MapB Protein is the Essential Methionine Aminopeptidase in Mycobacterium tuberculosis

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacteria and Growth Conditions
2.2. Gene Introduction and Deletion
2.3. Expression and Purification of Recombinant MapB Proteins
2.4. Activity of the Recombinant Proteins Assessment
3. Results
3.1. mapA (Rv0734, Mb0755) is Dispensible for Bacterial Viability
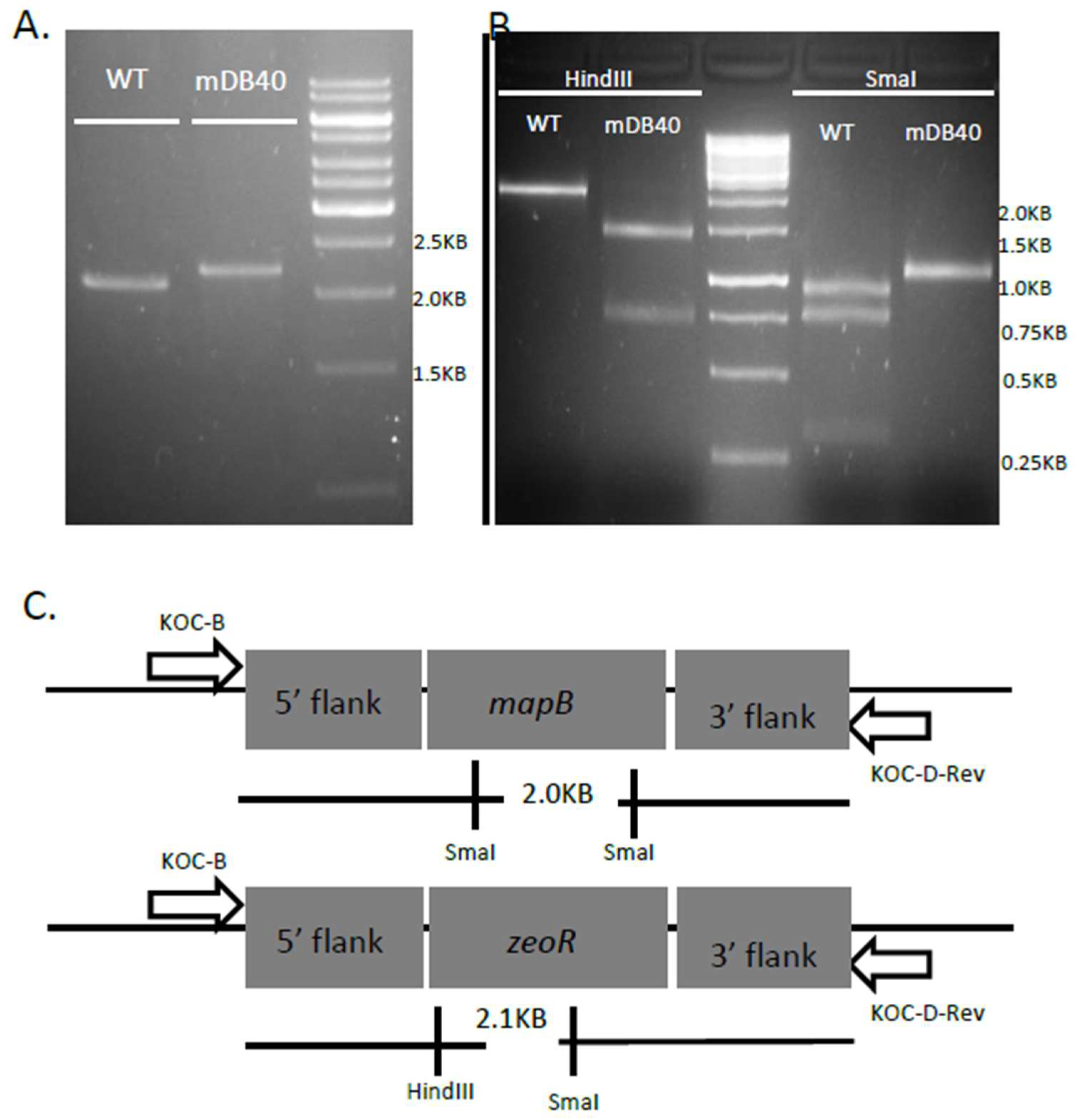
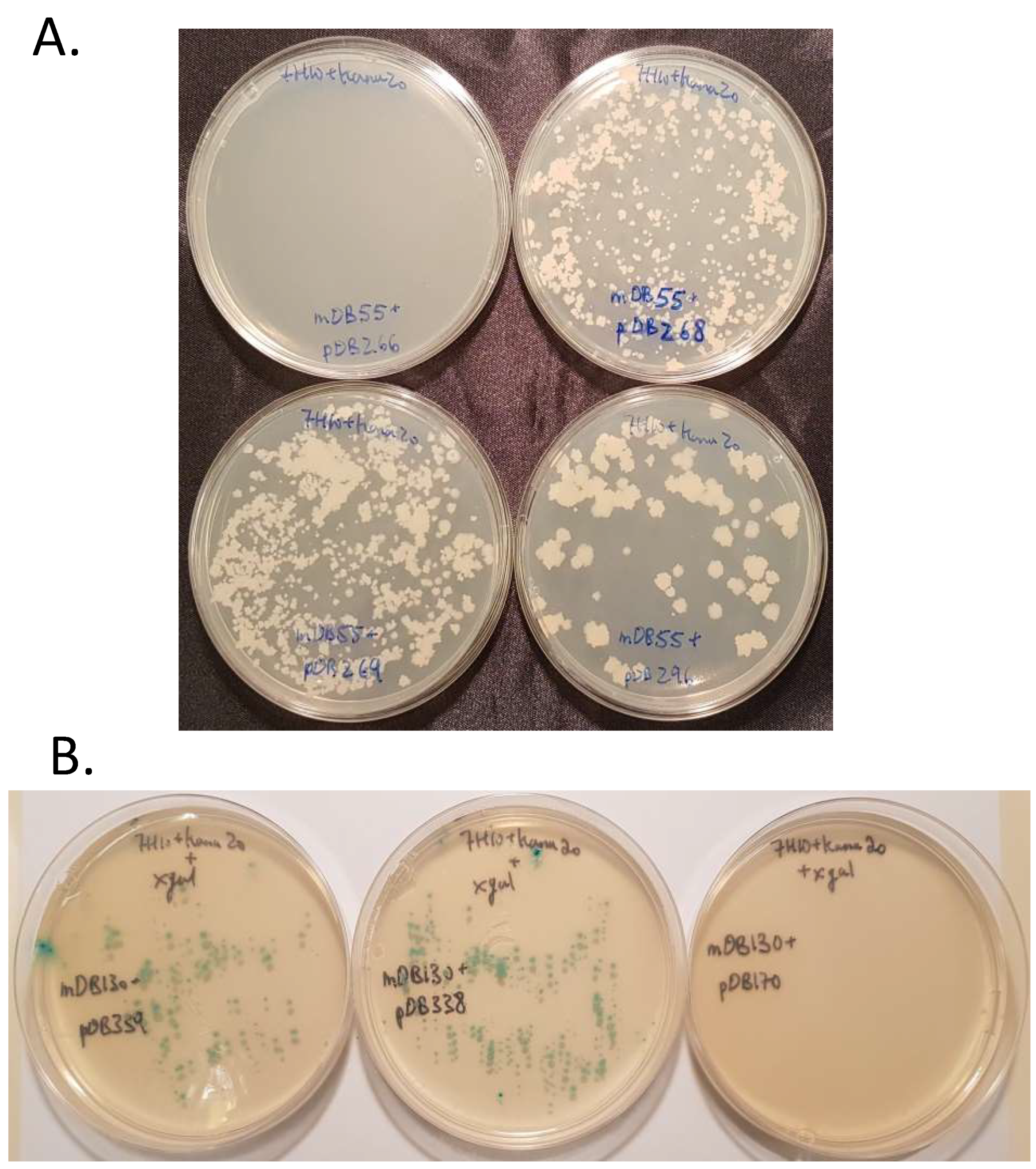
3.2. mapB (Rv 2861c, Mb2886c) is Genetically Essential for Bacterial Viability
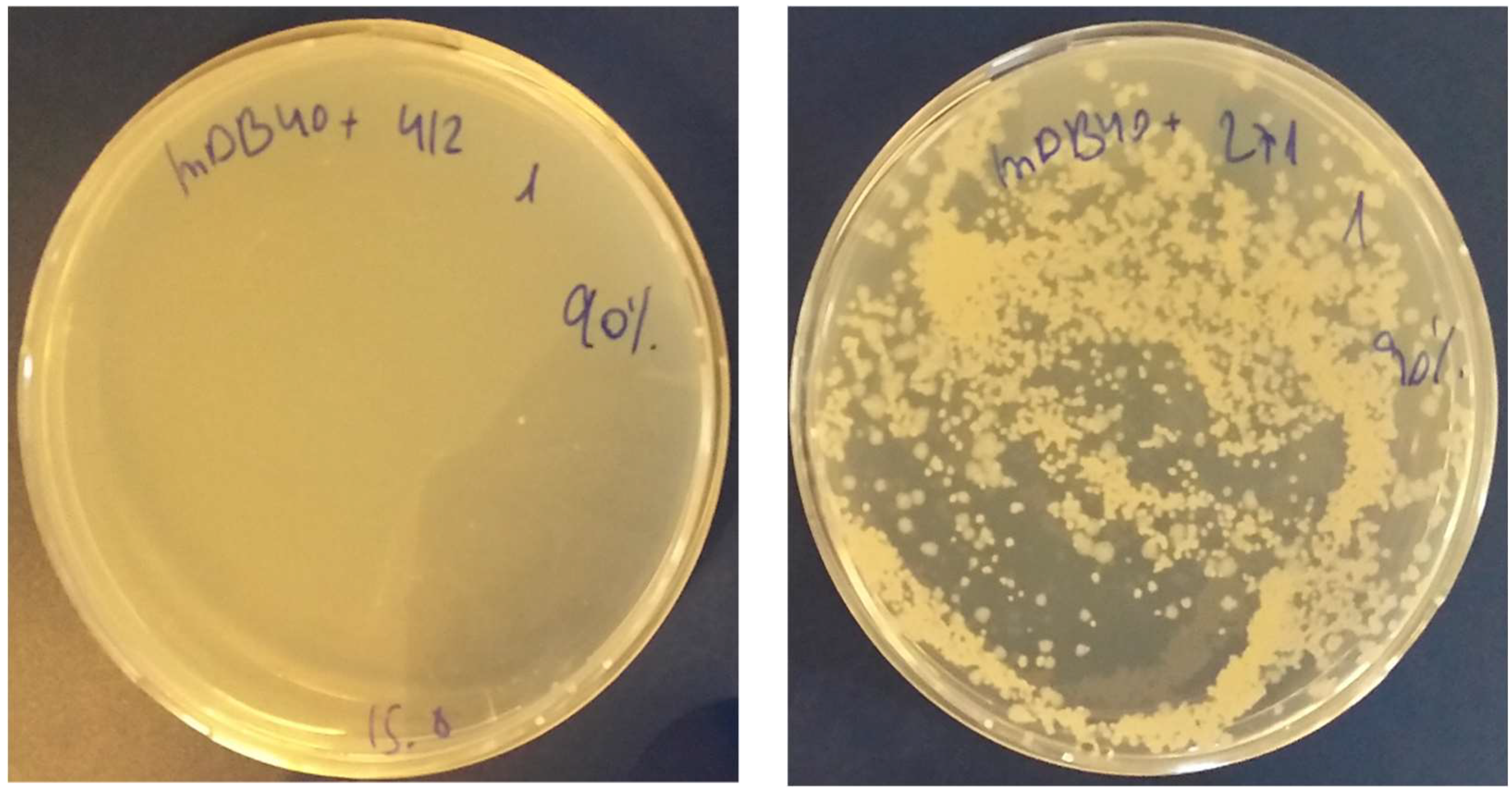
3.3. Creation of a Double Map Mutant
3.4. Complementation of ΔmapB Mutants with mapB with Reduced Enzymatic Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2016; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Zhang, Y. The magic bullets and tuberculosis drug targets. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 529–564. [Google Scholar] [CrossRef] [PubMed]
- Helgren, T.R.; Wangtrakuldee, P.; Staker, B.L.; Hagen, T.J. Advances in bacterial methionine aminopeptidase inhibition. Curr. Topics med. Chem. 2016, 16, 397–414. [Google Scholar] [CrossRef]
- Gonzales, T.; Robert-Baudouy, J. Bacterial aminopeptidases: properties and functions. FEMS Microbiol. Rev. 1996, 18, 319–344. [Google Scholar] [CrossRef]
- Chang, S.; McGARY, E.C.; Chang, S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J. Bacteriol. 1989, 171, 4071–4072. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, C.G.; Kukral, A.M.; Miller, J.L.; Movva, N.R. pepM is an essential gene in Salmonella typhimurium. J. Bacteriol. 1989, 171, 5215–5217. [Google Scholar] [CrossRef] [PubMed]
