-derived therapy of myocardial infarction. Theranostics 2018, 8, 2799–2813. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Technique | Cell Type | Mutation | Correction Strategy | Delivery Method | Reference |
|---|---|---|---|---|---|
| Meganucleases | Myoblasts | ΔEx45-52 | Knock-in Ex45-52 | lentiviral | [115] |
| ZFN | Myoblasts | ΔEx48–50 | Skip Ex51 | Electroporation | [107] |
| TALEN | Myoblasts | ΔEx45-52 | ΔEx44-54 | Adenoviral | [122,123] |
| iPSC | ΔEx44 | Knock-in Ex44 Skip Ex44-45 | Electroporation | [111] | |
| CRISPR/Cas9 | Myoblasts | ΔEx45-52 | ΔEx53 ΔEx44-54 ΔEx51 | Adenoviral | [122,123] |
| Myoblasts | ΔEx45-52 | Frameshift Ex51 | Adenoviral | [122,123] | |
| Myoblasts | Dupl. Ex2 | ΔDuplEx2 | Lentiviral | [124] | |
| Myoblasts | ΔEx51-53 | Reframing Ex50 and Ex54 | Lipofectamine | [114] | |
| iPSC | ΔEx44 | Knock-in Ex44 Skip Ex44-45 Frameshift Ex44 | Electroporation | [111] | |
| iPSC | Δ48-50 Dupl. Ex55-59 Pt Ex47 | Skip Ex47-52 ΔDuplEx55-59 Δmutated Ex47 | Lipofectamine | [112] | |
| iPSCs | ΔEx8-9 | ΔEx3–9, ΔEx6–9, or ΔEx7–11 | Electroporation | [108] | |
| iPSC | Dup. Ex50 ΔEx46-51 ΔEx46-47 | ΔEx45-55 | Electroporation | [113] | |
| CRISPR-Cpf1 | iPSC | ΔEx48-59 | Skip Ex51 | Electroporation | [125] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breuls, N.; Giacomazzi, G.; Sampaolesi, M. (Epi)genetic Modifications in Myogenic Stem Cells: From Novel Insights to Therapeutic Perspectives. Cells 2019, 8, 429. https://doi.org/10.3390/cells8050429
Breuls N, Giacomazzi G, Sampaolesi M. (Epi)genetic Modifications in Myogenic Stem Cells: From Novel Insights to Therapeutic Perspectives. Cells. 2019; 8(5):429. https://doi.org/10.3390/cells8050429
Chicago/Turabian StyleBreuls, Natacha, Giorgia Giacomazzi, and Maurilio Sampaolesi. 2019. "(Epi)genetic Modifications in Myogenic Stem Cells: From Novel Insights to Therapeutic Perspectives" Cells 8, no. 5: 429. https://doi.org/10.3390/cells8050429
APA StyleBreuls, N., Giacomazzi, G., & Sampaolesi, M. (2019). (Epi)genetic Modifications in Myogenic Stem Cells: From Novel Insights to Therapeutic Perspectives. Cells, 8(5), 429. https://doi.org/10.3390/cells8050429