- Addlagatta, A.; Quillin, M.L.; Omotoso, O.; Liu, J.O.; Matthews, B.W. Identification of an SH3-binding motif in a new class of methionine aminopeptidases from Mycobacterium tuberculosis suggests a mode of interaction with the ribosome. Biochemistry 2005, 44, 7166–7174. [Google Scholar] [CrossRef]
- Olaleye, O.; Raghunand, T.R.; Bhat, S.; He, J.; Tyagi, S.; Lamichhane, G.; Gu, P.; Zhou, J.; Zhang, Y.; Grosset, J.; et al. Methionine aminopeptidases from Mycobacterium tuberculosis as novel antimycobacterial targets. Chem. Bio. 2010, 17, 86–97. [Google Scholar] [CrossRef]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef]
- Olaleye, O.; Raghunand, T.R.; Bhat, S.; Chong, C.; Gu, P.; Zhou, J.; Zhang, Y.; Bishai, W.R.; Liu, J.O. Characterization of clioquinol and analogues as novel inhibitors of methionine aminopeptidases from Mycobacterium tuberculosis. Tuberculosis 2011, 91, S61–S65. [Google Scholar] [CrossRef]
- Otero, J.; Jacobs, W.R.; Glickman, M.S. Efficient allelic exchange and transposon mutagenesis in Mycobacterium avium by specialized transduction. Appl. Environ. Microbiol. 2003, 69, 5039–5044. [Google Scholar] [CrossRef]
- Ehrt, S.; Guo, X.V.; Hickey, C.M.; Ryou, M.; Monteleone, M.; Riley, L.W.; Schnappinger, D. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005, 33, e21. [Google Scholar] [CrossRef]
- Barkan, D.; Rao, V.; Sukenick, G.D.; Glickman, M.S. Redundant function of cmaA2 and mmaA2 in Mycobacterium tuberculosis cis cyclopropanation of oxygenated mycolates. J. Bacteriol. 2010, 192, 3661–3668. [Google Scholar] [CrossRef] [PubMed]
- Pashley, C.A.; Parish, T. Efficient switching of mycobacteriophage L5-based integrating plasmids in Mycobacterium tuberculosis. FEMS Microbiol. Lett. 2003, 229, 211–215. [Google Scholar] [CrossRef]
- Gill, W.P.; Harik, N.S.; Whiddon, M.R.; Liao, R.P.; Mittler, J.E.; Sherman, D.R. A replication clock for Mycobacterium tuberculosis. Nat. Med. 2009, 15, 211. [Google Scholar] [CrossRef]
- Barkan, D.; Hedhli, D.; Yan, H.G.; Huygen, K.; Glickman, M.S. Mycobacterium tuberculosis lacking all mycolic acid cyclopropanation is viable but highly attenuated and hyperinflammatory in mice. Infect. Immun. 2012, 80, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Kanudia, P.; Mittal, M.; Kumaran, S.; Chakraborti, P.K. Amino-terminal extension present in the methionine aminopeptidase type 1c of Mycobacterium tuberculosis is indispensible for its activity. BMC biochem. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.V.; Sharma, V.; Sacchettini, J.C. TB drug discovery: addressing issues of persistence and resistance. Tuberculosis 2004, 84, 45–55. [Google Scholar] [CrossRef]
- Boxem, M.; Tsai, C.W.; Zhang, Y.; Saito, R.M.; Liu, J.O. The C. elegans methionine aminopeptidase 2 analog map-2 is required for germ cell proliferation. FEBS Lett. 2004, 576, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Sin, N.; Meng, L.; Wang, M.Q.; Wen, J.J.; Bornmann, W.G.; Crews, C.M. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc. Natl. Acad. Sci. USA 1997, 94, 6099–6103. [Google Scholar] [CrossRef]
- Wang, J.; Lou, P.; Henkin, J. Selective inhibition of endothelial cell proliferation by fumagillin is not due to differential expression of methionine aminopeptidases. J. Cell. Biochem. 2000, 77, 465–473. [Google Scholar] [CrossRef]
- Giglione, C.; Fieulaine, S.; Meinnel, T. N-terminal protein modifications: Bringing back into play the ribosome. Biochimie 2015, 114, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Giglione, C.; Vallon, O.; Meinnel, T. Control of protein life-span by N-terminal methionine excision. EMBO J. 2003, 22, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Meinnel, T.; Serero, A.; Giglione, C. Impact of the N-terminal amino acid on targeted protein degradation. Bio. Chem. 2006, 387, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Chemical genetics: where genetics and pharmacology meet. Cell 2007, 128, 425–430. [Google Scholar] [CrossRef]
- Reddi, R.; Arya, T.; Kishor, C.; Gumpena, R.; Ganji, R.J.; Bhukya, S.; Addlagatta, A. Selective targeting of the conserved active site cysteine of Mycobacterium tuberculosis methionine aminopeptidase with electrophilic reagents. FEBS J. 2014, 281, 4240–4248. [Google Scholar] [CrossRef]
- Sandikci, A.; Gloge, F.; Martinez, M.; Mayer, M.P.; Wade, R.; Bukau, B.; Kramer, G. Dynamic enzyme docking to the ribosome coordinates N-terminal processing with polypeptide folding. Nat. Struct. Mol. Biol. 2013, 20, 843. [Google Scholar] [CrossRef]
- Vetro, J.A.; Dummitt, B.; Chang, Y.-H. Methionine Aminopeptidase. In Aminopeptidases in Biology and Disease; Springer: New York, NY, USA, 2004; pp. 17–44. [Google Scholar]
- Teo, J.W.; Thayalan, P.; Beer, D.; Yap, A.S.; Nanjundappa, M.; Ngew, X.; Duraiswamy, J.; Liung, S.; Dartois, V.; Schreiber, M.; et al. Peptide deformylase inhibitors as potent antimycobacterial agents. Antimicrob. Agents Chemother. 2006, 50, 3665–3673. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanunu, M.; Schall, P.; Reingewertz, T.-H.; Chakraborti, P.K.; Grimm, B.; Barkan, D. MapB Protein is the Essential Methionine Aminopeptidase in Mycobacterium tuberculosis. Cells 2019, 8, 393. https://doi.org/10.3390/cells8050393
Vanunu M, Schall P, Reingewertz T-H, Chakraborti PK, Grimm B, Barkan D. MapB Protein is the Essential Methionine Aminopeptidase in Mycobacterium tuberculosis. Cells. 2019; 8(5):393. https://doi.org/10.3390/cells8050393
Chicago/Turabian StyleVanunu, Miriam, Patrick Schall, Tali-Haviv Reingewertz, Pradip K. Chakraborti, Bernhard Grimm, and Daniel Barkan. 2019. "MapB Protein is the Essential Methionine Aminopeptidase in Mycobacterium tuberculosis" Cells 8, no. 5: 393. https://doi.org/10.3390/cells8050393
APA StyleVanunu, M., Schall, P., Reingewertz, T.-H., Chakraborti, P. K., Grimm, B., & Barkan, D. (2019). MapB Protein is the Essential Methionine Aminopeptidase in Mycobacterium tuberculosis. Cells, 8(5), 393. https://doi.org/10.3390/cells8050393